A Reversed-Phase HPLC Method for Determination of Osteopontin in Infant Formula



Abstract
Feature Application
Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation of OPN Standard
2.2. Preparation of IMF and IMF Spiked with OPN
2.3. Instrumental and Chromatographic Conditions
2.4. Method Validation
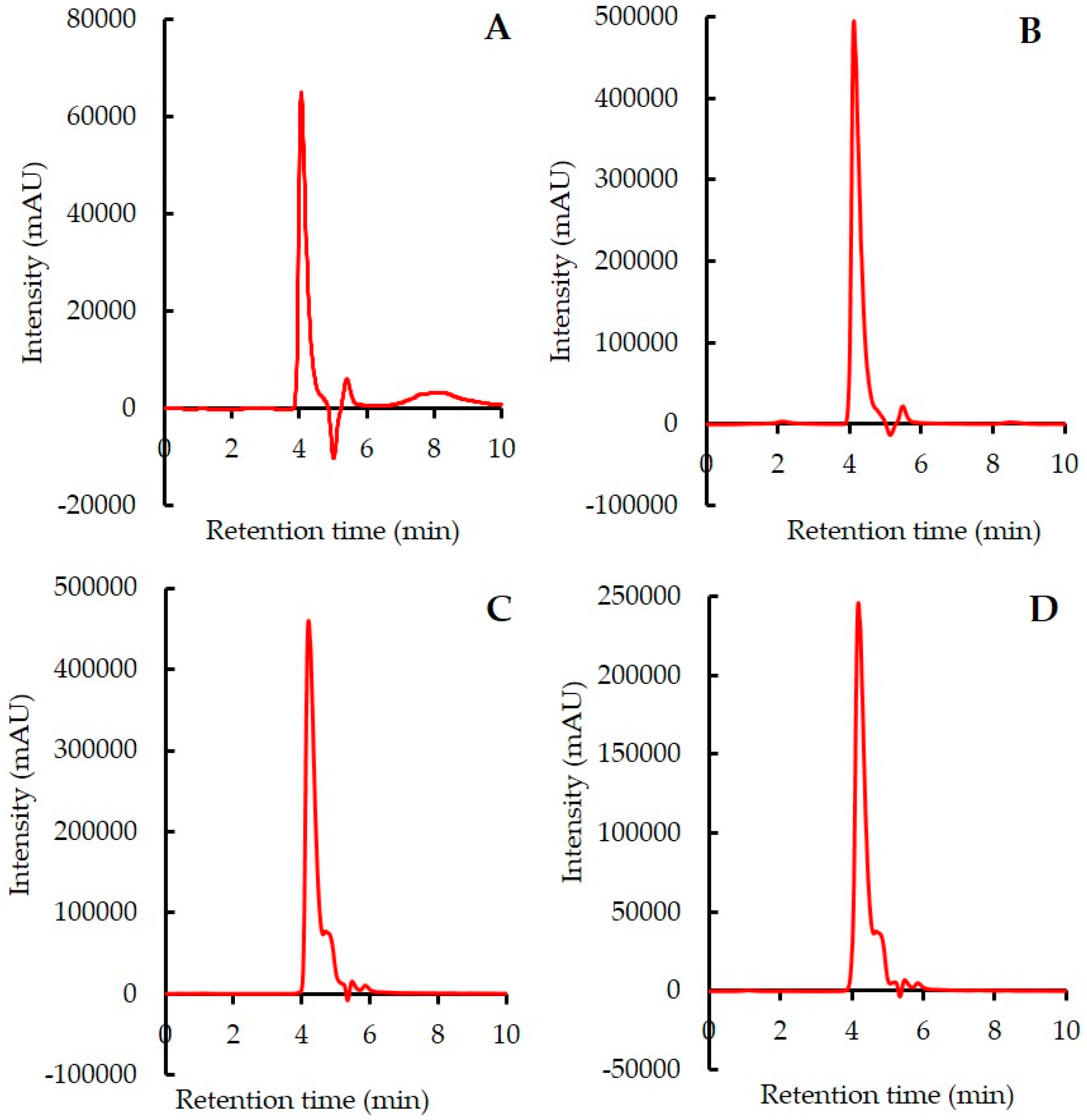
2.4.1. System Suitability (Specificity)
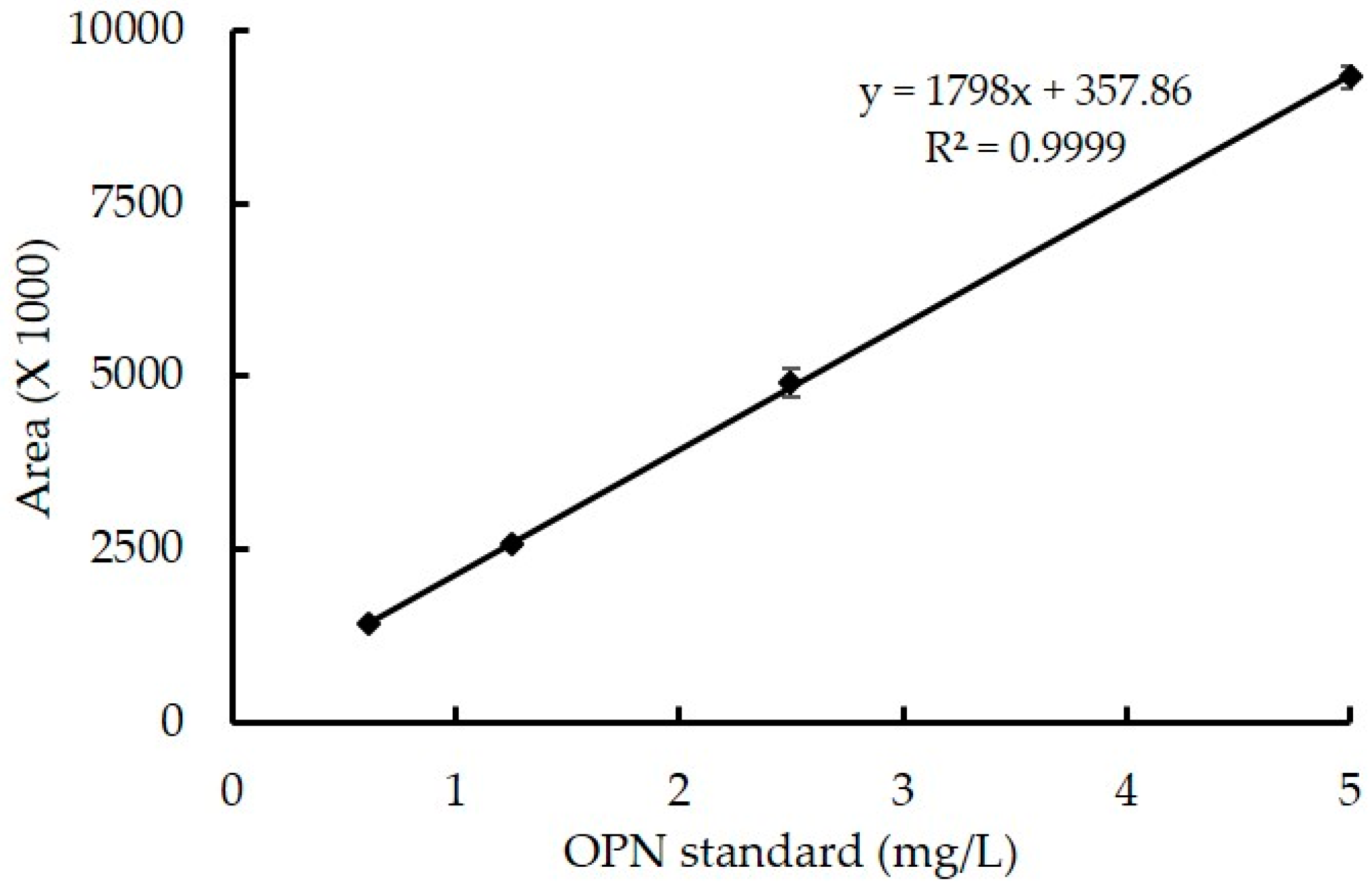
2.4.2. Linearity
2.4.3. Detection of Limit (Sensitivity)
2.4.4. Precision (Repeatability)
2.4.5. Accuracy (Recovery)
2.4.6. Method Robustness
3. Results and Discussion
3.1. Development of RP-HPLC Method
3.2. Method Validation
3.2.1. System Suitability
3.2.2. Linearity, LOD and LOQ
3.2.3. Precision and Accuracy
3.2.4. Robustness
3.2.5. Future Recommendation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sodek, J.; Ganss, B.; McKee, M.D. Osteopontin. Crit. Rev. Oral Biol. Med. 2000, 11, 279–303. [Google Scholar] [CrossRef] [PubMed]
- Schack, L.; Lange, A.; Kelsen, J.; Agnholt, J.; Christensen, B.; Petersen, T.; Sørensen, E. Considerable variation in the concentration of osteopontin in human milk, bovine milk, and infant formulas. J. Dairy Sci. 2009, 92, 5378–5385. [Google Scholar] [CrossRef] [PubMed]
- Demmelmair, H.; Prell, C.; Timby, N.; Lönnerdal, B. Benefits of Lactoferrin, Osteopontin and Milk Fat Globule Membranes for Infants. Nutrients 2017, 9, 817. [Google Scholar] [CrossRef] [PubMed]
- Lönnerdal, B. Bioactive Proteins in Human Milk: Health, Nutrition, and Implications for Infant Formulas. J. Pediatr. 2016, 173, S4–S9. [Google Scholar] [CrossRef] [PubMed]
- Azuma, N.; Maeta, A.; Fukuchi, K.; Kanno, C. A rapid method for purifying osteopontin from bovine milk and interaction between osteopontin and other milk proteins. Int. Dairy J. 2006, 16, 370–378. [Google Scholar] [CrossRef]
- Christensen, B.; Sørensen, E.S. Structure, function and nutritional potential of milk osteopontin. Int. Dairy J. 2016, 57, 1–6. [Google Scholar] [CrossRef]
- Cross, T.G.; Hornshaw, M.P. Can LC and LC-MS ever replace immunoassays? J. Appl. Bioanal. 2016, 2, 108–116. [Google Scholar] [CrossRef]
- Laskowska, E.; Kuczyńska-Wiśnik, D.; Lipińska, B. Proteomic analysis of protein homeostasis and aggregation. J. Proteom. 2019, 198, 98–112. [Google Scholar] [CrossRef]
- Faria, M. Quantitative Mass Spectrometric Investigations of Protein Biomarkers: Serum Thymidine Kinase 1 and Human Osteopontin. 2014. Available online: https://scholarscompass.vcu.edu/cgi/viewcontent.cgi?referer=https://scholar.google.co.nz/&httpsredir=1&article=4602&context=etd (accessed on 1 August 2019).
- Bordin, G.; Raposo, F.C.; De La Calle, B.; Rodriguez, A. Identification and quantification of major bovine milk proteins by liquid chromatography. J. Chromatogr. A 2001, 928, 63–76. [Google Scholar] [CrossRef]
- Tacoma, R.; Fields, J.; Ebenstein, D.B.; Lam, Y.-W.; Greenwood, S.L. Characterization of the bovine milk proteome in early-lactation Holstein and Jersey breeds of dairy cows. J. Proteom. 2016, 130, 200–210. [Google Scholar] [CrossRef]
- Christensen, B.; Sørensen, E. Osteopontin is highly susceptible to cleavage in bovine milk and the proteolytic fragments bind the αVβ3-integrin receptor. J. Dairy Sci. 2014, 97, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Kläning, E.; Christensen, B.; Bajic, G.; Hoffmann, S.V.; Jones, N.C.; Callesen, M.M.; Andersen, G.R.; Sørensen, E.S.; Vorup-Jensen, T. Multiple low-affinity interactions support binding of human osteopontin to integrin αXβ2. Biochim. Biophys. Acta Proteins Proteom. 2015, 1854, 930–938. [Google Scholar] [CrossRef] [PubMed]
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). Validation of Analytical Procedures: Text and Methodology Q2(R1). In Proceedings of the International Conference on Harmonization, Geneva, Switzerland, 24–28 Octobers 2005; pp. 11–12. Available online: https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf (accessed on 1 August 2019).
- Association of Analytical Communities (AOAC International). Guidelines for Single Laboratory Validation of Chemical Methods for Dietary Supplements and Botanicals. 2002. Available online: https://www.aoac.org/aoac_prod_imis/AOAC_Docs/StandardsDevelopment/SLV_Guidelines_Dietary_Supplements.pdf (accessed on 1 August 2019).
- Food and Drug Administration, US. (FDA). Guidance for Industry: Analytical Procedures and Methods Validation for Drugs and Biologics. 2015. Available online: https://www.fda.gov/media/87801/download (accessed on 1 August 2019).
- Chen, Z.G.; Buchanan, P.; Quek, S.Y. Development and validation of an HPLC-DAD-MS method for determination of four nucleoside compounds in the New Zealand native mushroom Hericium sp. Food Chem. 2019, 278, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.D.A.; Ozogul, F.; Quantick, P.C.; Ozogul, Y. A rapid HPLC-determination of ATP-related compounds and its application to herring stored under modified atmosphere. Int. J. Food Sci. Technol. 2000, 35, 549–554. [Google Scholar]
- Green, J.M. Peer Reviewed: A Practical Guide to Analytical Method Validation. Anal. Chem. 1996, 68, 305A–309A. [Google Scholar] [CrossRef]
- Thompson, M.; Wood, R. Harmonized guidelines for internal quality control in analytical chemistry laboratories (Technical Report). Pure Appl. Chem. 1995, 67, 649–666. [Google Scholar] [CrossRef]
- Greibrokk, T.; Andersen, T. High-temperature liquid chromatography. J. Chromatogr. A 2003, 1000, 743–755. [Google Scholar] [CrossRef]
- Ibrahim, F.A.; Elmansi, H.; Fathy, M.E. Green RP-HPLC method for simultaneous determination of moxifloxacin combinations: Investigation of the greenness for the proposed method. Microchem. J. 2019, 148, 151–161. [Google Scholar] [CrossRef]
- Yabré, M.; Ferey, L.; Somé, I.T.; Gaudin, K. Greening Reversed-Phase Liquid Chromatography Methods Using Alternative Solvents for Pharmaceutical Analysis. Molecules 2018, 23, 1065. [Google Scholar] [CrossRef]
- Micăle, F.; Albu, F.; Iorgulescu, E.-E.; Medvedovici, A.; Tache, F. Ethyl Lactate as a Greener Alternative to Acetonitrile in RPLC: A Realistic Appraisal. J. Chromatogr. Sci. 2015, 53, 1701–1707. [Google Scholar] [CrossRef][Green Version]
- Van Beek, T.A.; Shen, Y.; Chen, B. Alternative solvents can make preparative liquid chromatography greener. Green Chem. 2015, 17, 4073–4081. [Google Scholar]
- Tobiszewski, M.; Namieśnik, J. Greener organic solvents in analytical chemistry. Curr. Opin. Green Sustain. Chem. 2017, 5, 1–4. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| RP-HPLC Conditions | [9] | [12] | [13] | [10,11] | This Study |
|---|---|---|---|---|---|
| Purpose | Human OPN only as a biomarker | Determination of C-terminal of bovine OPN to investigate its susceptibility to proteolytic cleavage | Separation of N- and C-terminal of to evaluate binding of human OPN to integrin | Identification and characterization of bovine OPN fragments | Quantitative measurement of bovine OPN in infant formula |
| Column | C18 | C2/C18 | C18 | C4 | C18 |
| Mobile phase-A | 0.1% formic acid | 0.1% TFA | 0.1% TFA | 10% ACN in 0.1% TFA | 0.1% TFA |
| Mobile phase-B | 0.1% formic acid in ACN | 60% ACN in 0.1% TFA | 75% of 2-propanol in 0.1% TFA | 90% ACN in 0.1% TFA | 60% ACN in 0.1% TFA |
| Elution | Gradient, 7 min | Gradient, 54 min | Gradient, time NR * | Gradient (28 min) followed by isocratic (28 min); total 56 min | Isocratic, 10 min; 20% of mobile phase A and 80% of mobile phase B |
| Temperature | 40 °C | NR | NR | 40 °C | 40 °C |
| Absorbance | NR | 214 nm | NR | 214 nm | 214 nm |
| Sample injection | 25 µL | NR | NR | 4 µL | 10 µL |
| Flow rate (ml/min) | 0.3 | 0.15 | NR | NR | 0.3 |
| Injection of OPN (1 mg/L) | Intra-Day | Inter-Day | ||
|---|---|---|---|---|
| Retention Time (min) | RSD (%) | Retention Time (min) | RSD (%) | |
| 1 | 4.059 | 0.161 | 4.051 | 0.148 |
| 2 | 4.060 | 4.055 | ||
| 3 | 4.056 | 4.048 | ||
| 4 | 4.050 | 4.061 | ||
| 5 | 4.071 | 4.055 | ||
| 6 | 4.052 | 4.059 | ||
| 7 | 4.049 | 4.049 | ||
| 8 | 4.058 | 4.060 | ||
| 9 | 4.063 | 4.054 | ||
| 10 | 4.057 | 4.042 | ||
| Mean | 4.058 ± 0.007 | 4.053 ± 0.006 | ||
| Spiked Amount (mg/L) | Recovery (%) | RSD (%) |
|---|---|---|
| 1.25 | 100.48 ± 1.62 | 1.62 |
| 2.5 | 99.63 ± 2.73 | 2.74 |
| 5 | 102.39 ± 2.17 | 2.12 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wazed, M.A.; Farid, M. A Reversed-Phase HPLC Method for Determination of Osteopontin in Infant Formula. Appl. Sci. 2019, 9, 3711. https://doi.org/10.3390/app9183711
Wazed MA, Farid M. A Reversed-Phase HPLC Method for Determination of Osteopontin in Infant Formula. Applied Sciences. 2019; 9(18):3711. https://doi.org/10.3390/app9183711
Chicago/Turabian StyleWazed, Md Abdul, and Mohammed Farid. 2019. "A Reversed-Phase HPLC Method for Determination of Osteopontin in Infant Formula" Applied Sciences 9, no. 18: 3711. https://doi.org/10.3390/app9183711
APA StyleWazed, M. A., & Farid, M. (2019). A Reversed-Phase HPLC Method for Determination of Osteopontin in Infant Formula. Applied Sciences, 9(18), 3711. https://doi.org/10.3390/app9183711