Metal Carbides for Biomass Valorization



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Carbide Synthesis and Characterization
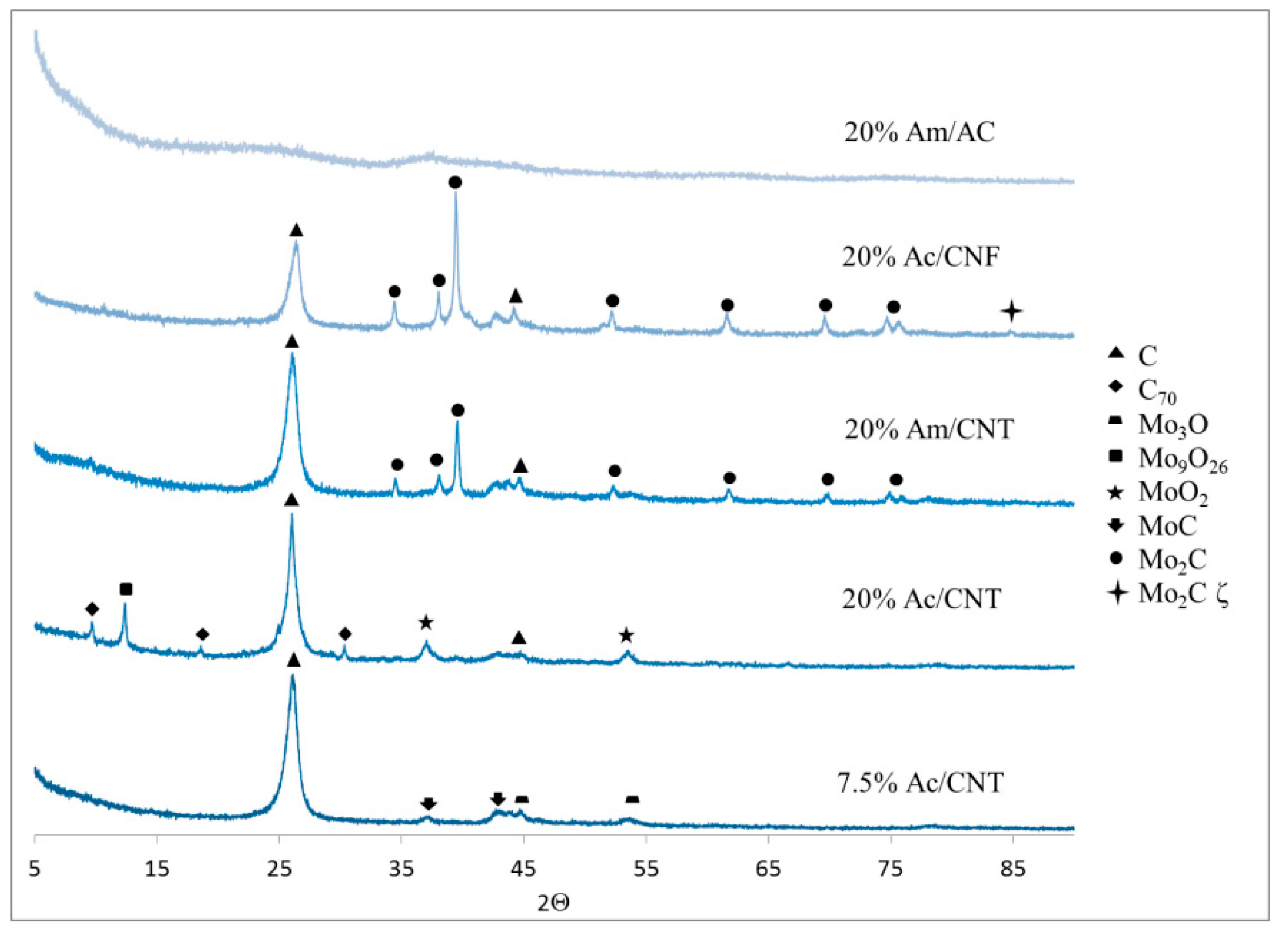
2.1. Temperature-Programmed Reduction (TPR)
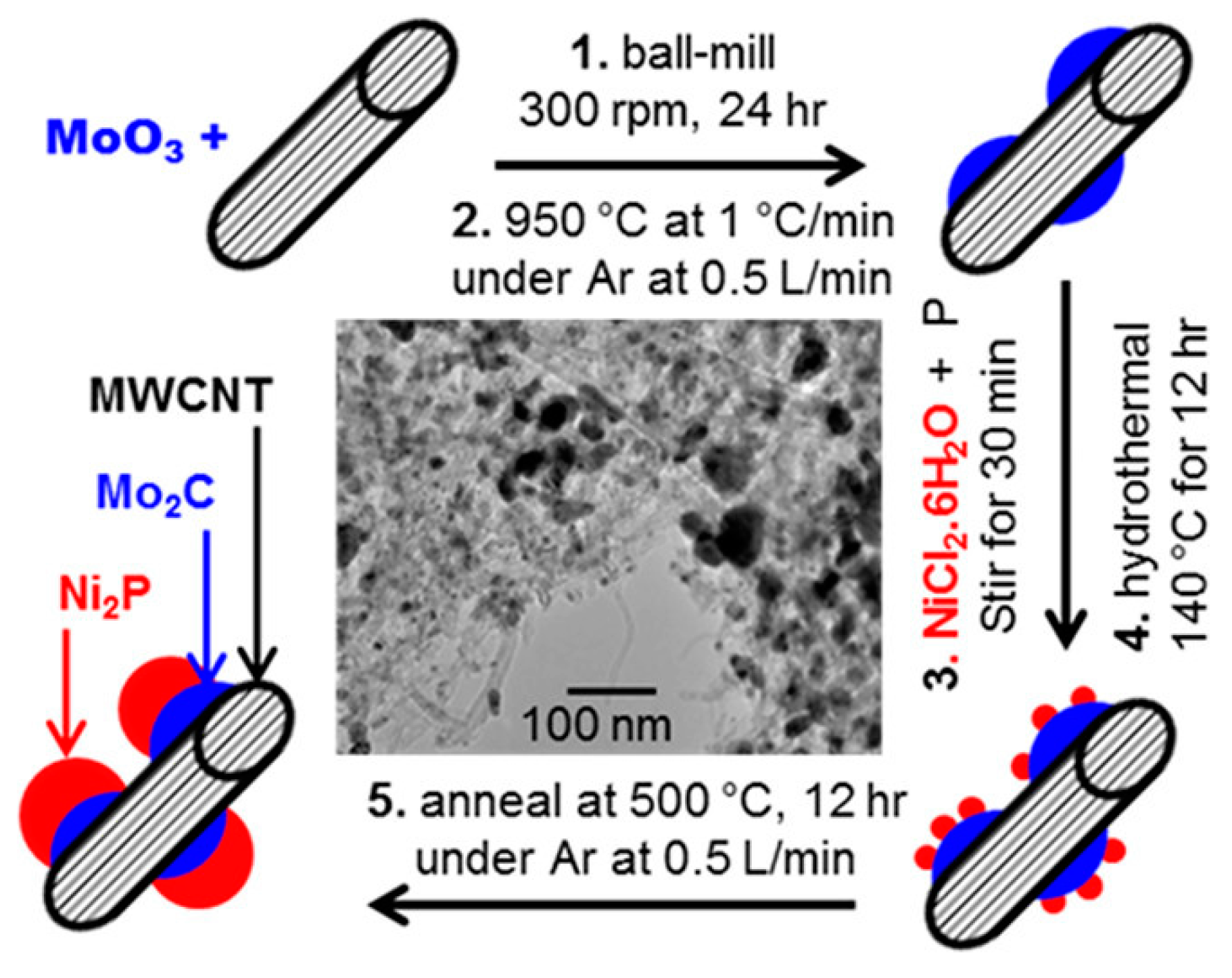
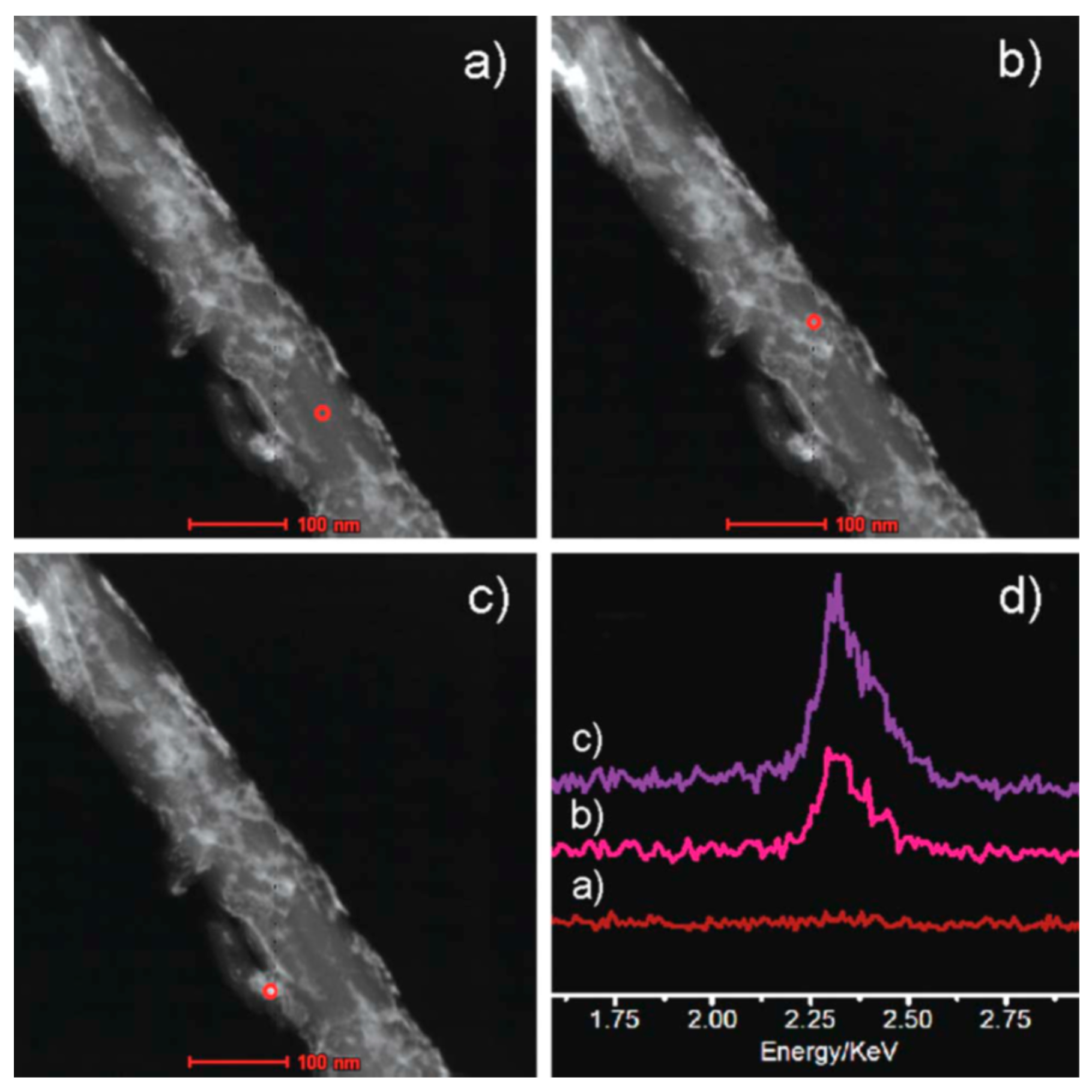
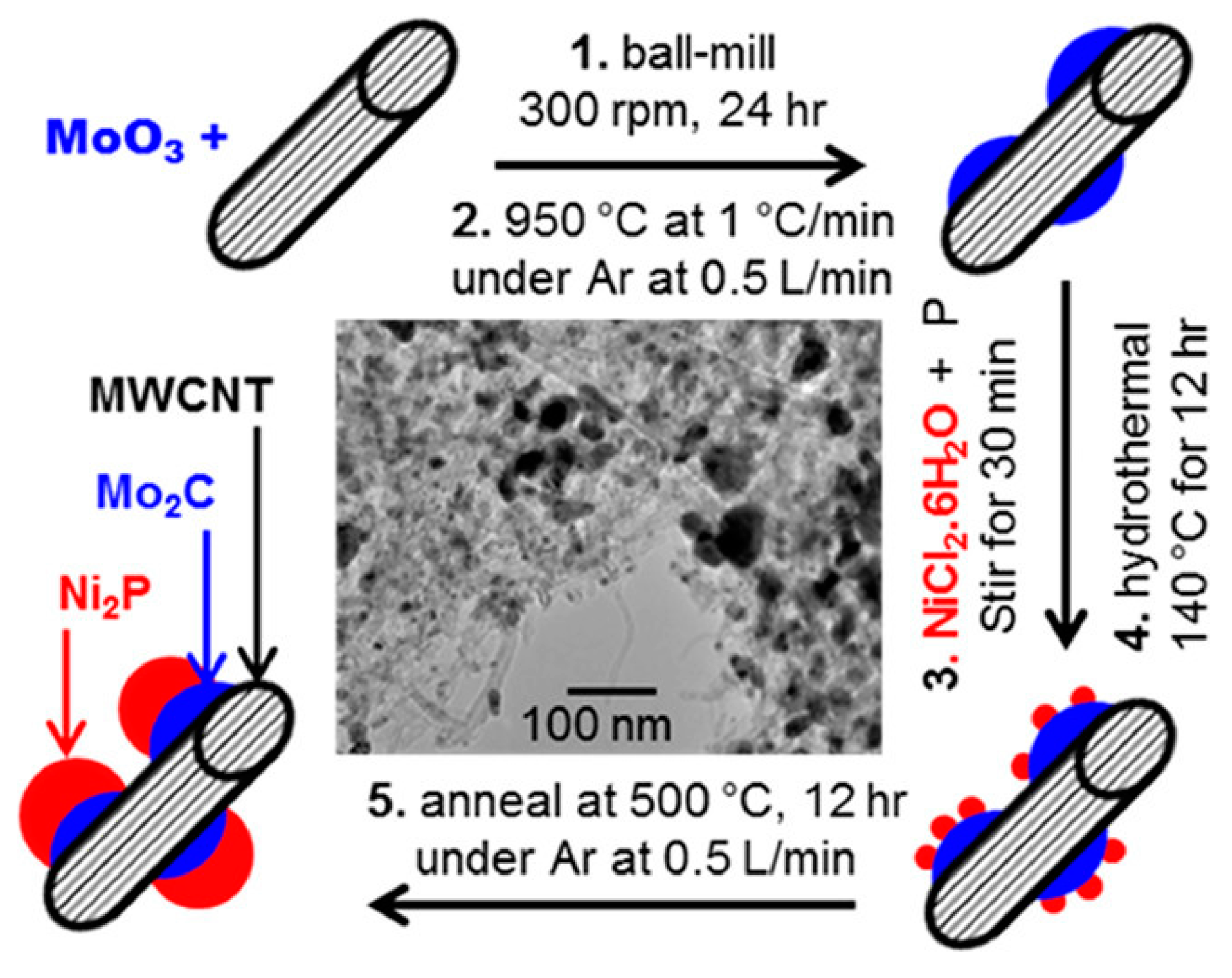
2.2. Ball Milling
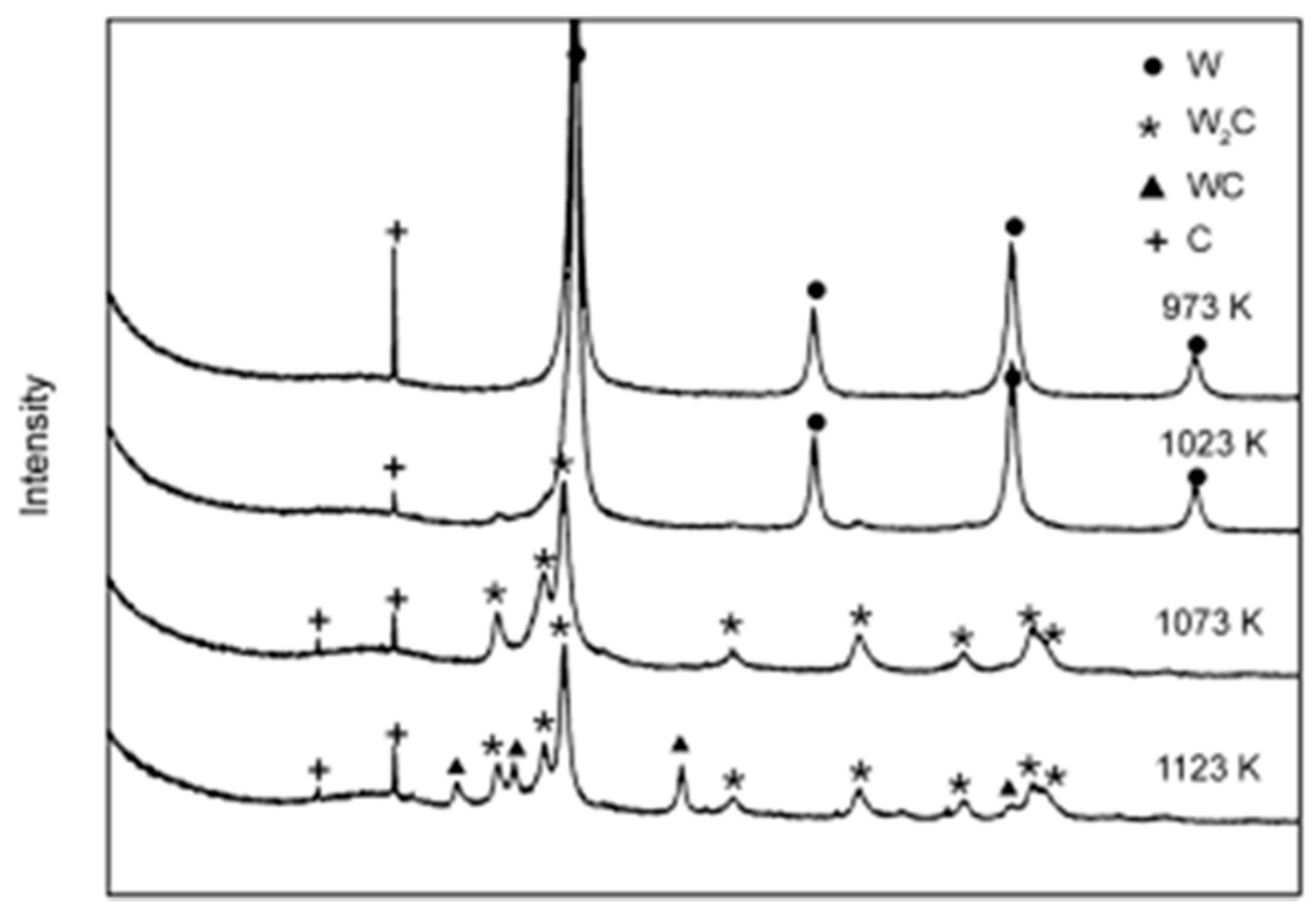
2.3. Carbothermal Hydrogen Reduction (CHR)
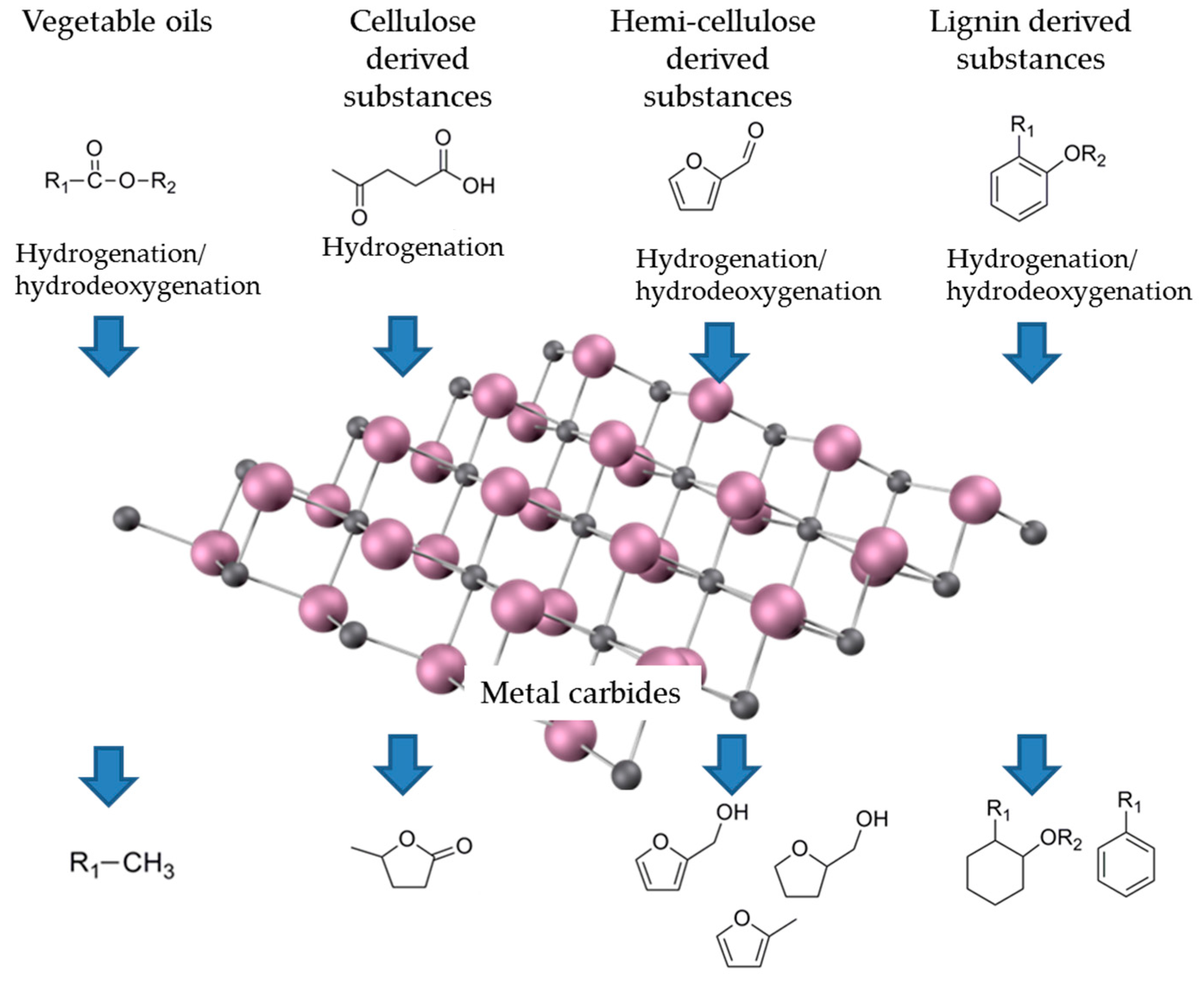
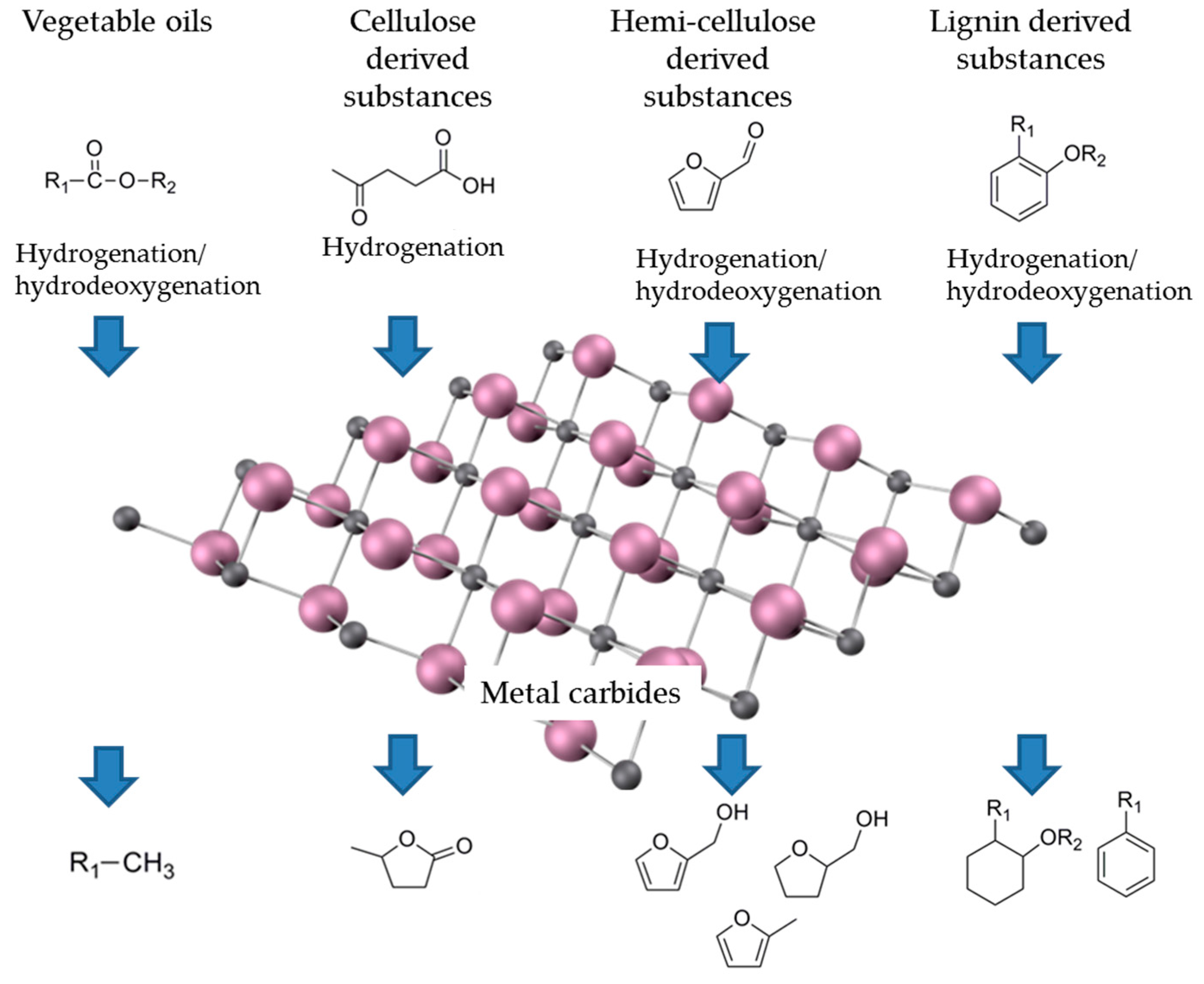
3. Application in Biomass Transformation
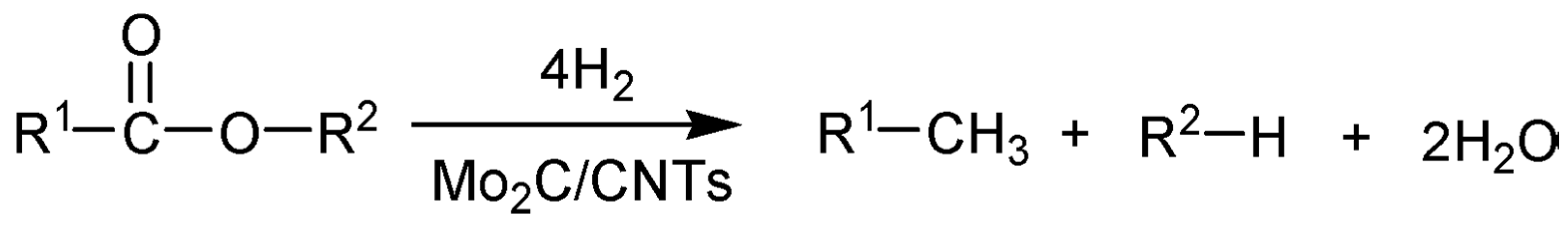
3.1. First-Generation Biomass
3.2. Second-Generation Biomass
3.2.1. Cellulose
3.2.2. Hemicellulose
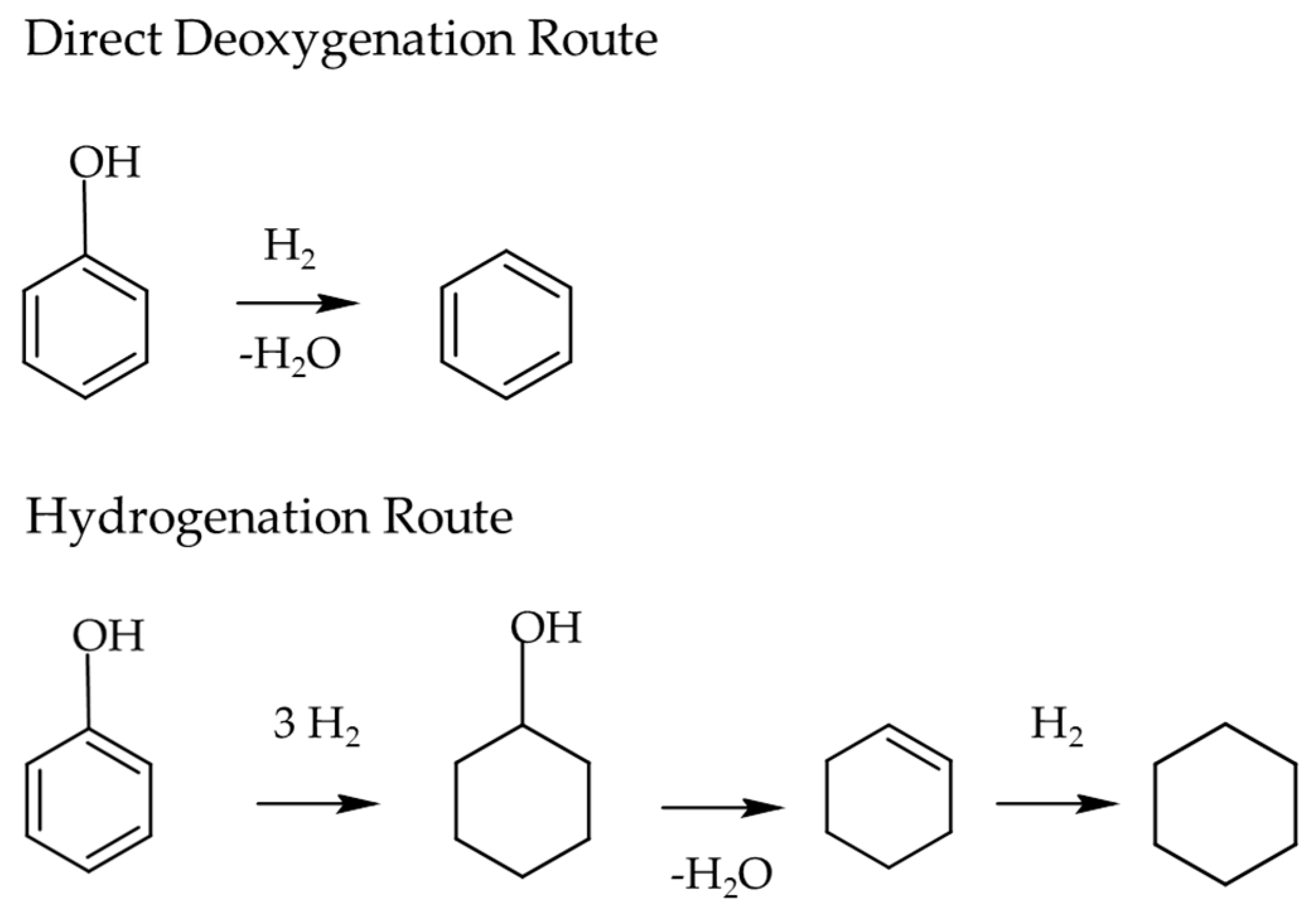
3.2.3. Lignin
3.2.4. Reforming Reactions: Selective C-C Bond Scission
4. Conclusions and Perspective
Author Contributions
Conflicts of Interest
References
- Navarro, R.M.; Peña, M.A.; Fierro, J.L.G. Hydrogen production reactions from carbon feedstocks: Fossil fuels and biomass. Chem. Rev. 2007, 107, 3952–3991. [Google Scholar] [CrossRef] [PubMed]
- Corma Canos, A.; Iborra, S.; Velty, A. Chemical routes for the transformation of biomass into chemicals. Chem. Rev. 2007, 107, 2411–2502. [Google Scholar] [CrossRef] [PubMed]
- Okkerse, C.; van Bekkum, H. From fossil to green. Green Chem. 1999, 1, 107–114. [Google Scholar] [CrossRef]
- Huber, G.W.; Shabaker, J.W.; Dumesic, J.A.; Dumesic, J.A. Raney Ni-Sn Catalyst for H2 Production from Biomass-Derived Hydrocarbons. Science 2003, 300, 2075–2077. [Google Scholar] [CrossRef] [PubMed]
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of transportation fuels from biomass: Chemistry, catalysts, and engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [PubMed]
- Gallezot, P. Conversion of biomass to selected chemical products. Chem. Soc. Rev. 2012, 41, 1538–1558. [Google Scholar] [CrossRef] [PubMed]
- Chheda, J.N.; Huber, G.W.; Dumesic, J.A. Liquid-Phase Catalytic Processing of Biomass-Derived Oxygenated Hydrocarbons to Fuels and Chemicals. Angew. Chem. Int. Ed. 2007, 46, 7164–7183. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Dutta, S.; Saha, B. Critical design of heterogeneous catalysts for biomass valorization: Current thrust and emerging prospects. Catal. Sci. Technol. 2016, 6, 7364–7385. [Google Scholar] [CrossRef]
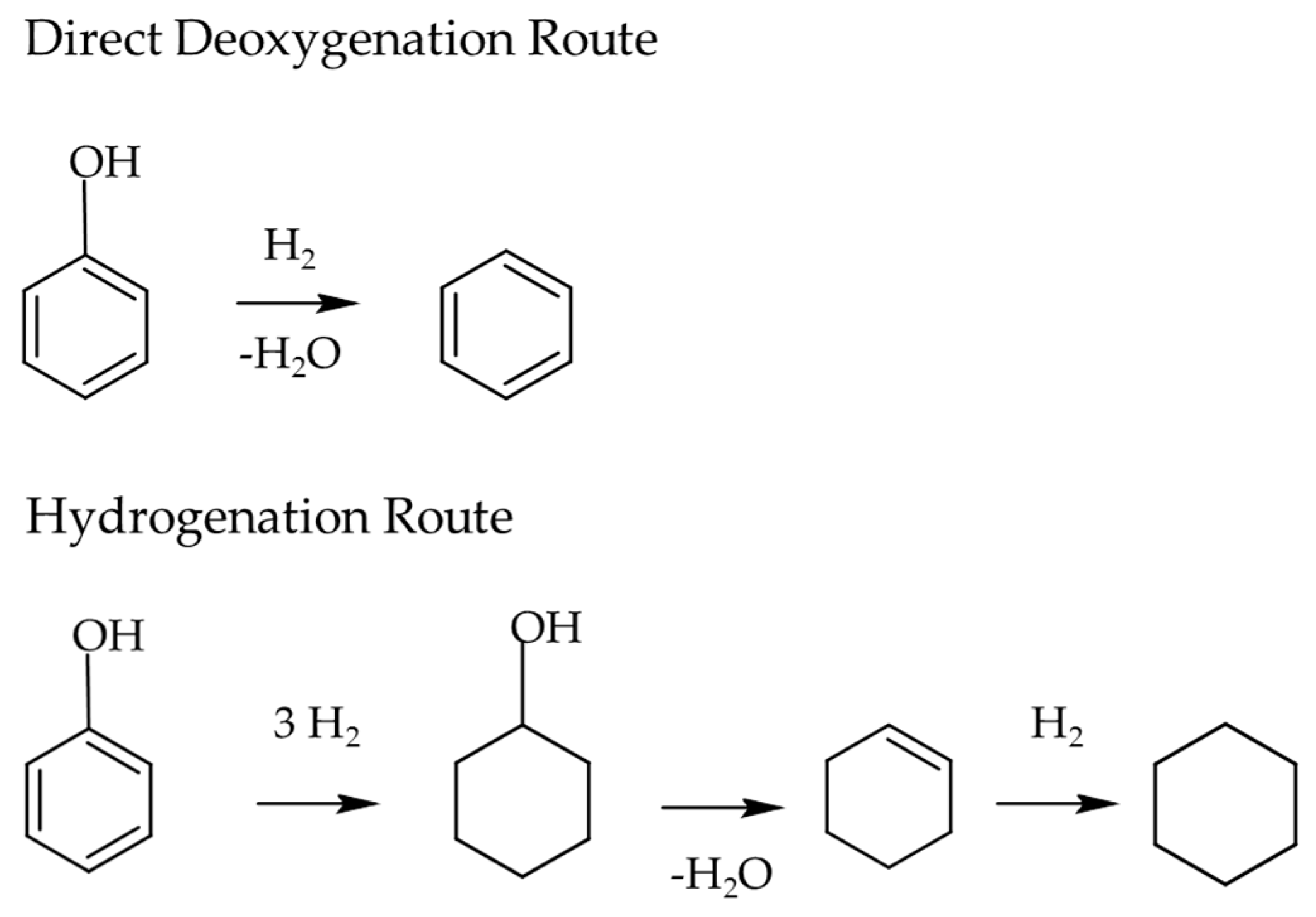
- Furimsky, E. Catalytic hydrodeoxygenation. Appl. Catal. A Gen. 2000, 199, 147–190. [Google Scholar] [CrossRef]
- Zhao, C.; Kou, Y.; Lemonidou, A.A.; Li, X.; Lercher, J.A. Highly Selective Catalytic Conversion of Phenolic Bio-Oil to Alkanes. Angew. Chem. Int. Ed. 2009, 48, 3987–3990. [Google Scholar] [CrossRef] [PubMed]
- Saidi, M.; Samimi, F.; Karimipourfard, D.; Nimmanwudipong, T.; Gates, B.C.; Rahimpour, M.R. Upgrading of lignin-derived bio-oils by catalytic hydrodeoxygenation. Energy Environ. Sci. 2014, 7, 103–129. [Google Scholar] [CrossRef]
- Serrano-Ruiz, J.C.; West, R.M.; Dumesic, J.A. Catalytic Conversion of Renewable Biomass Resources to Fuels and Chemicals. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 79–100. [Google Scholar] [CrossRef] [PubMed]
- Jae, J.; Zheng, W.; Karim, A.M.; Guo, W.; Lobo, R.F.; Vlachos, D.G. The Role of Ru and RuO2 in the Catalytic Transfer Hydrogenation of 5-Hydroxymethylfurfural for the Production of 2,5-Dimethylfuran. ChemCatChem 2014, 6, 848–856. [Google Scholar] [CrossRef]
- Gordon, R.B.; Bertram, M.; Graedel, T.E. Metal stocks and sustainability. Proc. Natl. Acad. Sci. USA 2006, 103, 1209–1214. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.; Masciangioli, T.; Olson, S. Replacing critical materials with abundant materials. In The Role of the Chemical Sciences in Finding Alternatives to Critical Resources: A Workshop Summary; National Academies Press (US): Washington, DC, USA, 2012; ISBN 978-0-309-25429-8. [Google Scholar]
- Hwu, H.H.; Chen, J.G. Surface chemistry of transition metal carbides. Chem. Rev. 2005, 105, 185–212. [Google Scholar] [CrossRef] [PubMed]
- Stottlemyer, A.L.; Kelly, T.G.; Meng, Q.; Chen, J.G. Reactions of oxygen-containing molecules on transition metal carbides: Surface science insight into potential applications in catalysis and electrocatalysis. Surf. Sci. Rep. 2012, 67, 201–232. [Google Scholar] [CrossRef]
- Levy, R.B.; Boudart, M. Platinum-like behavior of tungsten carbide in surface catalysis. Science 1973, 181, 547–549. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.G. Carbide and nitride overlayers on early transition metal surfaces: Preparation, characterization, and reactivities. Chem. Rev. 1996, 96, 1477–1498. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Campisi, S.; Giordano, C.; Otte, K.; Prati, L. Mo and W Carbide: Tunable Catalysts for Liquid Phase Conversion of Alcohols. ACS Catal. 2012, 2, 1377–1380. [Google Scholar] [CrossRef]
- Patel, M.A.; Baldanza, M.A.S.; Teixeira da Silva, V.; Bridgwater, A.V. In situ catalytic upgrading of bio-oil using supported molybdenum carbide. Appl. Catal. A Gen. 2013, 458, 48–54. [Google Scholar] [CrossRef]
- Sousa, L.A.; Zotin, J.L.; Teixeira da Silva, V. Hydrotreatment of sunflower oil using supported molybdenum carbide. Appl. Catal. A Gen. 2012, 449, 105–111. [Google Scholar] [CrossRef]
- Oyama, S.T. Preparation and catalytic properties of transition metal carbides and nitrides. Catal. Today 1992, 15, 179–200. [Google Scholar] [CrossRef]
- Márquez-Alvarez, C.; Calridge, J.B.; York, A.P.E.; Sloan, J.; Green, M.L.H. Benzene hydrogenation over transition metal carbides. Stud. Surf. Sci. Catal. 1997, 106, 485–490. [Google Scholar] [CrossRef]
- Ren, H.; Chen, Y.; Huang, Y.; Deng, W.; Vlachos, D.G.; Chen, J.G. Tungsten carbides as selective deoxygenation catalysts: Experimental and computational studies of converting C3 oxygenates to propene. Green Chem. 2014, 16, 761–769. [Google Scholar] [CrossRef]
- Iglesia, E.; Ribeiro, F.H.; Boudart, M.; Baumgartner, J.E. Synthesis, characterization, and catalytic properties of clean and oxygen-modified tungsten carbides. Catal. Today 1992, 15, 307–337. [Google Scholar] [CrossRef]
- Ribeiro, F.H.; Boudart, M.; Dalla Betta, R.A.; Iglesia, E. Catalytic reactions of n-Alkanes on β-W2C and WC: The effect of surface oxygen on reaction pathways. J. Catal. 1991, 130, 498–513. [Google Scholar] [CrossRef]
- Urzhuntsev, G.A.; Toktarev, A.V.; Echevskii, G.V.; Delii, I.V.; Vlasova, E.N.; Bukhtiyarova, G.A. Prospects for using Mo- and W-containing catalysts in hydroisomerization: A patent review. Part 1: Catalysts based on molybdenum and tungsten phosphides. Catal. Ind. 2016, 8, 32–39. [Google Scholar] [CrossRef]
- Ren, H.; Yu, W.; Salciccioli, M.; Chen, Y.; Huang, Y.; Xiong, K.; Vlachos, D.G.; Chen, J.G. Selective Hydrodeoxygenation of Biomass-Derived Oxygenates to Unsaturated Hydrocarbons using Molybdenum Carbide Catalysts. ChemSusChem 2013, 6, 798–801. [Google Scholar] [CrossRef] [PubMed]
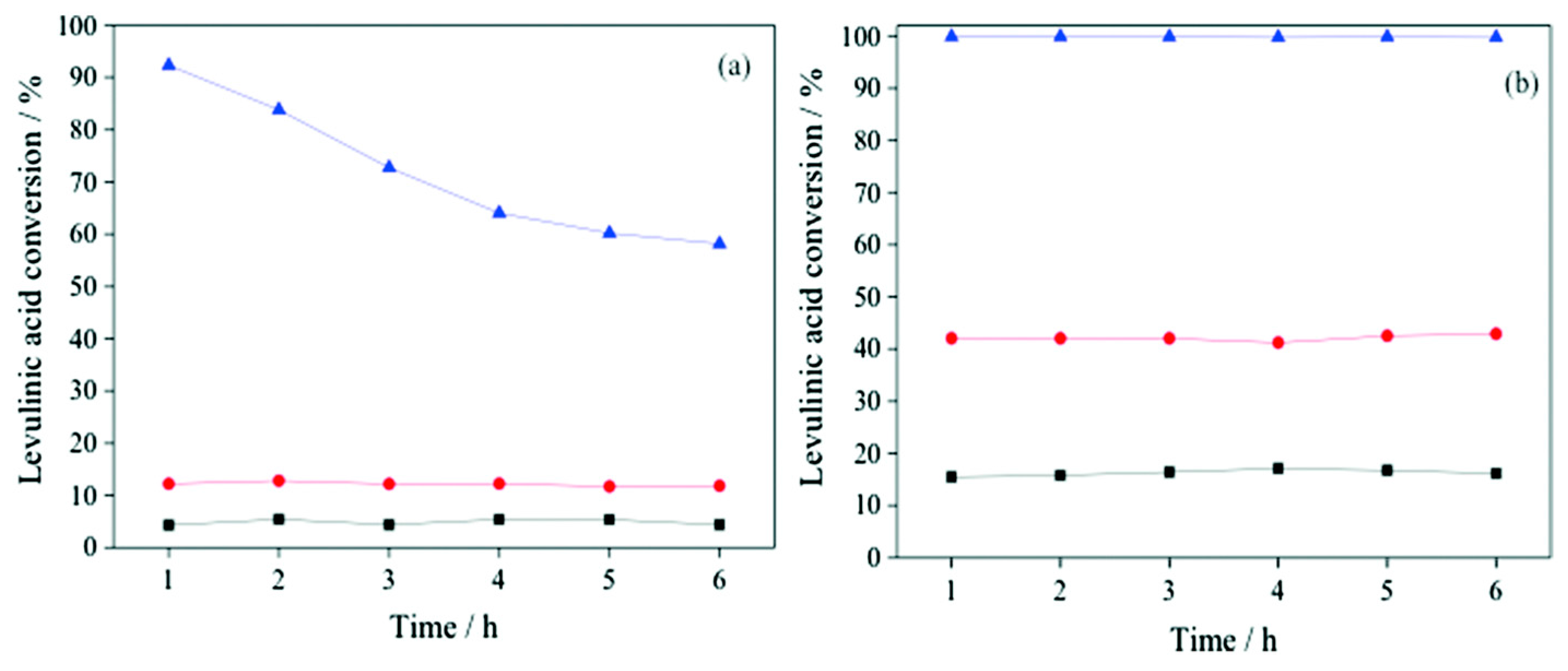
- Mai, E.F.; Machado, M.A.; Davies, T.E.; Lopez-Sanchez, J.A.; Teixeira da Silva, V. Molybdenum carbide nanoparticles within carbon nanotubes as superior catalysts for γ-valerolactone production via levulinic acid hydrogenation. Green Chem. 2014, 16, 4092–4097. [Google Scholar] [CrossRef]
- Lee, J.S.; Oyama, S.T.; Boudart, M. Molybdenum carbide catalysts: I. Synthesis of unsupported powders. J. Catal. 1987, 106, 125–133. [Google Scholar] [CrossRef]
- Lee, W.-S.; Wang, Z.; Zheng, W.; Vlachos, D.G.; Bhan, A. Vapor phase hydrodeoxygenation of furfural to 2-methylfuran on molybdenum carbide catalysts. Catal. Sci. Technol. 2014, 4, 2340–2352. [Google Scholar] [CrossRef]
- Ma, R.; Hao, W.; Ma, X.; Tian, Y.; Li, Y. Catalytic Ethanolysis of Kraft Lignin into High-Value Small-Molecular Chemicals over a Nanostructured α-Molybdenum Carbide Catalyst. Angew. Chem. Int. Ed. 2014, 53, 7310–7315. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-S.; Bugli, G.; Djéga-Mariadassou, G. Influence of the Degree of Carburization on the Density of Sites and Hydrogenating Activity of Molybdenum Carbides. J. Catal. 2000, 193, 238–247. [Google Scholar] [CrossRef]
- Quiroz, J.; Mai, E.F.; Teixeira da Silva, V. Synthesis of Nanostructured Molybdenum Carbide as Catalyst for the Hydrogenation of Levulinic Acid to γ-Valerolactone. Top. Catal. 2016, 59, 148–158. [Google Scholar] [CrossRef]
- Gosselink, R.W.; Stellwagen, D.R.; Bitter, J.H. Tungsten-Based Catalysts for Selective Deoxygenation. Angew. Chem. Int. Ed. 2013, 52, 5089–5092. [Google Scholar] [CrossRef] [PubMed]
- Jongerius, A.L.; Gosselink, R.W.; Dijkstra, J.; Bitter, J.H.; Bruijnincx, P.C.A.; Weckhuysen, B.M. Carbon Nanofiber Supported Transition-Metal Carbide Catalysts for the Hydrodeoxygenation of Guaiacol. ChemCatChem 2013, 5, 2964–2972. [Google Scholar] [CrossRef]
- Regmi, Y.N.; Rogers, B.R.; Labbé, N.; Chmely, S.C. Scalable and Tunable Carbide–Phosphide Composite Catalyst System for the Thermochemical Conversion of Biomass. ACS Sustain. Chem. Eng. 2017, 5, 7751–7758. [Google Scholar] [CrossRef]
- Santillan-Jimenez, E.; Perdu, M.; Pace, R.; Morgan, T.; Crocker, M. Activated Carbon, Carbon Nanofiber and Carbon Nanotube Supported Molybdenum Carbide Catalysts for the Hydrodeoxygenation of Guaiacol. Catalysts 2015, 5, 424–441. [Google Scholar] [CrossRef]
- Han, J.; Duan, J.; Chen, P.; Lou, H.; Zheng, X.; Hong, H. Nanostructured molybdenum carbides supported on carbon nanotubes as efficient catalysts for one-step hydrodeoxygenation and isomerization of vegetable oils. Green Chem. 2011, 13, 2561–2568. [Google Scholar] [CrossRef]
- Sun, J.; Zheng, M.; Wang, X.; Wang, A.; Cheng, R.; Li, T.; Zhang, T. Catalytic Performance of Activated Carbon Supported Tungsten Carbide for Hydrazine Decomposition. Catal. Lett. 2008, 123, 150–155. [Google Scholar] [CrossRef]
- Ji, N.; Zhang, T.; Zheng, M.; Wang, A.; Wang, H.; Wang, X.; Chen, J.G. Direct Catalytic Conversion of Cellulose into Ethylene Glycol Using Nickel-Promoted Tungsten Carbide Catalysts. Angew. Chem. Int. Ed. 2008, 47, 8510–8513. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, X.; Zhao, X.; Zheng, M.; Cheng, R.; Zhou, L.; Zhang, T. Microcalorimetric studies of the iridium catalyst for hydrazine decomposition reaction. Thermochim. Acta 2005, 434, 119–124. [Google Scholar] [CrossRef]
- Han, J.; Duan, J.; Chen, P.; Lou, H.; Zheng, X. Molybdenum Carbide-Catalyzed Conversion of Renewable Oils into Diesel-like Hydrocarbons. Adv. Synth. Catal. 2011, 353, 2577–2583. [Google Scholar] [CrossRef]
- Hollak, S.A.W.; Gosselink, R.W.; van Es, D.S.; Bitter, J.H. Comparison of Tungsten and Molybdenum Carbide Catalysts for the Hydrodeoxygenation of Oleic Acid. ACS Catal. 2013, 3, 2837–2844. [Google Scholar] [CrossRef]
- Shi, Y.; Yang, Y.; Li, Y.-W.; Jiao, H. Theoretical study about Mo2C(101)-catalyzed hydrodeoxygenation of butyric acid to butane for biomass conversion. Catal. Sci. Technol. 2016, 6, 4923–4936. [Google Scholar] [CrossRef]
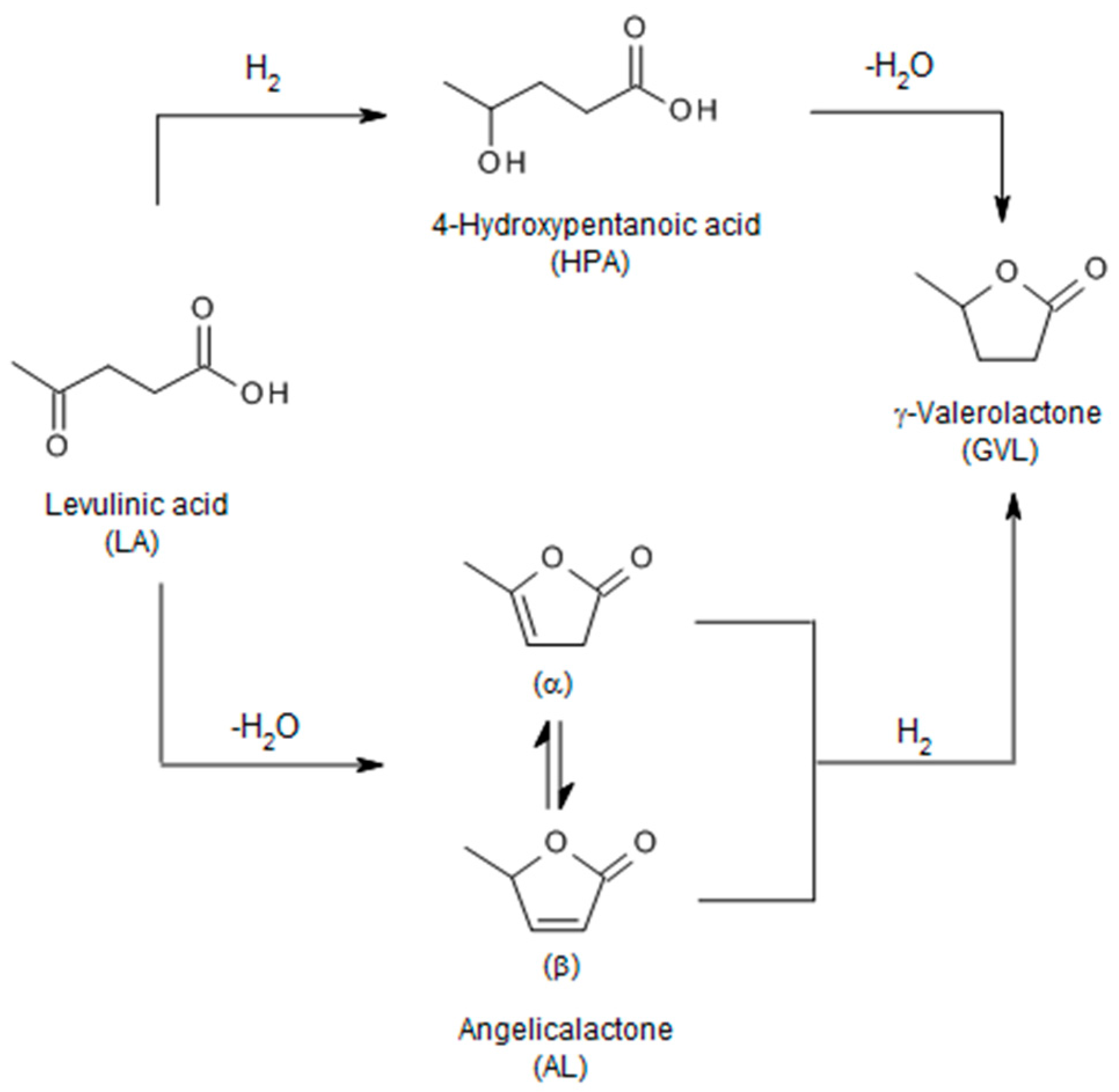
- Yan, K.; Jarvis, C.; Gu, J.; Yan, Y. Production and catalytic transformation of levulinic acid: A platform for speciality chemicals and fuels. Renew. Sustain. Energy Rev. 2015, 51, 986–997. [Google Scholar] [CrossRef]
- Alonso, D.M.; Wettstein, S.G.; Dumesic, J.A. Gamma-valerolactone, a sustainable platform molecule derived from lignocellulosic biomass. Green Chem. 2013, 15, 584–595. [Google Scholar] [CrossRef]
- Leclercq, L.; Imura, K.; Yoshida, S.; Barbee, T.; Boudart, M. Synthesis of New Catalytic Materials: Metal Carbides of the Group VI B Elements. Stud. Surf. Sci. Catal. 1979, 3, 627–639. [Google Scholar] [CrossRef]
- Chen, W.-F.; Wang, C.-H.; Sasaki, K.; Marinkovic, N.; Xu, W.; Muckerman, J.T.; Zhu, Y.; Adzic, R.R. Highly active and durable nanostructured molybdenum carbide electrocatalysts for hydrogen production. Energy Environ. Sci. 2013, 6, 943–951. [Google Scholar] [CrossRef]
- Rodella, C.B.; Barrett, D.H.; Moya, S.F.; Figueroa, S.J.A.; Pimenta, M.T.B.; Curvelo, A.A.S.; Teixeira da Silva, V. Physical and chemical studies of tungsten carbide catalysts: Effects of Ni promotion and sulphonated carbon. RSC Adv. 2015, 5, 23874–23885. [Google Scholar] [CrossRef]
- Yue, H.; Zhao, Y.; Ma, X.; Gong, J. Ethylene glycol: Properties, synthesis, and applications. Chem. Soc. Rev. 2012, 41, 4218–4244. [Google Scholar] [CrossRef] [PubMed]
- Ji, N.; Zhang, T.; Zheng, M.; Wang, A.; Wang, H.; Wang, X.; Shu, Y.; Stottlemyer, A.L.; Chen, J.G. Catalytic conversion of cellulose into ethylene glycol over supported carbide catalysts. Catal. Today 2009, 147, 77–85. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, A.; Zhang, T. A new 3D mesoporous carbon replicated from commercial silica as a catalyst support for direct conversion of cellulose into ethylene glycol. Chem. Commun. 2010, 46, 862–864. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Wang, S.; Liu, H. Cellulose Conversion into Polyols Catalyzed by Reversibly Formed Acids and Supported Ruthenium Clusters in Hot Water. Angew. Chem. Int. Ed. 2007, 46, 7636–7639. [Google Scholar] [CrossRef] [PubMed]
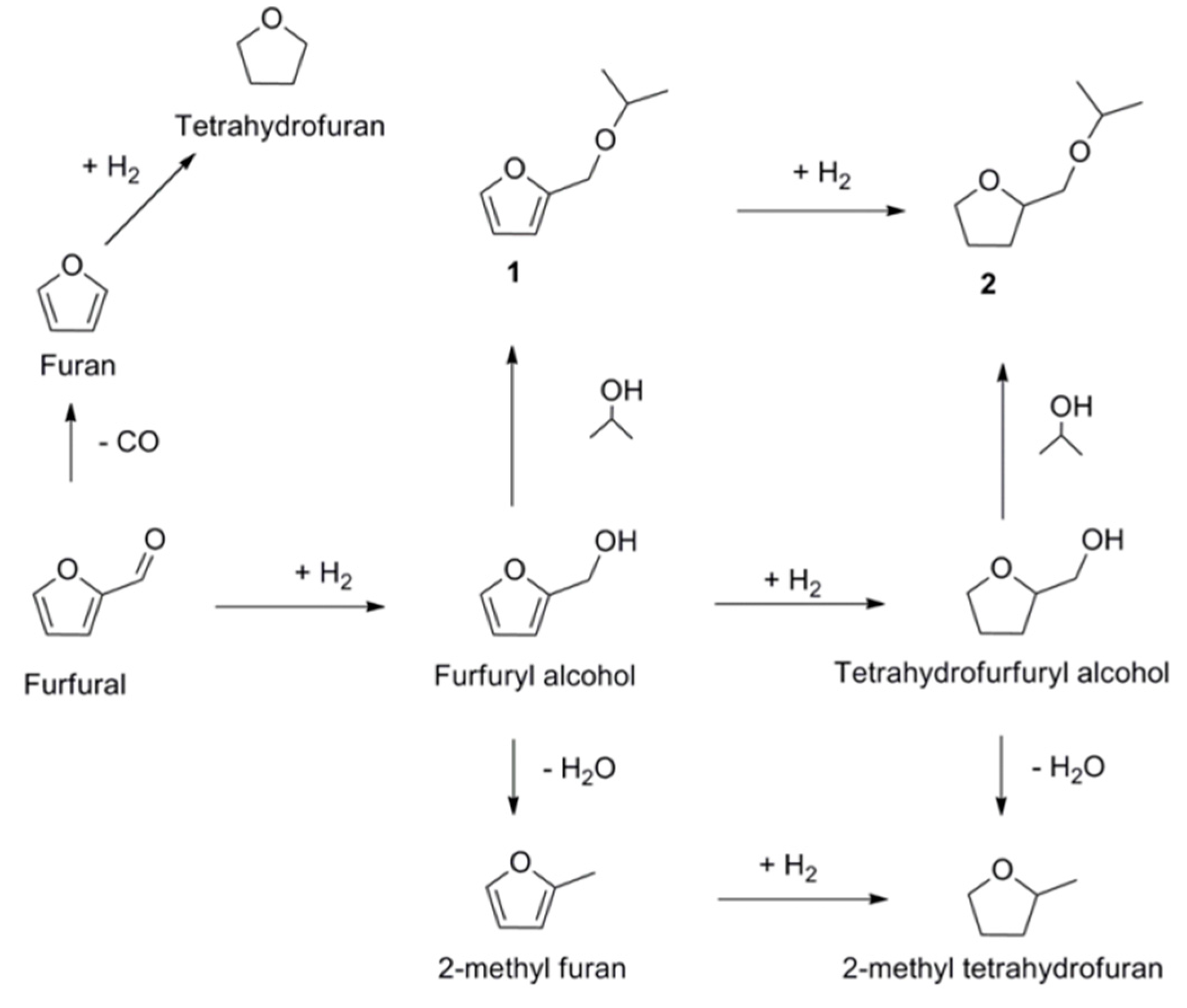
- Xiong, K.; Lee, W.-S.; Bhan, A.; Chen, J.G. Molybdenum Carbide as a Highly Selective Deoxygenation Catalyst for Converting Furfural to 2-Methylfuran. ChemSusChem 2014, 7, 2146–2149. [Google Scholar] [CrossRef] [PubMed]
- Lange, J.-P.; van der Heide, E.; van Buijtenen, J.; Price, R. Furfural-A Promising Platform for Lignocellulosic Biofuels. ChemSusChem 2012, 5, 150–166. [Google Scholar] [CrossRef] [PubMed]
- Román-Leshkov, Y.; Barrett, C.J.; Liu, Z.Y.; Dumesic, J.A. Production of dimethylfuran for liquid fuels from biomass-derived carbohydrates. Nature 2007, 447, 982–985. [Google Scholar] [CrossRef] [PubMed]
- McManus, J.R.; Vohs, J.M. Deoxygenation of glycolaldehyde and furfural on Mo2C/Mo(100). Surf. Sci. 2014, 630, 16–21. [Google Scholar] [CrossRef]
- Xiong, K.; Yu, W.; Chen, J.G. Selective deoxygenation of aldehydes and alcohols on molybdenum carbide (Mo2C) surfaces. Appl. Surf. Sci. 2014, 323, 88–95. [Google Scholar] [CrossRef]
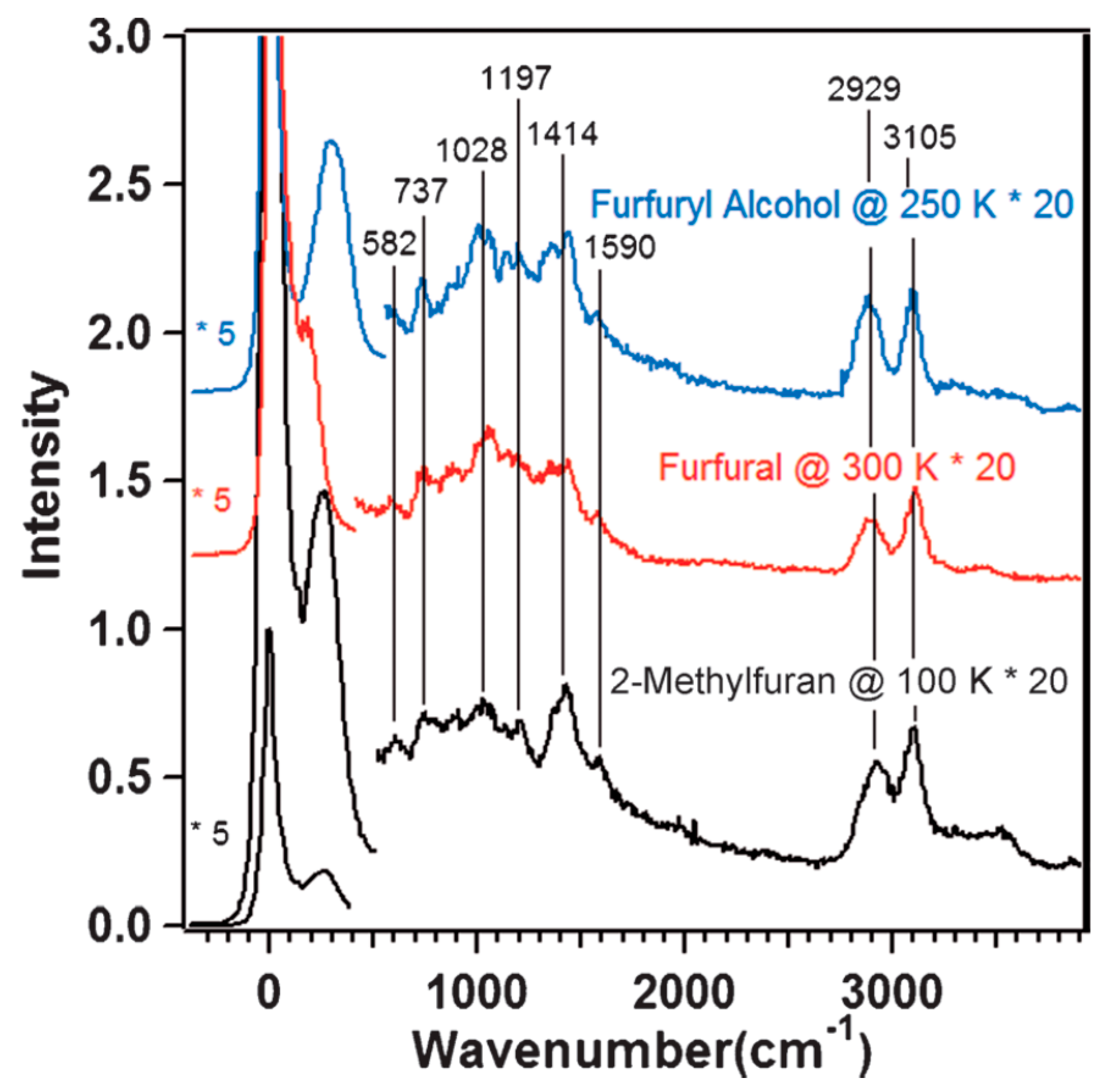
- Pang, S.H.; Medlin, J.W. Adsorption and Reaction of Furfural and Furfuryl Alcohol on Pd(111): Unique Reaction Pathways for Multifunctional Reagents. ACS Catal. 2011, 1, 1272–1283. [Google Scholar] [CrossRef]
- Li, G.; Li, N.; Wang, Z.; Li, C.; Wang, A.; Wang, X.; Cong, Y.; Zhang, T. Synthesis of High-Quality Diesel with Furfural and 2-Methylfuran from Hemicellulose. ChemSusChem 2012, 5, 1958–1966. [Google Scholar] [CrossRef] [PubMed]
- Subrahmanyam, A.V.; Thayumanavan, S.; Huber, G.W. C-C Bond Formation Reactions for Biomass-Derived Molecules. ChemSusChem 2010, 3, 1158–1161. [Google Scholar] [CrossRef] [PubMed]
- Bej, S.K.; Bennett, C.A.; Thompson, L.T. Acid and base characteristics of molybdenum carbide catalysts. Appl. Catal. A Gen. 2003, 250, 197–208. [Google Scholar] [CrossRef]
- Shi, Y.; Yang, Y.; Li, Y.-W.; Jiao, H. Mechanisms of Mo2C(101)-Catalyzed Furfural Selective Hydrodeoxygenation to 2-Methylfuran from Computation. ACS Catal. 2016, 6, 6790–6803. [Google Scholar] [CrossRef]
- Li, C.; Zhao, X.; Wang, A.; Huber, G.W.; Zhang, T. Catalytic Transformation of Lignin for the Production of Chemicals and Fuels. Chem. Rev. 2015, 115, 11559–11624. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Ma, R.; Hao, W.; Chen, M.; Yan, F.; Cui, K.; Tian, Y.; Li, Y. Common Pathways in Ethanolysis of Kraft Lignin to Platform Chemicals over Molybdenum-Based Catalysts. ACS Catal. 2015, 5, 4803–4813. [Google Scholar] [CrossRef]
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The Catalytic Valorization of Lignin for the Production of Renewable Chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-J.; Lee, W.-S.; Bhan, A. Mo2C catalyzed vapor phase hydrodeoxygenation of lignin-derived phenolic compound mixtures to aromatics under ambient pressure. Appl. Catal. A Gen. 2016, 510, 42–48. [Google Scholar] [CrossRef]
- Cheng, S.; Wilks, C.; Yuan, Z.; Leitch, M.; Xu, C. (Charles) Hydrothermal degradation of alkali lignin to bio-phenolic compounds in sub/supercritical ethanol and water–ethanol co-solvent. Polym. Degrad. Stab. 2012, 97, 839–848. [Google Scholar] [CrossRef]
- Engelhardt, J.; Lyu, P.; Nachtigall, P.; Schüth, F.; García, Á.M. The Influence of Water on the Performance of Molybdenum Carbide Catalysts in Hydrodeoxygenation Reactions: A Combined Theoretical and Experimental Study. ChemCatChem 2017, 9, 1985–1991. [Google Scholar] [CrossRef]
- Guo, H.; Zhang, B.; Li, C.; Peng, C.; Dai, T.; Xie, H.; Wang, A.; Zhang, T. Tungsten Carbide: A Remarkably Efficient Catalyst for the Selective Cleavage of Lignin C−O Bonds. ChemSusChem 2016, 9, 3220–3229. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Zhang, B.; Qi, Z.; Li, C.; Ji, J.; Dai, T.; Wang, A.; Zhang, T. Valorization of Lignin to Simple Phenolic Compounds over Tungsten Carbide: Impact of Lignin Structure. ChemSusChem 2017, 10, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.C.C.; Chang, H.-F.; Lin, F.-J.; Lin, K.-H.; Chen, C.-H. Biomass gasification for hydrogen production. Int. J. Hydrogen Energy 2011, 36, 14252–14260. [Google Scholar] [CrossRef]
- Kaewpanha, M.; Guan, G.; Ma, Y.; Hao, X.; Zhang, Z.; Reubroychareon, P.; Kusakabe, K.; Abudula, A. Hydrogen production by steam reforming of biomass tar over biomass char supported molybdenum carbide catalyst. Int. J. Hydrogen Energy 2015, 40, 7974–7982. [Google Scholar] [CrossRef]
- Darujati, A.R.; LaMont, D.C.; Thomson, W.J. Oxidation stability of Mo2C catalysts under fuel reforming conditions. Appl. Catal. A Gen. 2003, 253, 397–407. [Google Scholar] [CrossRef]

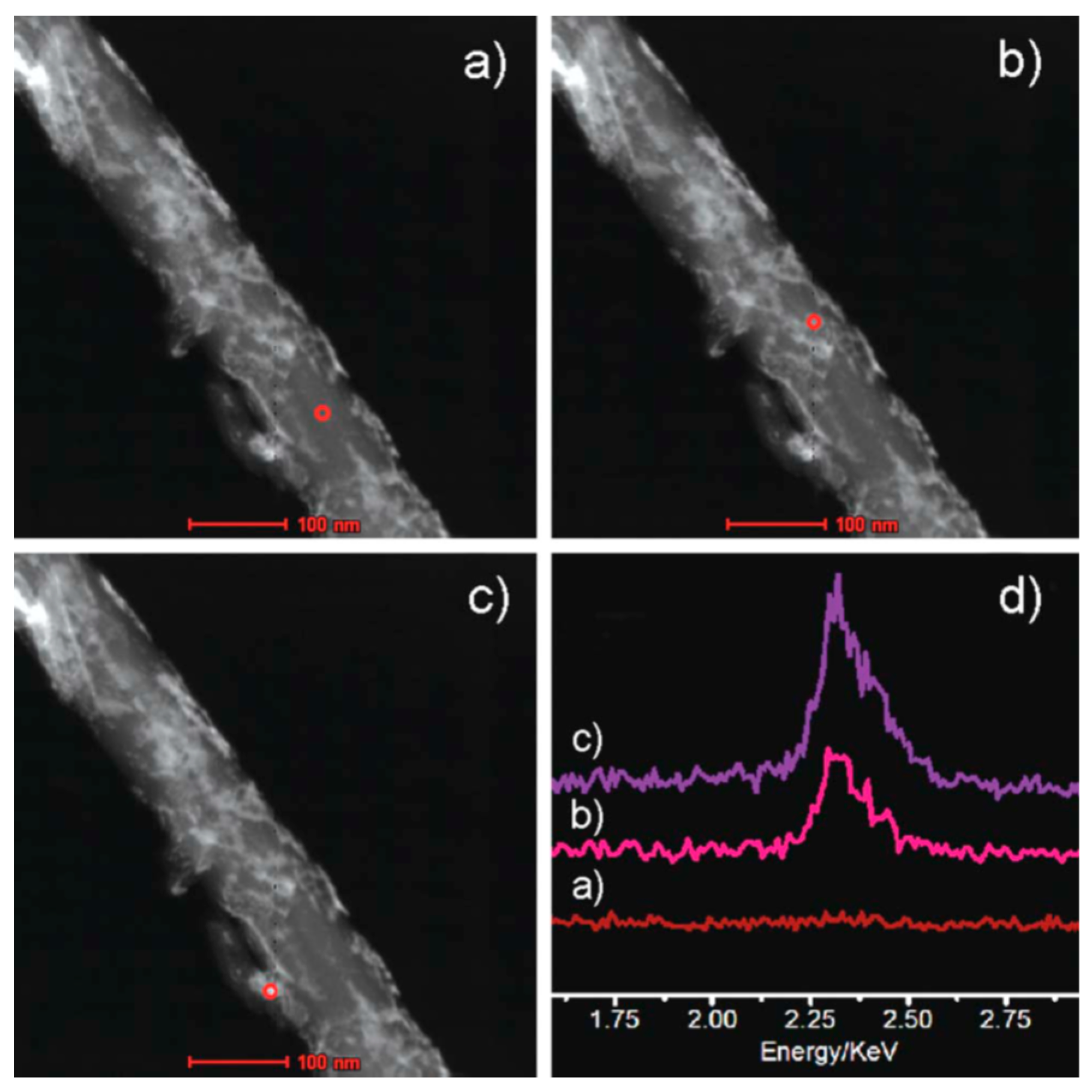
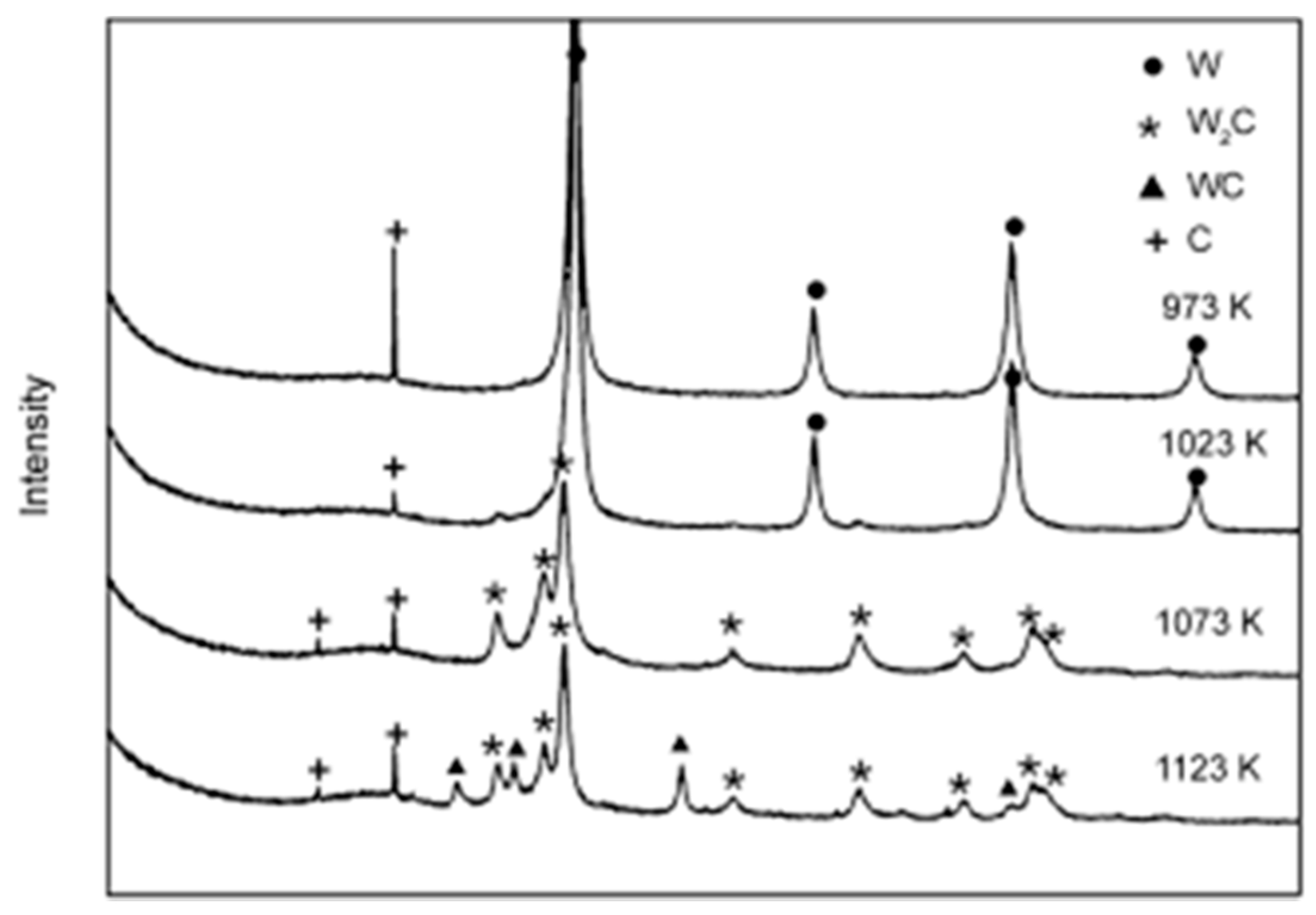
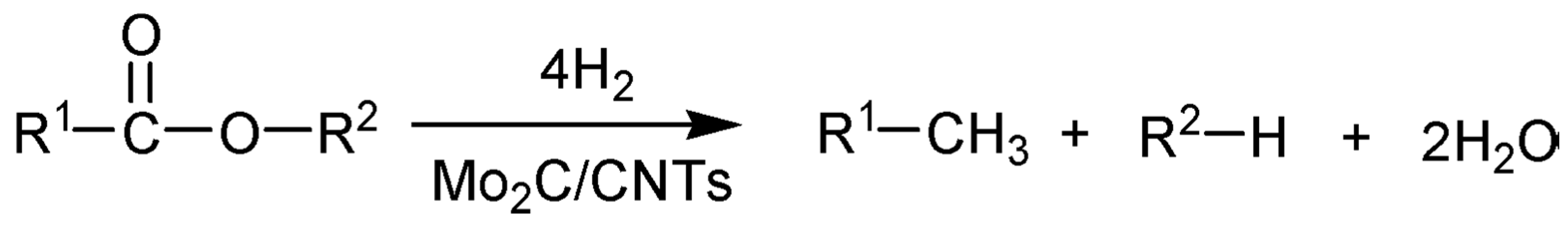
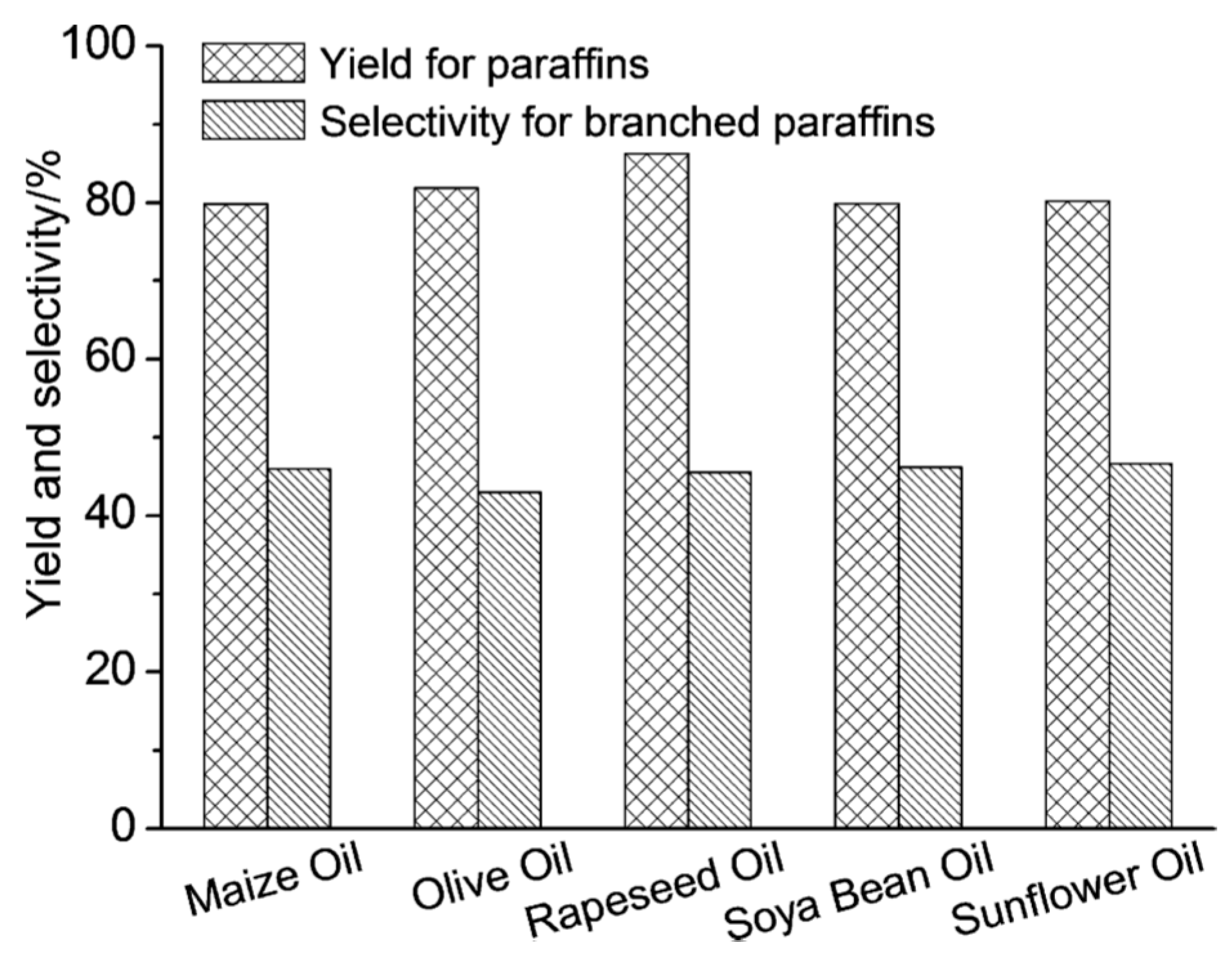
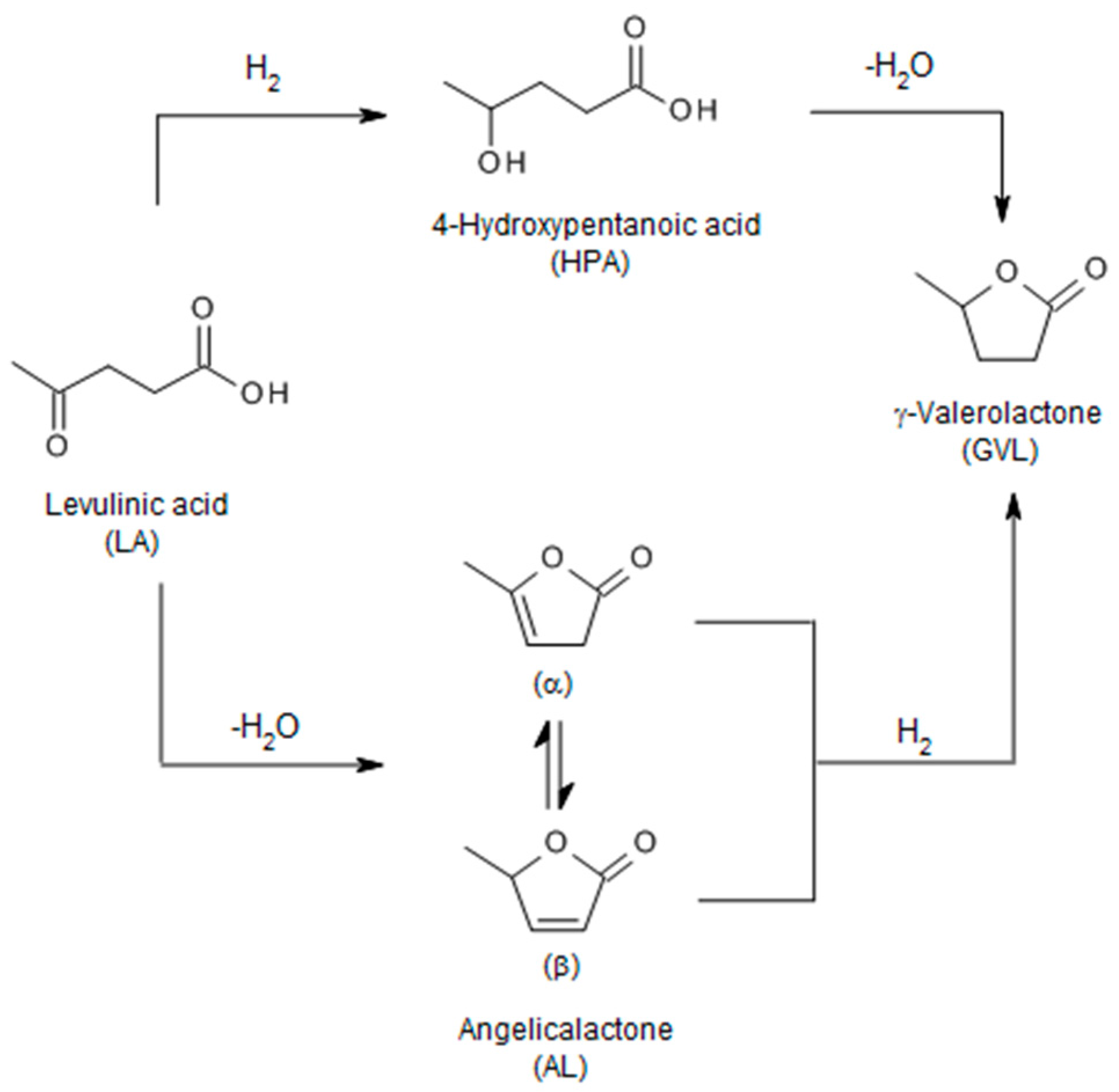
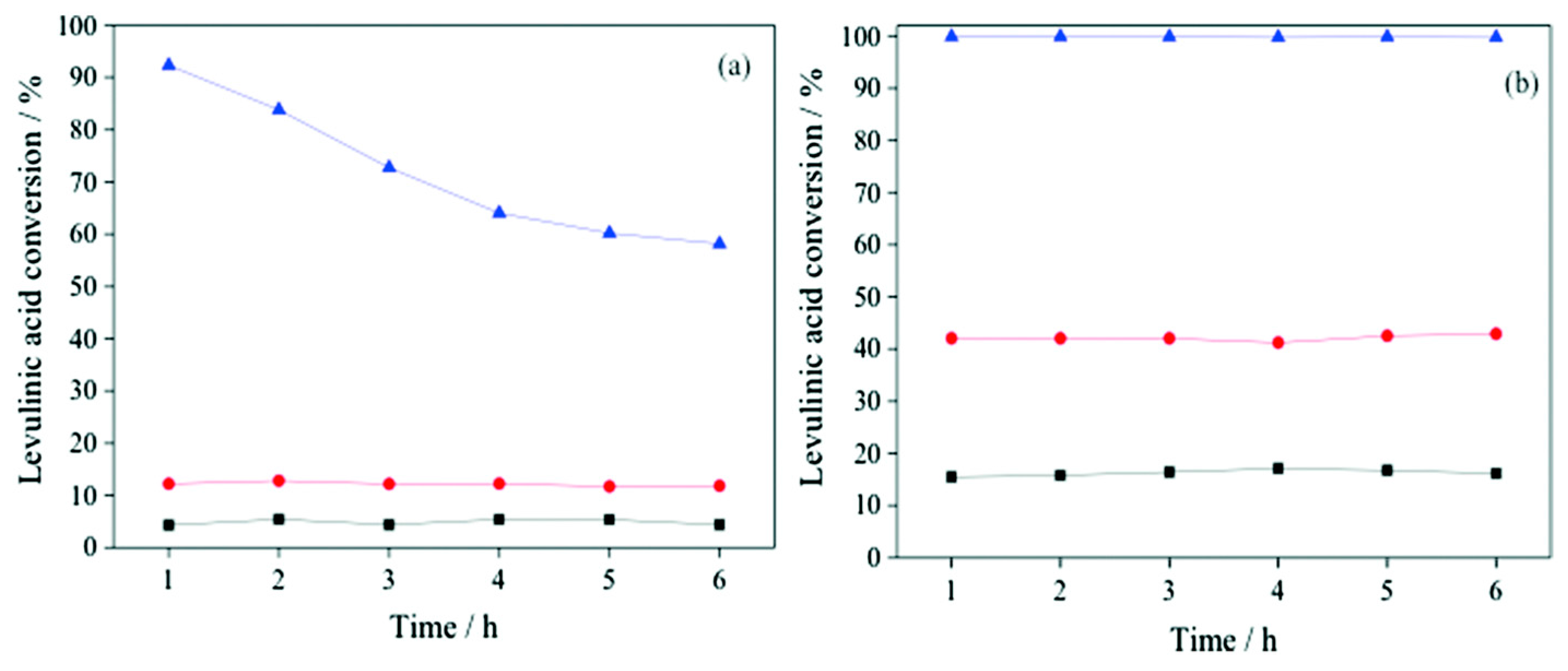

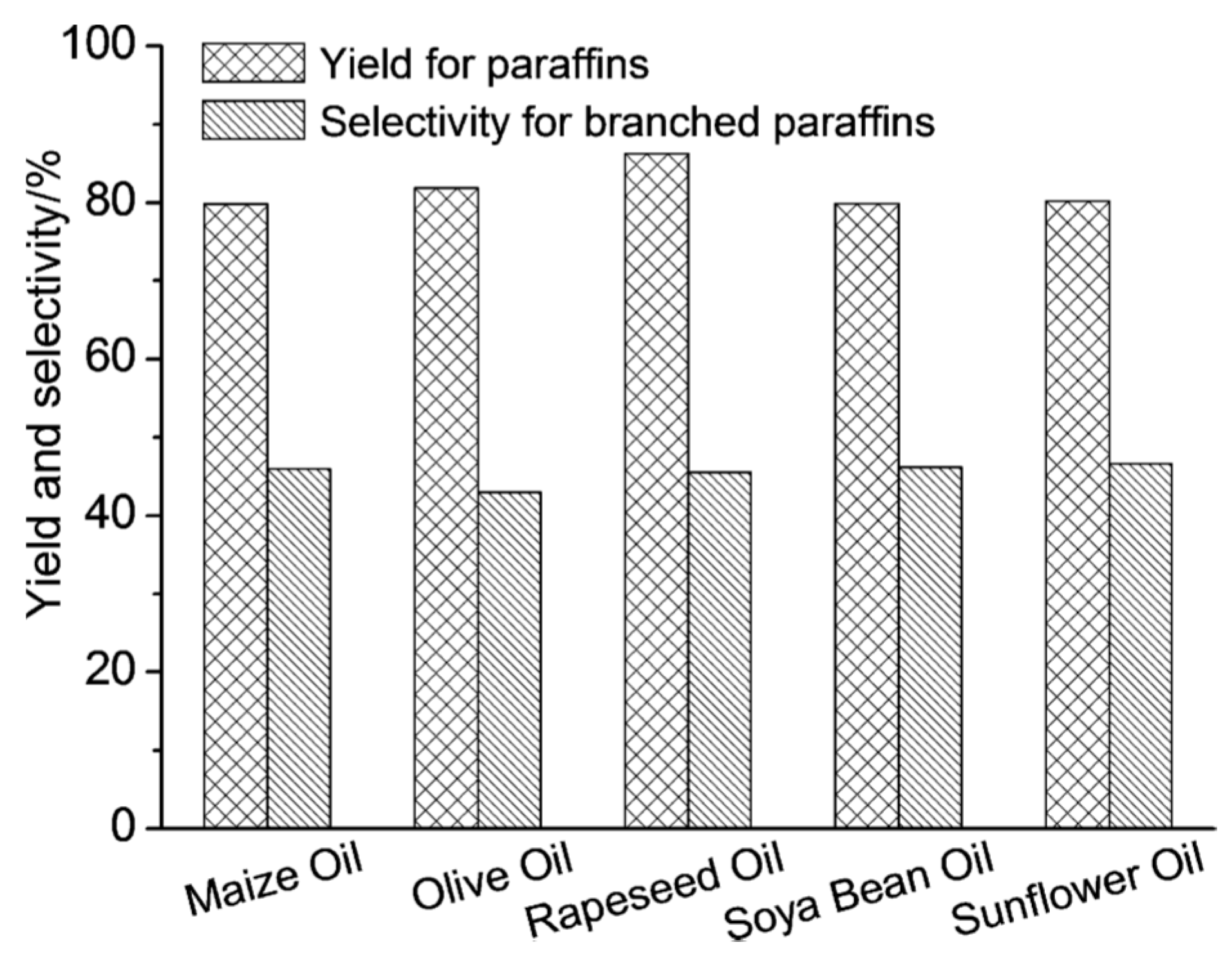
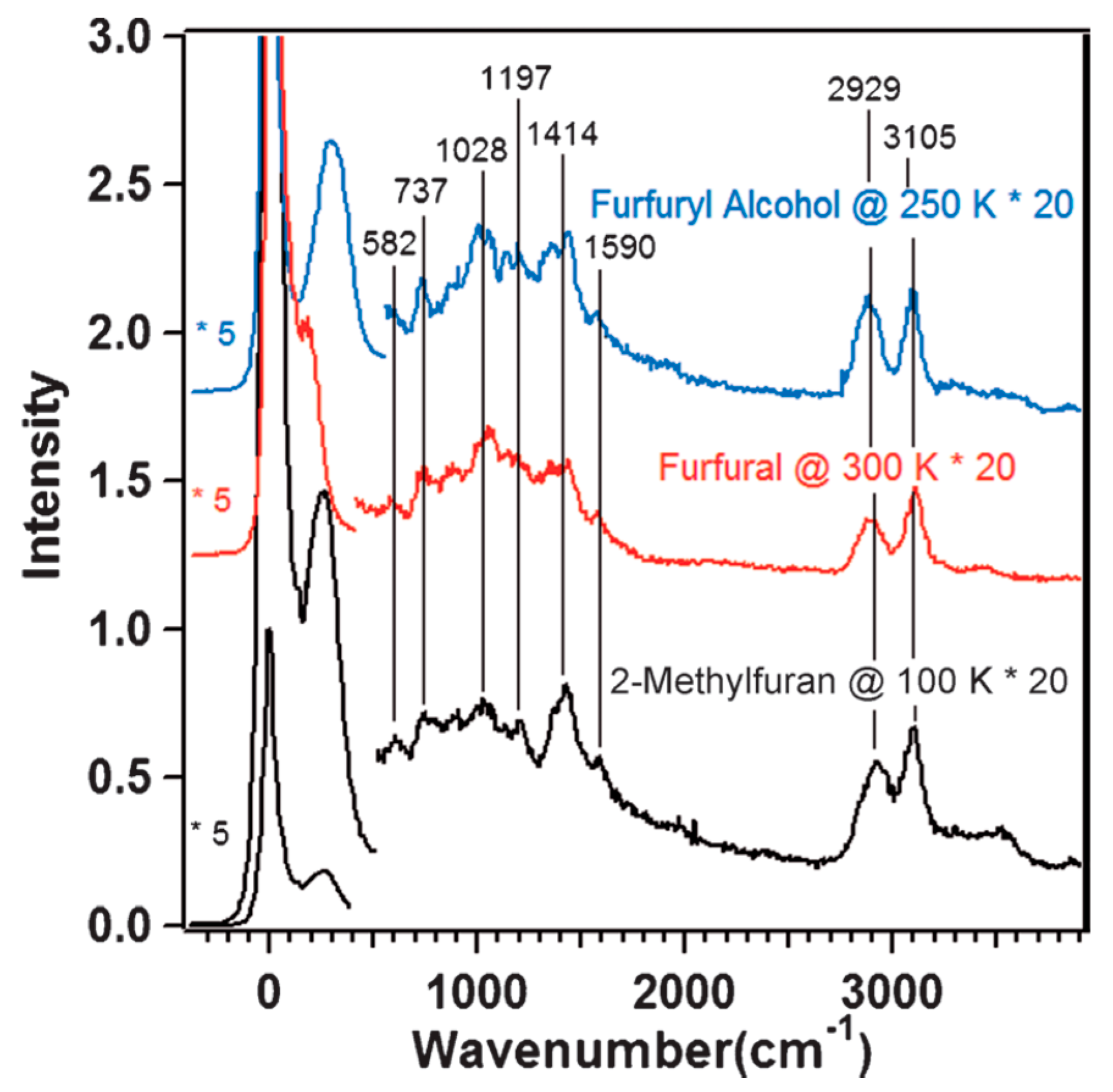
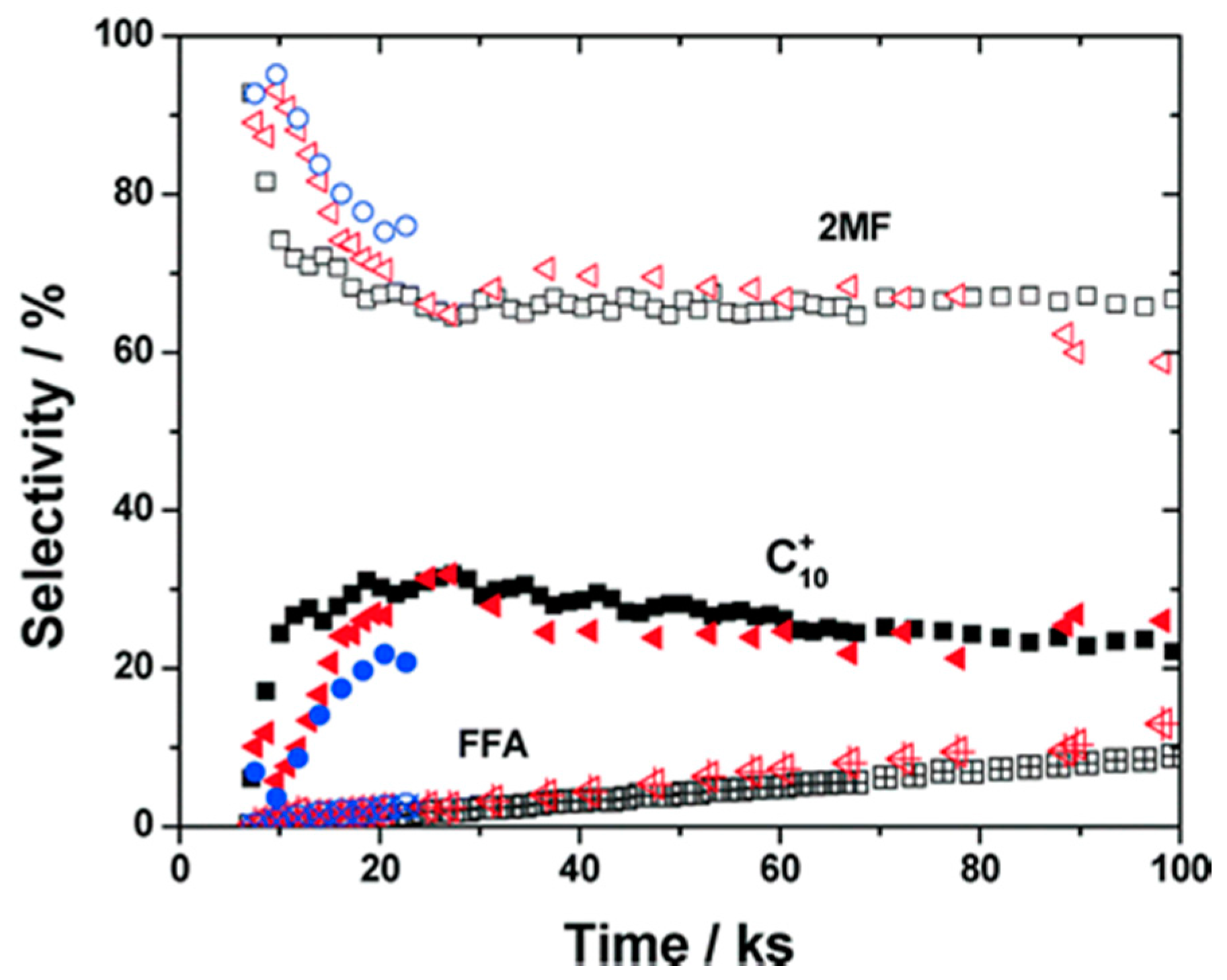
 ) and second (
) and second ( ) regeneration cycle. Reproduced with permission from [32]. Copyright The Royal Society of Chemistry, 2014.
) and second () regeneration cycle. Reproduced with permission from [32]. Copyright The Royal Society of Chemistry, 2014.
) regeneration cycle. Reproduced with permission from [32]. Copyright The Royal Society of Chemistry, 2014.
) and second () regeneration cycle. Reproduced with permission from [32]. Copyright The Royal Society of Chemistry, 2014.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan-Thaw, C.E.; Villa, A. Metal Carbides for Biomass Valorization. Appl. Sci. 2018, 8, 259. https://doi.org/10.3390/app8020259
Chan-Thaw CE, Villa A. Metal Carbides for Biomass Valorization. Applied Sciences. 2018; 8(2):259. https://doi.org/10.3390/app8020259
Chicago/Turabian StyleChan-Thaw, Carine E., and Alberto Villa. 2018. "Metal Carbides for Biomass Valorization" Applied Sciences 8, no. 2: 259. https://doi.org/10.3390/app8020259
APA StyleChan-Thaw, C. E., & Villa, A. (2018). Metal Carbides for Biomass Valorization. Applied Sciences, 8(2), 259. https://doi.org/10.3390/app8020259