Abstract
Microstructure and mechanical properties are key parameters influencing the performance of structural multi-phase alloys such as those based on intermetallic TiAl compounds. There, the main constituent, a -TiAl phase, is derived from a face-centered cubic structure. Consequently, the dissociation of dislocations and generation of stacking faults (SFs) are important factors contributing to the overall deformation behavior, as well as mechanical properties, such as tensile/creep strength and, most importantly, fracture elongation below the brittle-to-ductile transition temperature. In this work, SFs on the plane in -TiAl are revisited by means of ab initio calculations, finding their energies in agreement with previous reports. Subsequently, stacking fault energies are evaluated for eight ternary additions, namely group IVB–VIB elements, together with Ti off-stoichiometry. It is found that the energies of superlattice intrinsic SFs, anti-phase boundaries (APBs), as well as complex SFs decrease by 20–40% with respect to values in stoichiometric -TiAl once an alloying element X is present in the fault plane having thus a composition of Ti-50Al-12.5X. In addition, Mo, Ti and V stabilize the APB on the (111) plane, which is intrinsically unstable at in stoichiometric -TiAl.
1. Introduction
Titanium aluminides are intermetallic compounds and alloys with a wide reach for high-temperature applications. These range from low-pressure turbine blades in the aircraft industry to turbocharger turbine wheels and valves in the automotive industry [1,2,3]. Their outstanding properties include low mass density, high specific strength and stiffness and good creep properties up to 750 . They outperform titanium alloys by their good oxidation behavior and burn resistance [2]. In contrast to ceramic materials, titanium aluminides also exhibit the ability to plastically deform at room temperature [4].
Current state-of-the-art TiAl alloys consist of a close to face-centered cubic (fcc) -TiAl phase, a hexagonal -TiAl phase, a body-centered cubic (bcc) -TiAl phase, and, occasionally, additional minor phases [1]. However, the main constituent is the -TiAl phase, which fundamentally influences the alloy properties during processing, e.g., hot-forging and application.
The -TiAl phase has an L1 structure which is a tetragonally strained fcc lattice with planes occupied alternatively by Ti and Al atoms. Therefore, stacking faults (SFs) become a topic of a great importance, similar to fcc metals, in which a stacking fault is a deviation from the normal stacking sequence of the planes. The stacking fault energy (SFE), , an energy stored by the stacking fault defect, has a huge impact on a plastic deformation behavior of fcc metals. It determines the spreading of dissociated partial dislocations and, therefore, influences the cross-slip properties of screw dislocations. It is observed that metals with smaller stacking fault energies exhibit more mechanical twinning whereby they possess an additional deformation mechanism [5]. Especially, this effect was utilized in the development of novel high-strength -TiAl-based alloys exhibiting a certain ductility at room temperature. Here, the reader is referred to [1,2] and the papers cited therein.
Measuring the stacking fault energies experimentally is, nevertheless, non-trivial. Most commonly, they are measured indirectly using transmission electron microscopy (TEM) from the separation of partial dislocations. Such an experimental procedure is, however, time consuming and deals with all the difficulties related to a multi-phase complex alloy (non-homogeneous concentrations, stresses, sample preparation, to name a few). Consequently, experimental works dealing with this topic in -TiAl are scarce [6,7,8,9]. Hence, theoretical studies of stacking fault energies comprise a welcome alternative to examine stacking faults. These included both ab initio methods [10,11,12,13,14], as well as empirical atomistic modeling [15,16,17].
Nonetheless, the “real” alloys go beyond the simple binary Ti-Al system [1,2,4]. It is therefore of immense interest to know what impact alloying has on the SFEs and, consequently, on the expected deformation mechanisms. A rare example of such modeling effort is the study of Woodward and MacLaren [18], who used the coherent potential approximation (CPA) to investigate the impact of Nb and Cr on SFEs in -TiAl. This formalism, however, does not allow for any relaxations of the local atomic environments. Moreover, information on other alloying elements, namely the group IVB, VB, and VIB transition metal (TM) elements, commonly used experimentally, is still missing. The aim of the present study is to fill this gap by investigating the impact of ternary alloying elements on planar faults in -TiAl using ab initio techniques.
2. Methods
2.1. Geometry of Planar Defects in -TiAl
The stacking faults in fcc materials are irregularities of stacking the planes. Let A, B and C denote the three configurations of the close-packed planes, being mutually displaced by a vector in coordinates related to the conventional cubic cell (containing four atoms). The perfect stacking could change to , where the denotes the position of a twin boundary (a twin mirror plane). Other faults include a missing or an extra plane without a mirror. The former is called intrinsic stacking fault (ISF) and is described by a stacking sequence , while the latter is called extrinsic stacking fault (ESF) and corresponds to .
The situation is somewhat more complicated for the L structure as in the case of -TiAl. Its lattice is a slightly tetragonally-deformed fcc lattice (; hence, the direction is not equivalent with or any more). Moreover, the planes are alternatively occupied by Al and Ti atoms. Consequently, out of the three displacement vectors , and , being equivalent in the fcc structure (and all producing ISF), only creates a fault-preserving local chemical neighborhood of the atoms. The resulting fault is called superlattice ISF (SISF). and , in addition to the stacking fault, also alter the chemical occupation of the sites, yielding so-called complex stacking faults (CSFs). Finally, a displacement results in an undistorted lattice, but altered lattice occupations, so-called anti-phase, and the corresponding fault plane is therefore called the anti-phase boundary (APB).
SFE expresses the energy difference between the faulted, , and perfect, , configurations per unit area, A:
This energy can be calculated not only for the special translations corresponding to SFs as described above, but for any displacement vector , hence yielding the generalized stacking fault energy (GSFE) surface [19]. In addition, to the actual values of SFEs, the GSFE surface contains also energy barriers that need to be overcome when an SF is created. In this work, we discuss SFs on the planes; hence, GSFE surfaces are evaluated along u and v coordinates decomposing . It is worth noting that generalization of ESF for the L structure yields the superlattice extrinsic stacking fault (SESF), which is, however, not compatible with a single shear plane. Since it is not contained by the simple GSFE surface, it will not be discussed any further in this work. For similar reasons, we do not include twin boundaries or APBs on the planes.
2.2. Modeling of SFs
One possible approach for simulating SFs is to build a supercell of cells having the fault planes perpendicular to the lattice vector. Subsequently, a section of the supercell is displaced in the plane (fault plane) so as to produce an SF with , and if necessary, a layer is removed in order to restore the periodic boundary conditions (Figure 1). In some cases (e.g., APB), this approach implies that there are two SFs per supercell.
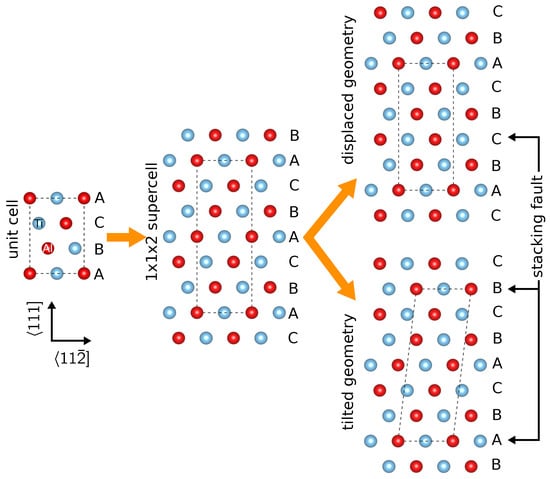
Figure 1.
Schematic drawing demonstrating the two possible modeling approaches to SFs in the -TiAl with the L structure.
In this work, we assumed another approach, which allows for a straightforward evaluation of GSFE. The Cartesian positions of all atoms remain unchanged in the whole supercell, and instead, the supercell vector is tilted to become . We note that, strictly speaking, these two approaches are inequivalent if the atomic relaxations are performed along the supercell lattice vector directions. In particular, to calculate SFE, one typically fixes the atomic positions in the SF plane and relaxes the positions (forces) in the direction. This direction is perpendicular to the SF plane in the former approach with the displaced geometry, while it is slightly tilted away in the latter approach. The actual difference on the predicted results will be discussed later in the text and will be shown to be negligible in the present case provided the supercell size n is large enough (the tilt decreases with increasing n).
2.3. Computational Details
The present quantum mechanical calculations are based on density functional theory (DFT) [20,21] as implemented in the Vienna Ab initio Simulation Package (VASP) [22,23]. The basis set contained plane waves corresponding to energies lower than . Our convergence tests showed that together with the Monkhorst–Pack mesh sampling the Brillouin zone of the -TiAl conventional cell with 2 Ti and 2 Al atoms, this cut-off energy should guarantee total energy accuracy in the range of a few meV/at. The exchange and correlation (xc) effects were treated within the generalized gradient approximation as parametrized by Perdew and Wang (GGA-PW91) [24]. The structural relaxations were carried out until the forces changed less than Å, while each electronic loop was converged until the total energy changes were smaller than (per simulation box). SFEs for the stoichiometric -TiAl were evaluated for (at least 15 planes), while the alloying studies, which required laterally larger cells, were performed for (in total, six planes).
3. Results and Discussion
3.1. SFE in -TiAl
The calculated SFEs are summarized in Table 1. In addition to the method based on tilting the supercell vectors, also a complementary method based on displacing a rigid block of the supercell [13,14] was employed. The differences are negligible. Similarly, our calculations predict only a small decrease of the SFE values (below 6%) when the local density approximation (LDA) is used instead of GGA. Test calculations with respect to the supercell size also suggest that the used models are large enough to yield converged results. The values calculated here lie in the range of previously published predictions based on DFT. Consequently, the GGA-PW91 xc potential together with the tilted supercell geometry were used for all subsequently discussed results.

Table 1.
Calculated SF energies (in mJ/m) compared with available literature data for -TiAl. Differences in calculation methods are noted.
Figure 2 shows the calculated GSFE surface with its profiles along dissociation paths for and superdislocations [1]:
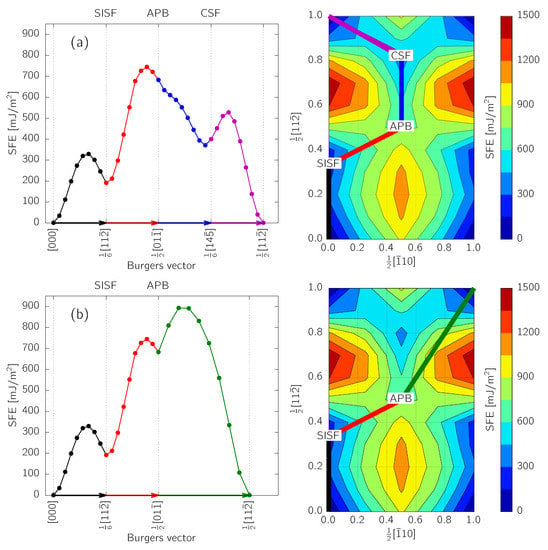
Figure 2.
Energy profile along the dissociation path according to (a) Equation (2) and (b) Equation (3). The right panels show the dissociation path overlayed on the calculated GSFE surface of the -TiAl lattice.
The APB turns out to be an unstable fault at as is does not correspond to a local minimum on the GSFE surface. This is in agreement with previous reports [10,14]. On the contrary, both SISF and CSF are stable; in order to produce them, however, barriers of ≈ and ≈, respectively, must be overcome when starting from a perfect stacking. We note, that these barriers correspond to the GSFE energy profiles along rigid pathways (as visualized in Figure 2), without attempting to estimate exact lowest-energy pathways. Even more importantly, it becomes apparent that the displacement vectors, , of the planar defects (i.e., positions of the local minima on the GSFE surface) do not correspond with the geometrically-determined ones based on the hard-sphere model. In particular, the CSF is shifted by approximately (), which is clearly visible also in the profile in Figure 2a. This actually lowers the from down to . Such an effect with similar magnitudes of displacements was previously predicted [10,11].
During the construction of the GSFE surface, the atomic coordinates in the plane were fixed, while a relaxation along the direction was allowed. Nonetheless, the SF energy can be decreased even more by fully relaxing the atomic positions once the geometry of the faulted material is trapped near the local energy minimum. This leads to a further decrease of SFEs to , and (see Table 1).
3.2. Impact of Alloying Elements
In order to tune various application-related properties, alloying elements are introduced to -TiAl-based alloys. This section thus presents predictions of the impact of early transition metals (TMs, group IVB–VIB elements) on SFEs of -TiAl. Experimentally, the most relevant are Ti-rich compositions [1,3,25]. Moreover, the early TMs preferably occupy the Ti sublattice in -TiAl [26,27]. Therefore, a scenario with a Ti anti-site (Ti atom on the Al sublattice) and a TM substitutional atom on the Ti sublattice was considered. The supercells consisted of six planes, each containing eight atoms (a supercell laterally), and the calculations were performed using the tilted geometry (Figure 1). Both the alloying element (having a concentration ) and the Ti anti-site were put in the fault plane. The results are summarized in Figure 3.
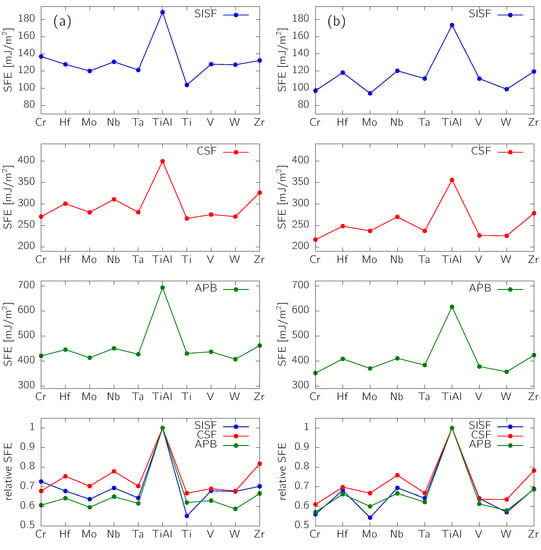
Figure 3.
Impact of alloying on -TiAl with a composition Ti-48Al-2X (X = transition metal element): (a) SFs fixed to their geometrically-dictated configurations and relaxed only in the perpendicular direction and (b) atomic positions fully relaxed.
The SFEs are predicted to significantly decrease by 20–40% as a result of the alloying (see Figure 3a; atomic positions were relaxed only in the direction perpendicular to the fault plane). Regarding the SISF, the biggest impact has Ti simply leading to a bigger Ti/Al non-stoichiometry () in comparison with the ternary systems (). From the ternary additions, the most pronounced effect has Mo ( drops by 36%) closely followed by Ta. The largest reduction of the CSF energy is caused by Cr, V and W, while the APB energy is most significantly decreased by Mo and W. The relative reduction of SFEs is even more pronounced when the full atomic relaxation is performed (Figure 3b).
The SFEs presented in (Figure 3 contained the alloying element directly in the fault plane. In order to see how localized the alloying impact is, we calculated the SFEs for the off-stoichiometric TiAl, i.e., 52Ti-48Al, as a function of the distance of the substitutional Ti atom from the fault plane. It turns out that the huge SFE reduction happens only in the case when Ti is directly in the SF ( in Figure 4). When positioned in the neighboring layer (), SFE is still decreased with respect to the stoichiometric value (most significantly in the SISF case). For , the SFEs stay practically constant, though slightly lower than in the stoichiometric case.
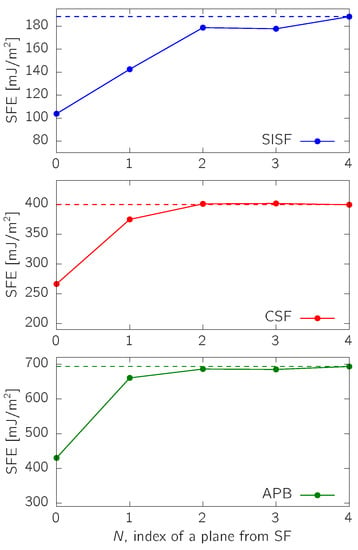
Figure 4.
Dependence of SFEs on the location of the substitutional atom (here, Ti). N labels the layer number away from the fault plane (located at ), and the dashed line represents a reference value for the stoichiometric TiAl (Table 1).
Alloying does not only influence the SFE, but the whole GSFE surface including its topology. Figure 5 gives an example of the GSFE profiles along the two dissociation paths, Equations (2) and (3), for -TiAl + Nb. When compared with analogous profiles in Figure 2, two observations can be made. Firstly, in addition to lower values of the local minima, also the local maxima (transformation barriers) are lower, leading to an overall easier creation of SFs. Secondly, while the APB was predicted to be unstable for stoichiometric -TiAl, a shallow local minimum is developed in the case of TiAl + Nb, suggesting that Nb stabilizes this fault geometry. A similar effect was predicted also for the V and Ti off-stoichiometry (52Ti-48Al).
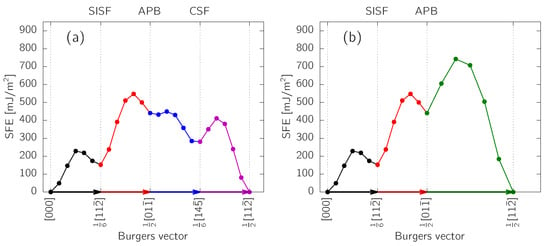
Figure 5.
GSFE profile for Ti-48Al-2Nb along the (a) Equation (2) and (b) Equation (3) dissociation paths.
3.3. Comparison with Experiment
A critical point for validating theoretical predictions is a comparison with available experimental data. The reported values for SISF energies decrease with Ti content from ≈ for Ti-54Al [6,7] to ≈ for Ti-49.6Al [8] to for Ti-48Al [8]. While these values are lower than those predicted by the ab initio methods (which, however, are all mutually consistent, cf. Table 1), the theory and the experiment agree on ordering the SFE as ([6,7] and Table 1), as well as on the decreasing value of SISF energy with increasing Ti content ([8] and Figure 3 and Figure 4). Seemingly better agreement is obtained between experiment and semi-empirical atomistic simulations yielding SISF energies in the range from 3 to (see [7,10] and the references therein). This is, however, not surprising since SFEs are often contained in the set of data used for fitting the interatomic potentials. Moreover, our own molecular dynamics calculations yielded an apparent disagreement between the SFEs’ hierarchy using two embedded-atom method potentials [28]: while the one parametrized by Zope and Mishin [29] yields , the one by Farkas and Jones [30] results in the same ordering as the ab initio calculations.
SISF energies for Nb-containing -TiAl were experimentally measured to be for Ti-48Al-1Nb [9] and for Ti-45Al-10Nb [8]. Using the value of reported for almost stoichiometric TiAl [8], Nb causes to drop by ≈, a value in an excellent agreement with our predictions (cf. Figure 3). It is worth noting that since we estimated that Nb impacts SFE only when present directly in the fault plane (or a plane next to it) and since each of the planes in our supercells contained eight atoms, we predict a drop by ≈ effectively for Ti-50Al-12.5Nb.
Consequently, we conclude that the ab initio predicted SFE values agree only semi-quantitatively with the experimental observations, but are expected to be representative of the prevailing trend: early TMs (group IVB–VIB elements) decrease SFE in Ti-rich -TiAl alloys. They increase valence electron concentration, which according to Thornton’s semi-empirical observation, leads to a decrease of the SFE [31]. Therefore, alloying these elements in -TiAl is expected to lead to various effects, which can be utilized for advanced alloy design. For example, a decreased SFE is accompanied by a dissociation of dislocations and, hence, decreasing their mobility (climb rate) at elevated temperatures. On the other hand, a lower SFE enhances the propensity for mechanical twinning, thus increasing the ductility at temperatures below the brittle-to-ductile transition temperature. However, it should be noted that an alloying element, before being eventually selected, must fulfil also other criteria, e.g., influence on oxidation behavior, etc.
4. Conclusions
In this work, we presented the results of ab initio calculations of stacking fault energies in -TiAl and ternary Ti-rich -Ti-Al-X alloys (X being a transition metal element). The energy of the anti-phase boundary of binary -TiAl was found to have the highest energy (), followed by the complex stacking fault () and a superlattice intrinsic stacking fault (). Cross-checking various methodological aspects (exchange-correlation potential, geometry of the supercell, type of relaxation) revealed that the results from various methods are consistent with each other, with the only exception being the scheme for the relaxation of atomic positions (only in the direction perpendicular to the fault plane or full).
Ternary additions, Cr, Hf, Mo, Nb, Ta, V, W and Zr, as well as Ti anti-sites were found to lead to a significant decrease of the SFEs by 20–40%. Such a strong impact is predicted only in the case when the ternary addition is directly in the fault plane, hence corresponding to a composition Ti-50Al-12.5X. The reduction of SFE is much smaller when the alloying element is localized in the plane next to the SF, while it is practically negligible when is it further from the SF plane. In addition to the reduction of the SFEs, our calculations predict stabilization of the APB(111) by Nb, Ti and V, a defect predicted to be unstable in stoichiometric -TiAl at .
Acknowledgments
Financial support from the German BMBF Project O3X3530A and the Austrian Science Fund (FWF) Project Number P29731 is greatly acknowledged.The computational results presented have been achieved in part using the Vienna Scientific Cluster (VSC).The authors are also thankful to Prof. Jörg Neugebauer and his group at the Max-Planck-Institut für Eisenforschung, Düsseldorf, Germany, for their technical support.
Author Contributions
P.D. performed all the ab initio calculations, evaluated results, and did the initial discussion. D.H. supervised the work and prepared this manuscript. H.C. and S.M. initiated the work and contributed to the discussion. All authors participated in writing and shaping this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| APB | anti-phase boundary |
| CASTEP | Cambridge Serial Total Energy Package |
| CPA | coherent potential approximation |
| CSF | complex stacking fault |
| DFT | density functional theory |
| ESF | extrinsic stacking fault |
| FP-LAPW | full-potential linearized augmented plane-wave (method) |
| FP-LMTO | full-potential linear muffin-tin orbital (method) |
| GGA | generalized gradient approximation |
| GSFE | generalized stacking fault energy |
| LDA | local density approximation |
| ISF | intrinsic stacking fault |
| SF | stacking fault |
| SFE | stacking fault energy |
| SISF | superlattice intrinsic stacking fault |
| TEM | transmission electron microscopy |
| TM | transition metal |
| VASP | Vienna Ab initio Simulation Package |
| xc | exchange and correlation (potential, effects) |
References
- Appel, F.; Paul, J.; Oehring, M. Gamma Titanium Aluminide Alloys: Science and Technology; Wiley: Hoboken, NJ, USA, 2011. [Google Scholar]
- Clemens, H.; Mayer, S. Design, Processing, Microstructure, Properties, and Applications of Advanced Intermetallic TiAl Alloys. Adv. Eng. Mater. 2013, 15, 191–215. [Google Scholar] [CrossRef]
- Mayer, S.; Erdely, P.; Fischer, F.D.; Holec, D.; Kastenhuber, M.; Klein, T.; Clemens, H. Intermetallic β-Solidifying γ-TiAl Based Alloys—From Fundamental Research to Application. Adv. Eng. Mater. 2017, 19, 1600735. [Google Scholar] [CrossRef]
- Dimiduk, D.M. Gamma titanium aluminide alloys—An assessment within the competition of aerospace structural materials. Mater. Sci. Eng. A 1999, 263, 281–288. [Google Scholar] [CrossRef]
- Tadmor, E.B.; Bernstein, N. A first-principles measure for the twinnability of FCC metals. J. Mech. Phys. Solids 2004, 52, 2507–2519. [Google Scholar] [CrossRef]
- Wiezorek, J.M.K.; Humphreys, C.J. On the hierarchy of planar fault energies in TiAl. Scr. Metall. Mater. 1995, 33, 451–458. [Google Scholar] [CrossRef]
- Yoo, M.H.; Fu, C.L. Physical constants, deformation twinning, and microcracking of titanium aluminides. Metall. Mater. Trans. A 1998, 29, 49–63. [Google Scholar] [CrossRef]
- Zhang, W.J.; Appel, F. Effect of Al content and Nb addition on the strength and fault energy of TiAl alloys. Mater. Sci. Eng. A 2002, 329–331, 649–652. [Google Scholar] [CrossRef]
- Yuan, Y.; Liu, H.W.; Zhao, X.N.; Meng, X.K.; Liu, Z.G.; Boll, T.; Al-Kassab, T. Dissociation of super-dislocations and the stacking fault energy in TiAl based alloys with Nb-doping. Phys. Lett. A 2006, 358, 231–235. [Google Scholar] [CrossRef]
- Vitek, V.; Ito, K.; Siegl, R.; Znam, S. Structure of interfaces in the lamellar TiAl: Effects of directional bonding and segregation. Mater. Sci. Eng. A 1997, 239, 752–760. [Google Scholar] [CrossRef]
- Ehmann, J.; Fähnle, M. Generalized stacking-fault energies for TiAl: Mechanical instability of the (111) antiphase boundary. Philos. Mag. A 1998, 77, 701–714. [Google Scholar] [CrossRef]
- Liu, Y.L.; Liu, L.M.; Wang, S.Q.; Ye, H.Q. First-principles study of shear deformation in TiAl and Ti3Al. Intermetallics 2007, 15, 428–435. [Google Scholar] [CrossRef]
- Wen, Y.F.; Sun, J. Generalized planar fault energies and mechanical twinning in gamma TiAl alloys. Scr. Mater. 2013, 68, 759–762. [Google Scholar] [CrossRef]
- Kanani, M.; Hartmaier, A.; Janisch, R. Interface properties in lamellar TiAl microstructures from density functional theory. Intermetallics 2014, 54, 154–163. [Google Scholar] [CrossRef]
- Simmons, J.P.; Rao, S.I.; Dimiduk, D.M. Atomistics simulations of structures and properties of 1/2 dislocations using three different embedded-atom method potentials fit to γ-TiAl. Philos. Mag. A 1997, 75, 1299–1328. [Google Scholar] [CrossRef]
- Mahapatra, R.; Girshick, A.; Pope, D.P.; Vitek, V. Deformation mechanisms of near-stoichiometric single phase TiAl single crystals: A combined experimental and atomistic modeling study. Scr. Metall. Mater. 1995, 33, 1921–1927. [Google Scholar] [CrossRef]
- Kanani, M.; Hartmaier, A.; Janisch, R. Stacking fault based analysis of shear mechanisms at interfaces in lamellar TiAl alloys. Acta Mater. 2016, 106, 208–218. [Google Scholar] [CrossRef]
- Woodward, C.; Maclaren, J.M. Planar fault energies and sessile dislocation configurations in substitutionally disordered Ti-Al with Nb and Cr ternary additions. Philos. Mag. A 1996, 74, 337–357. [Google Scholar] [CrossRef]
- Vitek, V. Theory of the Core Structures of Dislocations in Body-Centered-Cubic Metals. Cryst. Lattice Defects 1974, 5, 1–34. [Google Scholar]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B Condens. Matter 1992, 45, 13244–13249. [Google Scholar] [CrossRef] [PubMed]
- Clemens, H.; Mayer, S. Intermetallic titanium aluminides in aerospace applications—Processing, microstructure and properties. Mater. High Temp. 2016, 33, 560–570. [Google Scholar] [CrossRef]
- Jiang, C. First-principles study of site occupancy of dilute 3d, 4d and 5d transition metal solutes in L10 TiAl. Acta Mater. 2008, 56, 6224–6231. [Google Scholar] [CrossRef]
- Holec, D.; Reddy, R.K.; Klein, T.; Clemens, H. Preferential site occupancy of alloying elements in TiAl-based phases. J. Appl. Phys. 2016, 119, 205104. [Google Scholar] [CrossRef]
- Dumitraschkewitz, P. Planar Faults in γ-TiAl: An Atomistic Study. Master’s Thesis, Montanuniversität Leoben, Leoben, Austria, 2015. [Google Scholar]
- Zope, R.R.; Mishin, Y. Interatomic potentials for atomistic simulations of the Ti-Al system. Phys. Rev. B Condens. Matter 2003, 68, 024102. [Google Scholar] [CrossRef]
- Farkas, D.; Jones, C. Interatomic potentials for ternary Nb-Ti-Al alloys. Modell. Simul. Mater. Sci. Eng. 1999, 4, 23. [Google Scholar] [CrossRef]
- Thornton, P.R.; Mitchell, T.E.; Hirsch, P.B. The dependence of cross-slip on stacking-fault energy in face-centered cubic metals and alloys. Philos. Mag. 1962, 7, 1349–1369. [Google Scholar] [CrossRef]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).