Photoelectron Angular Distribution and Phase in Two-Photon Single Ionization of H and He by a Femtosecond and Attosecond Extreme-Ultraviolet Pulse


Abstract
:1. Introduction
2. Gaussian Pulse
2.1. Time-Dependent Perturbation Theory
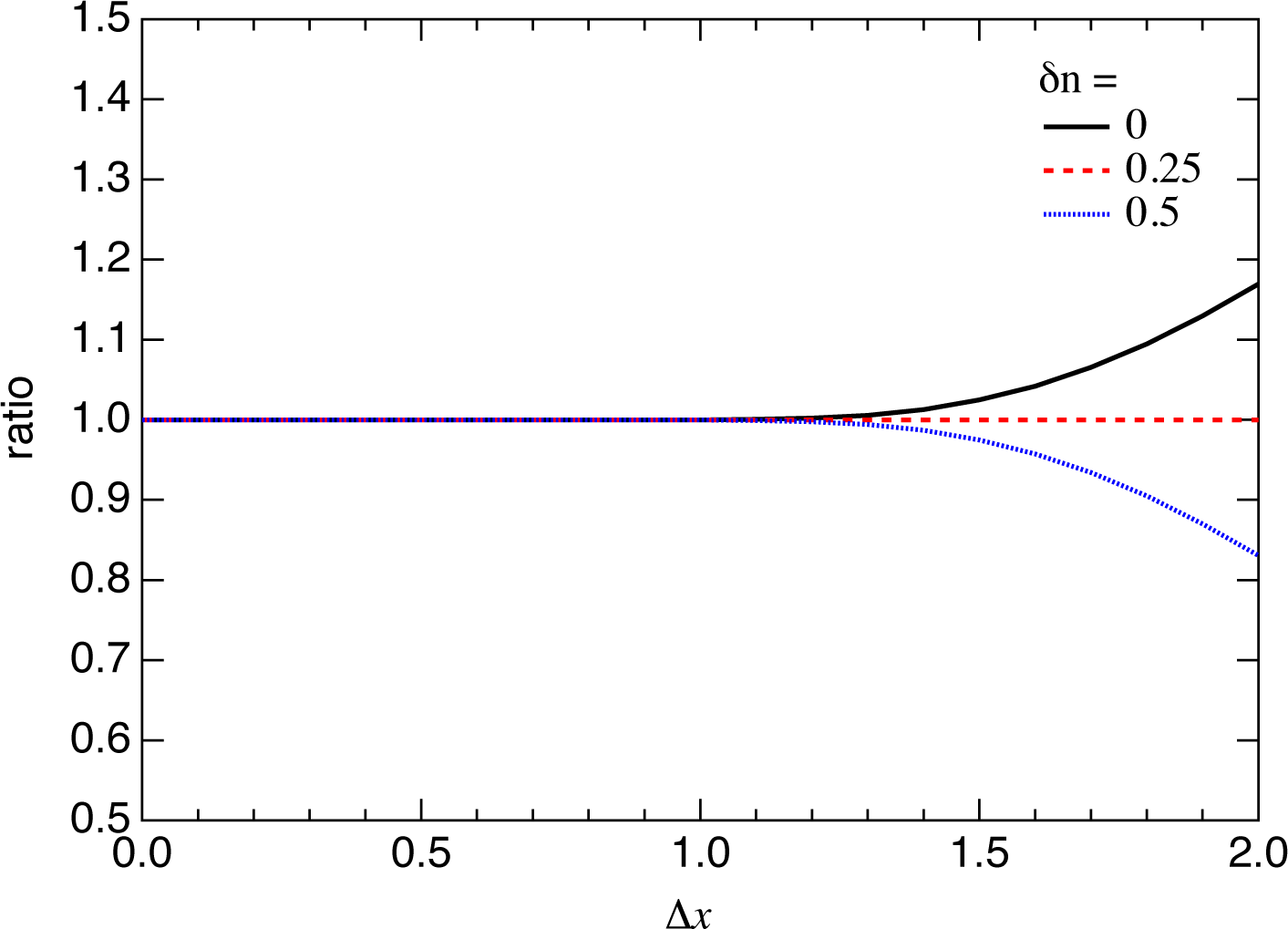
2.2. Photoelectron Angular Distribution
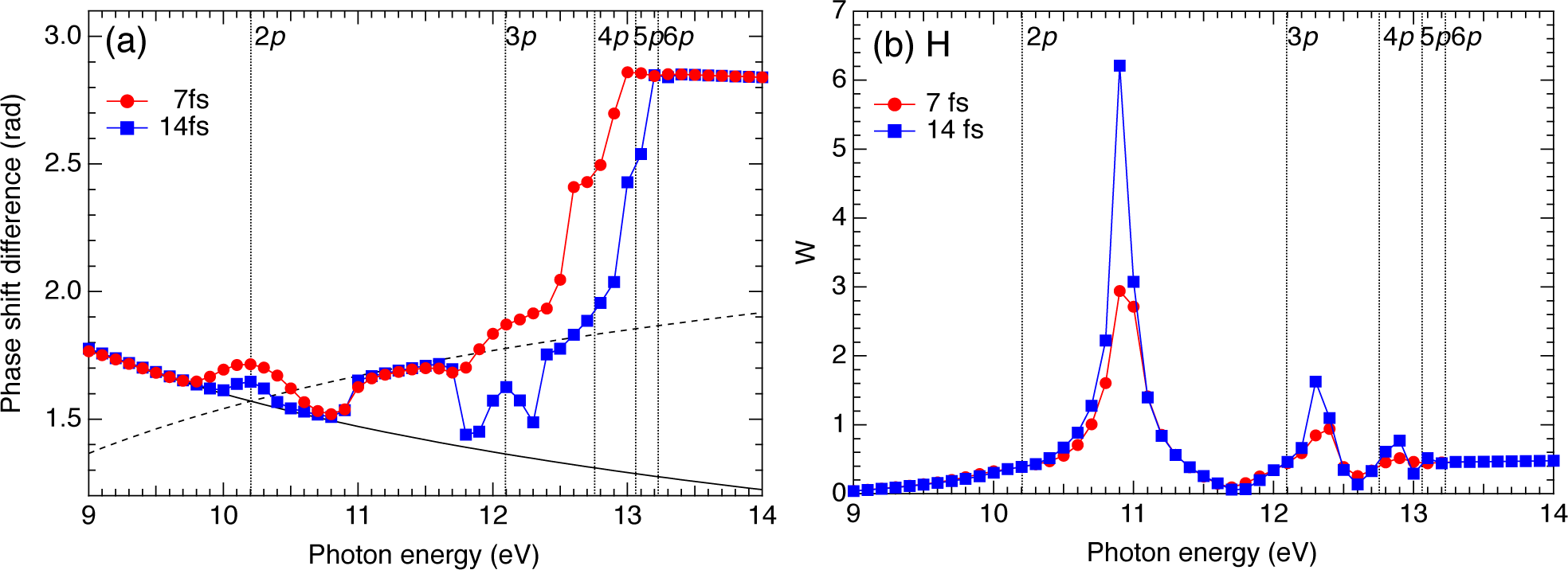
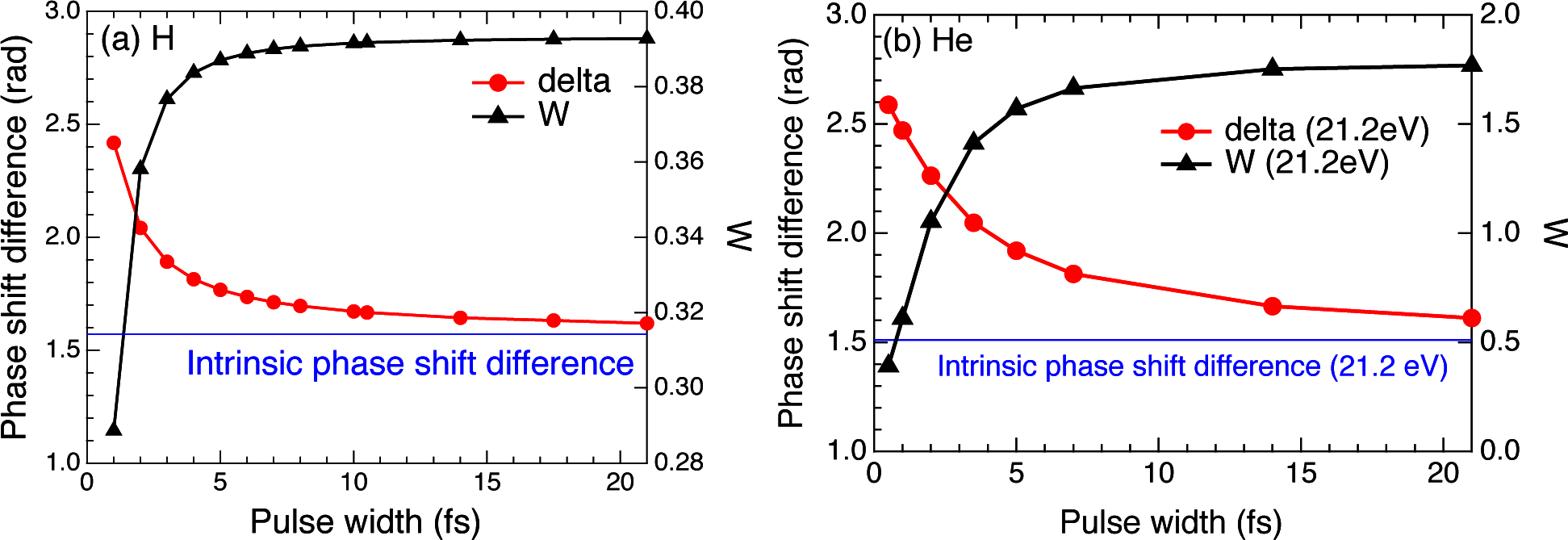
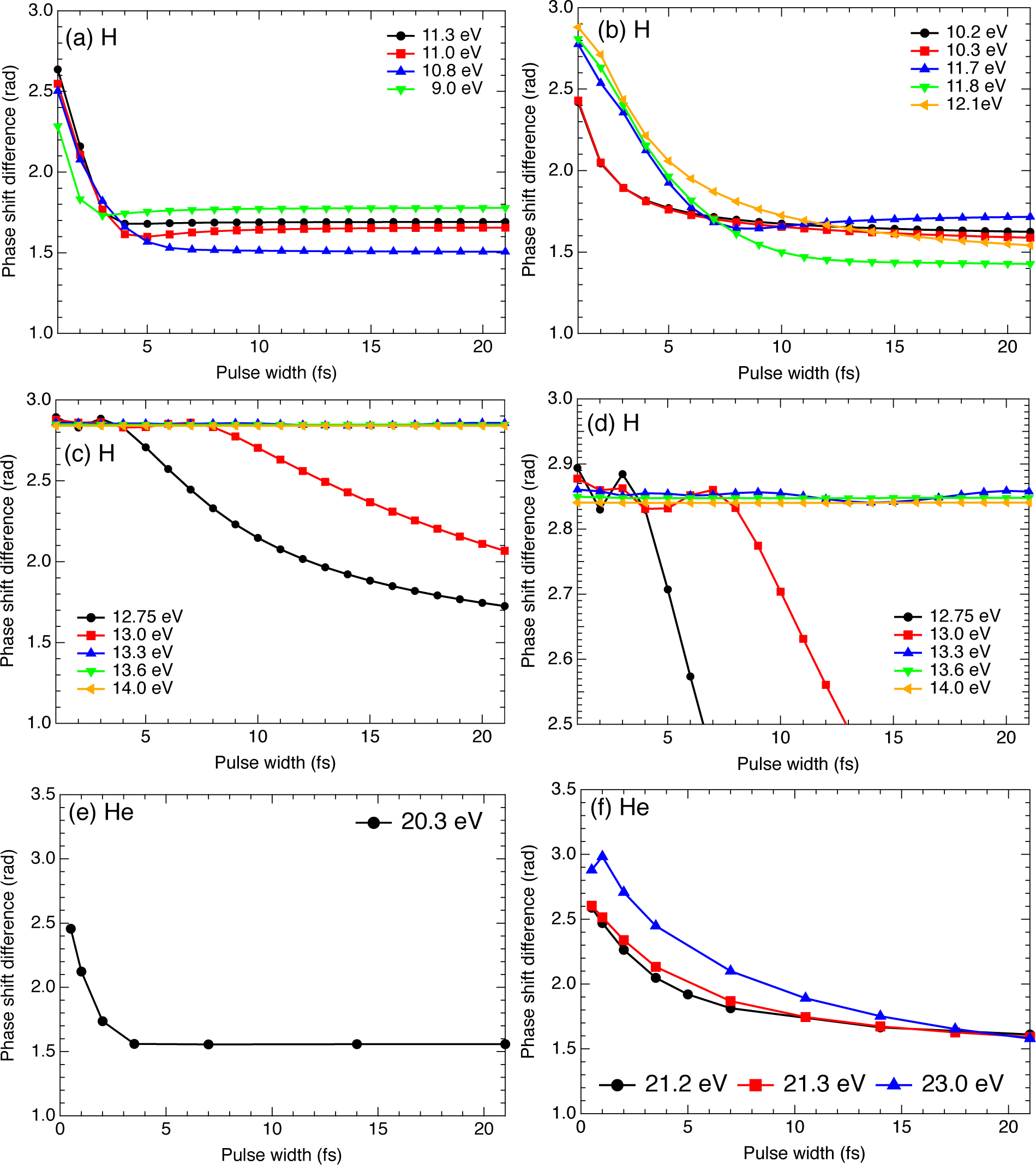
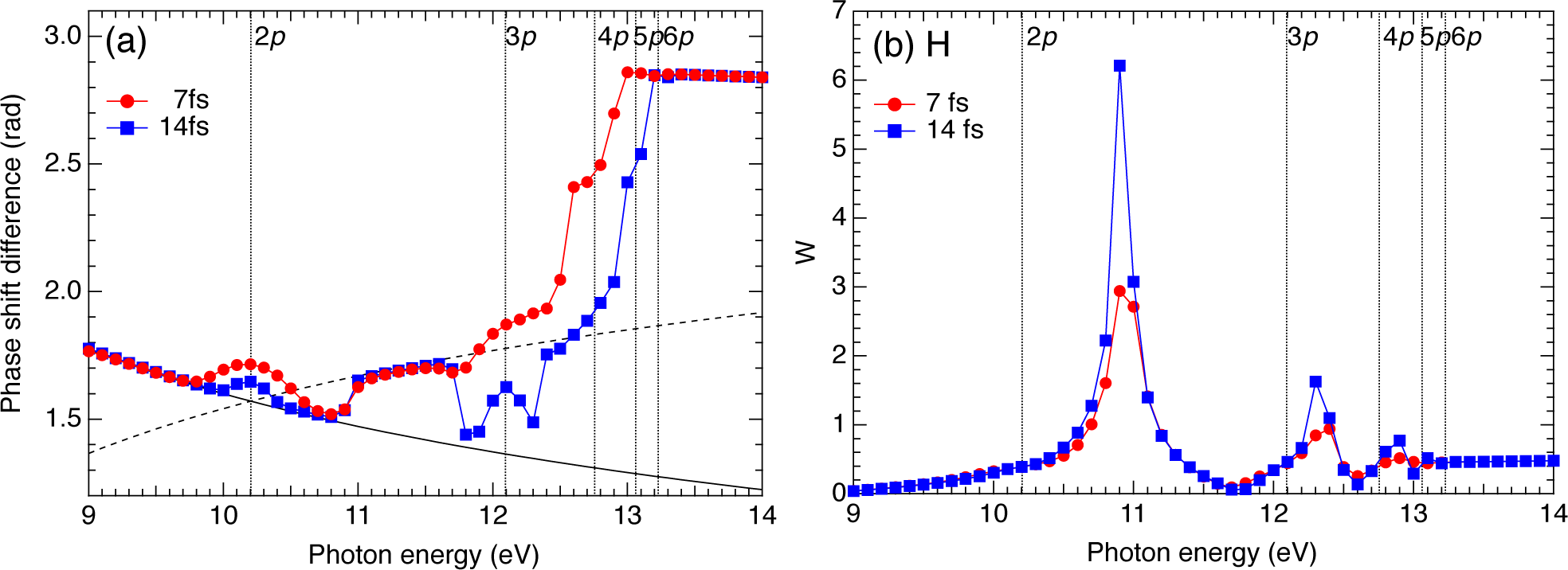
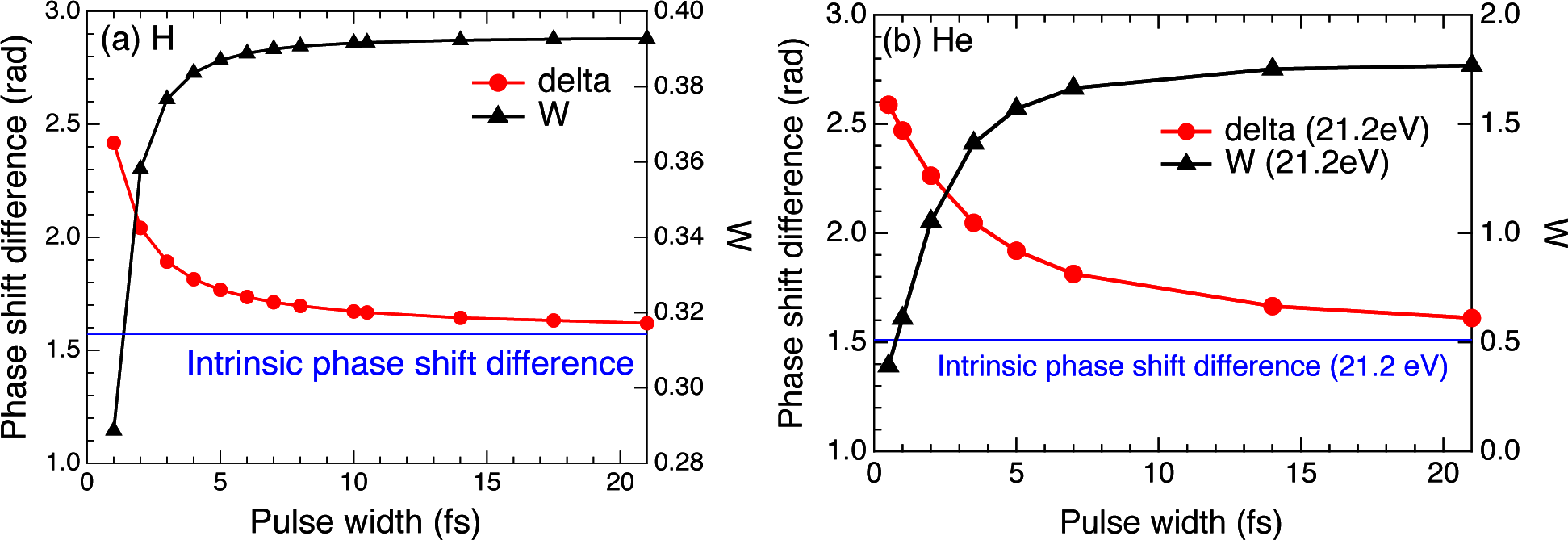
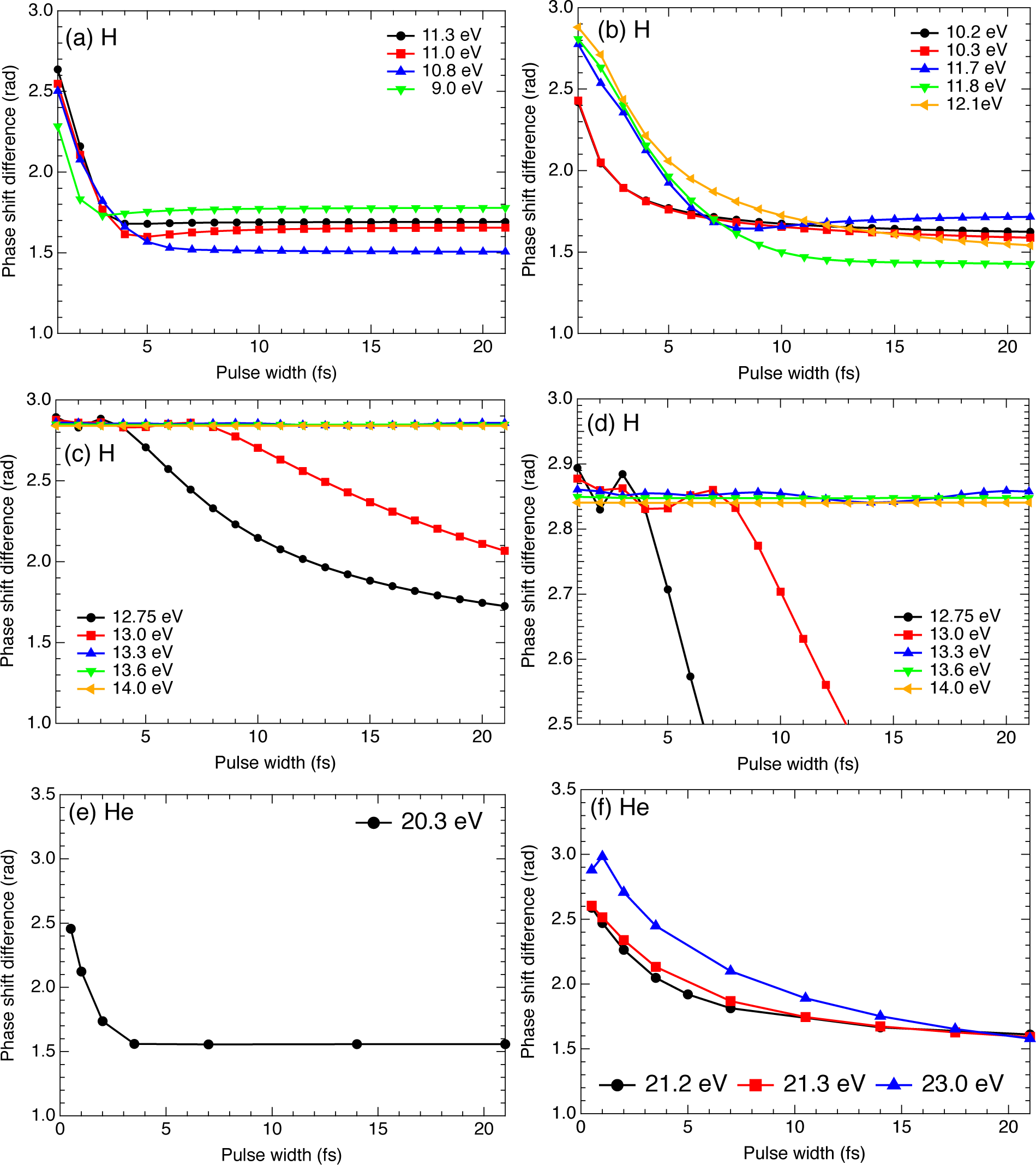
2.3. Hydrogen Atom
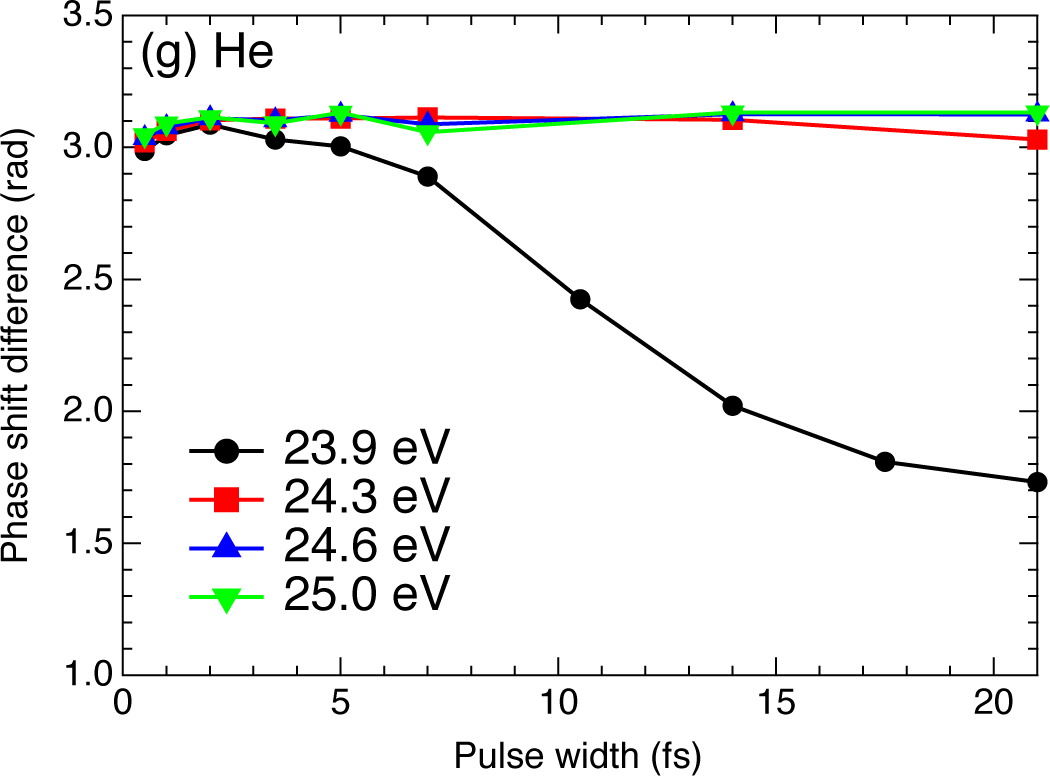
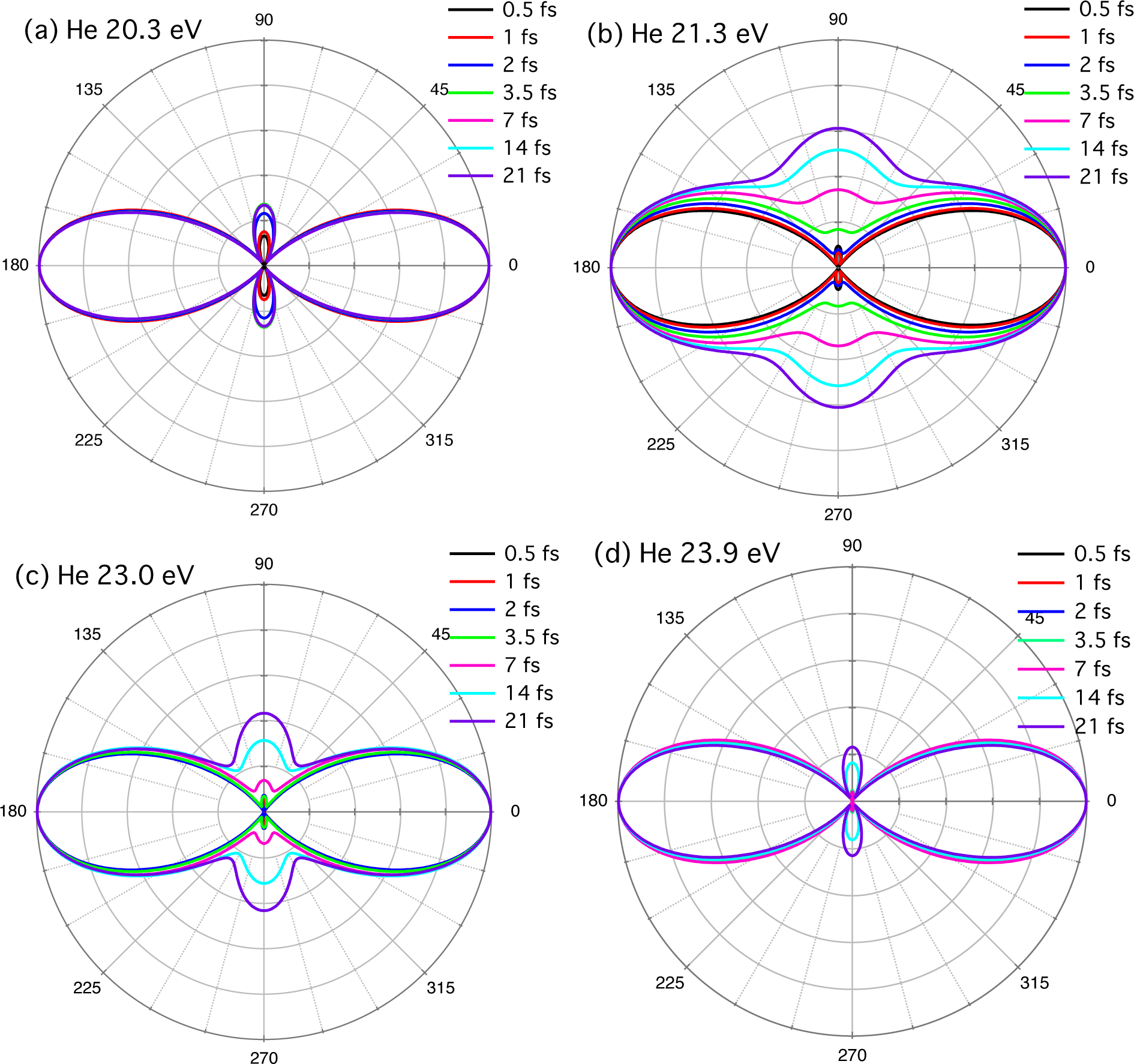
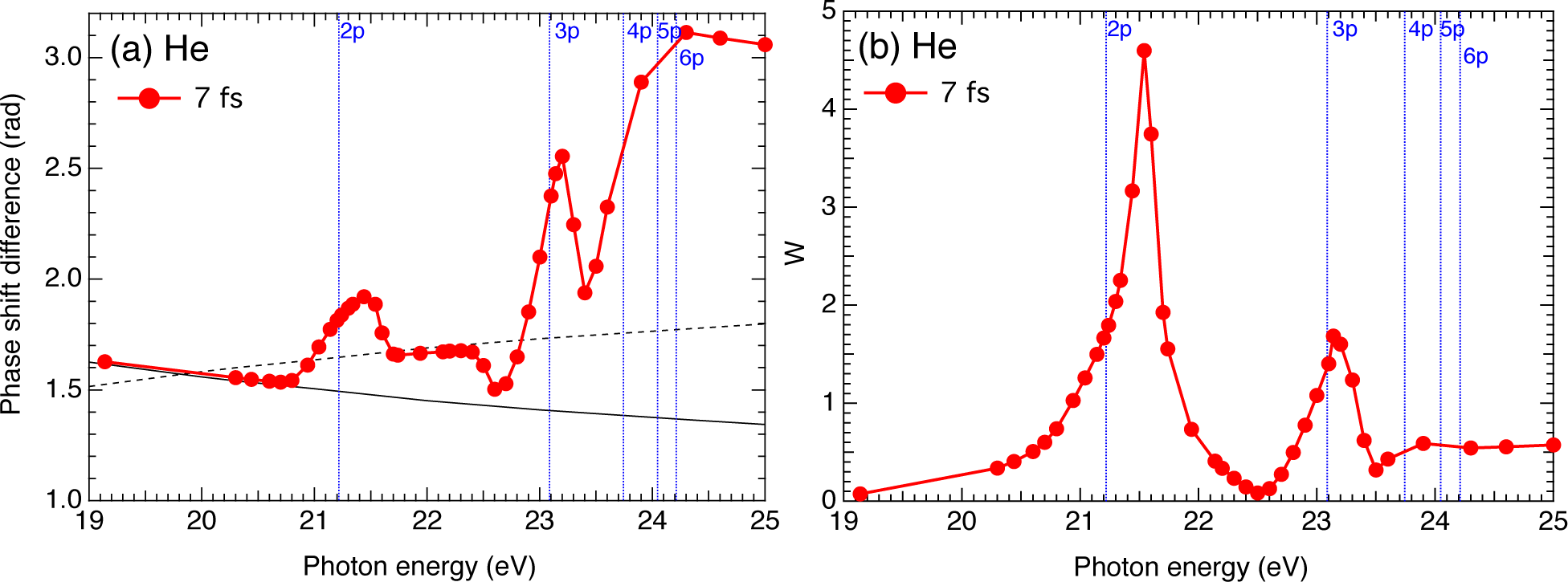
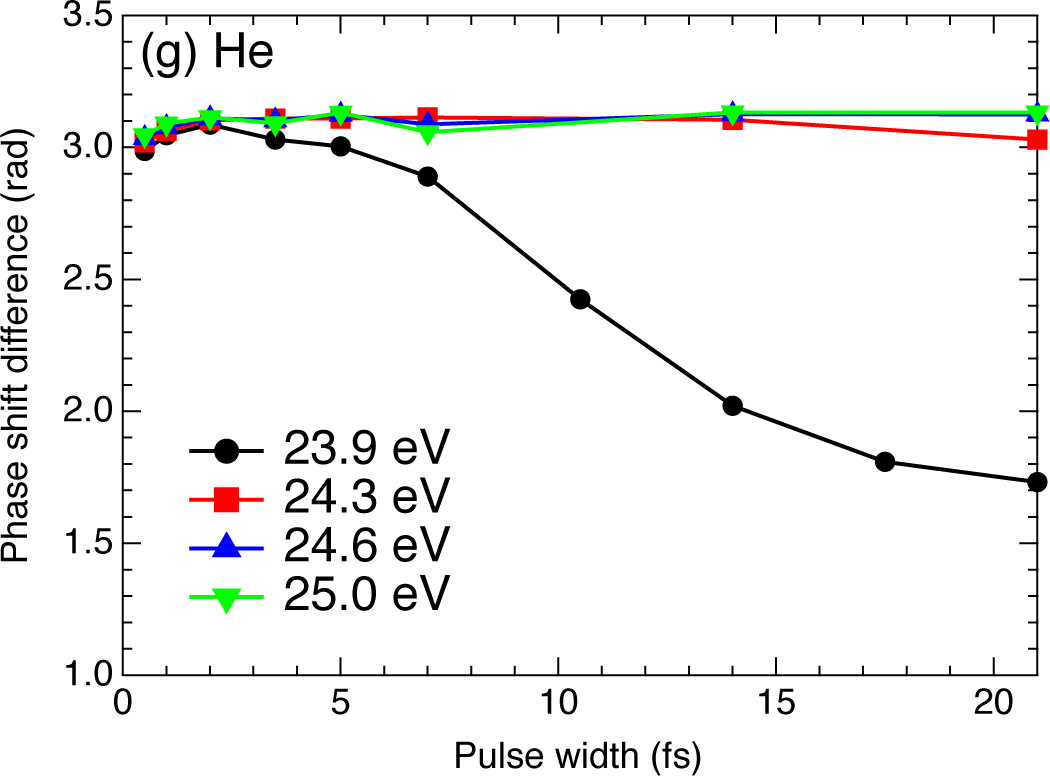
2.4. Helium Atom
3. Pulse Shapes Typical of Experiments
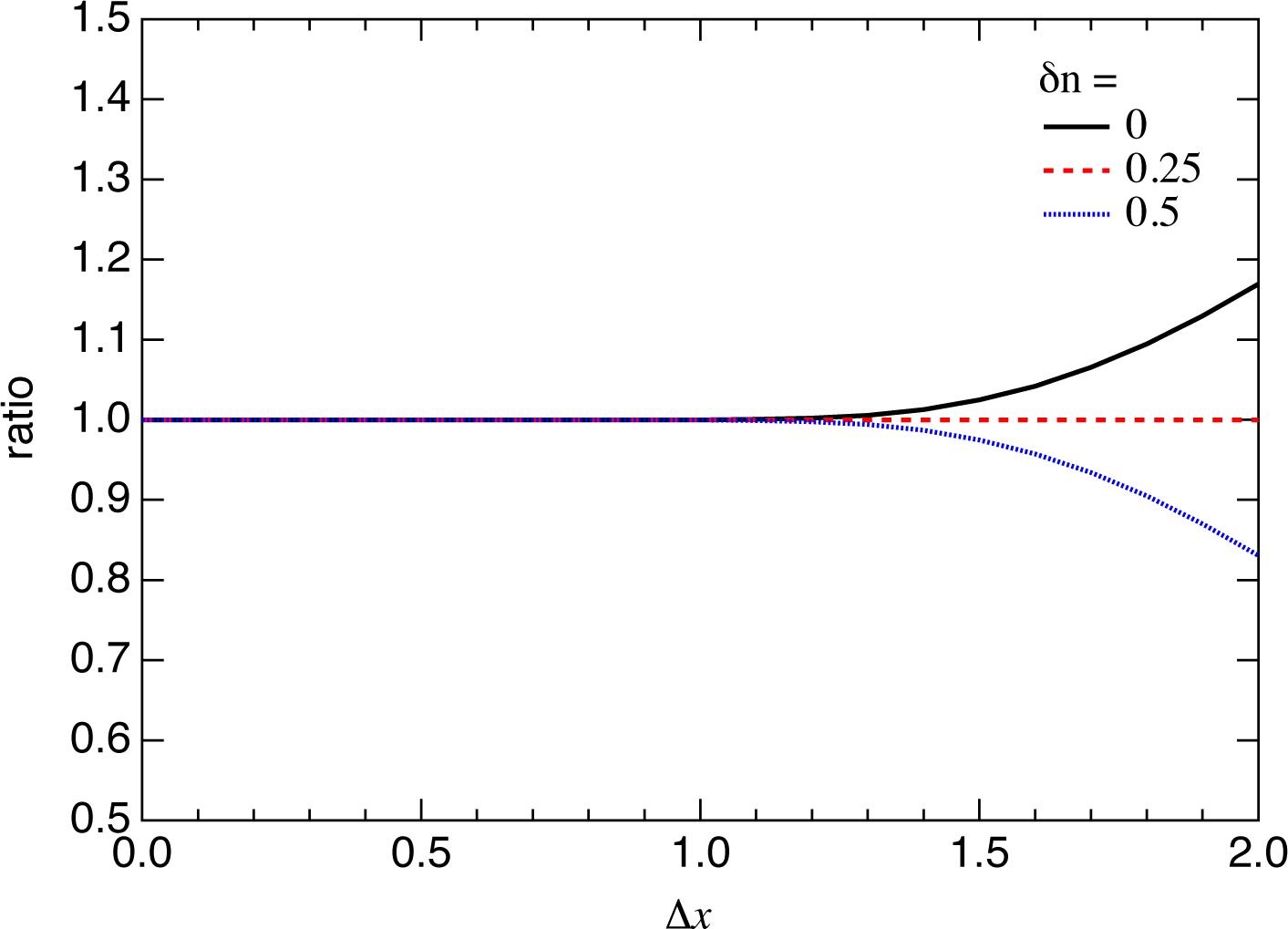
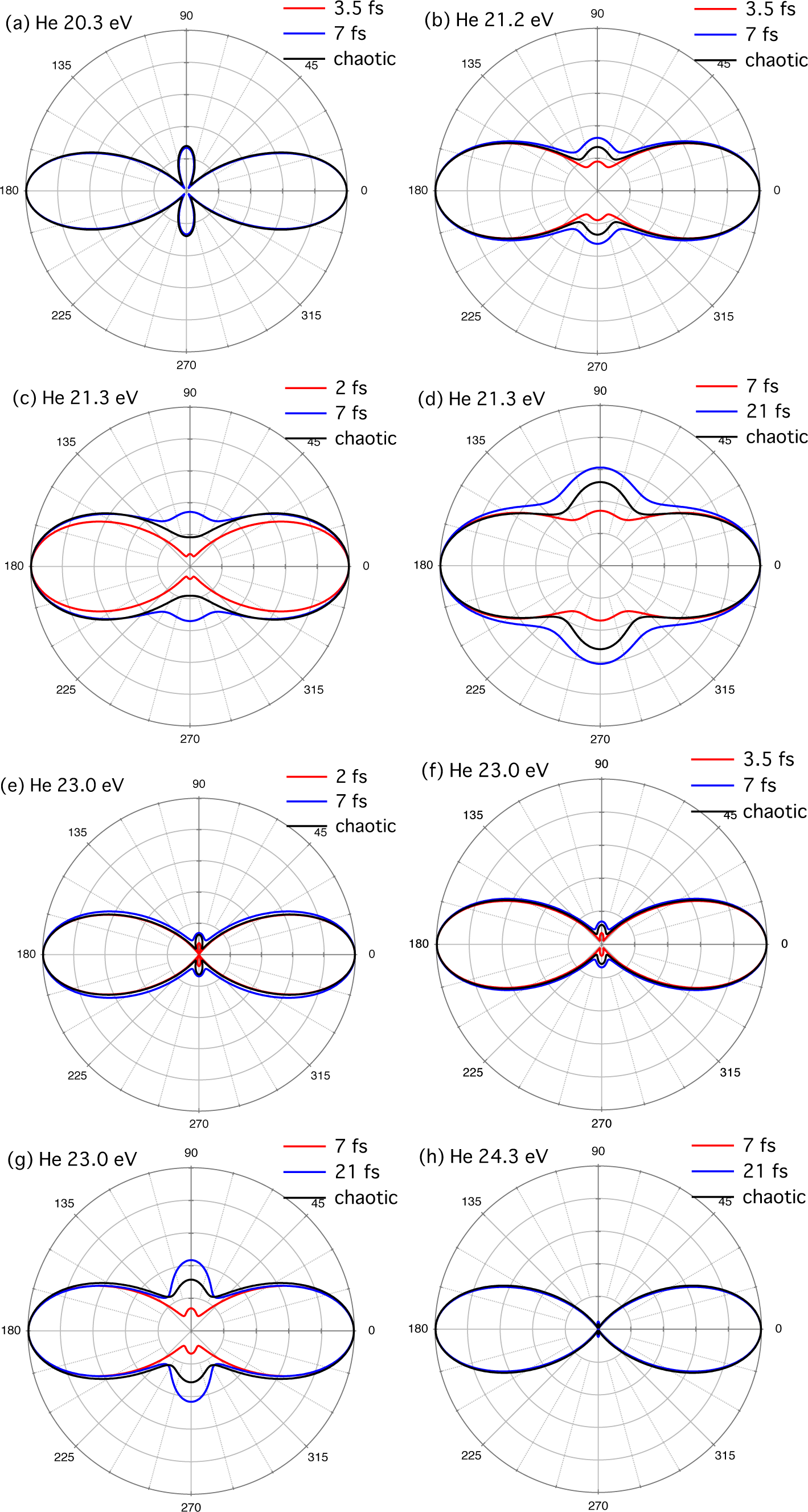
3.1. SASE-FEL Pulses
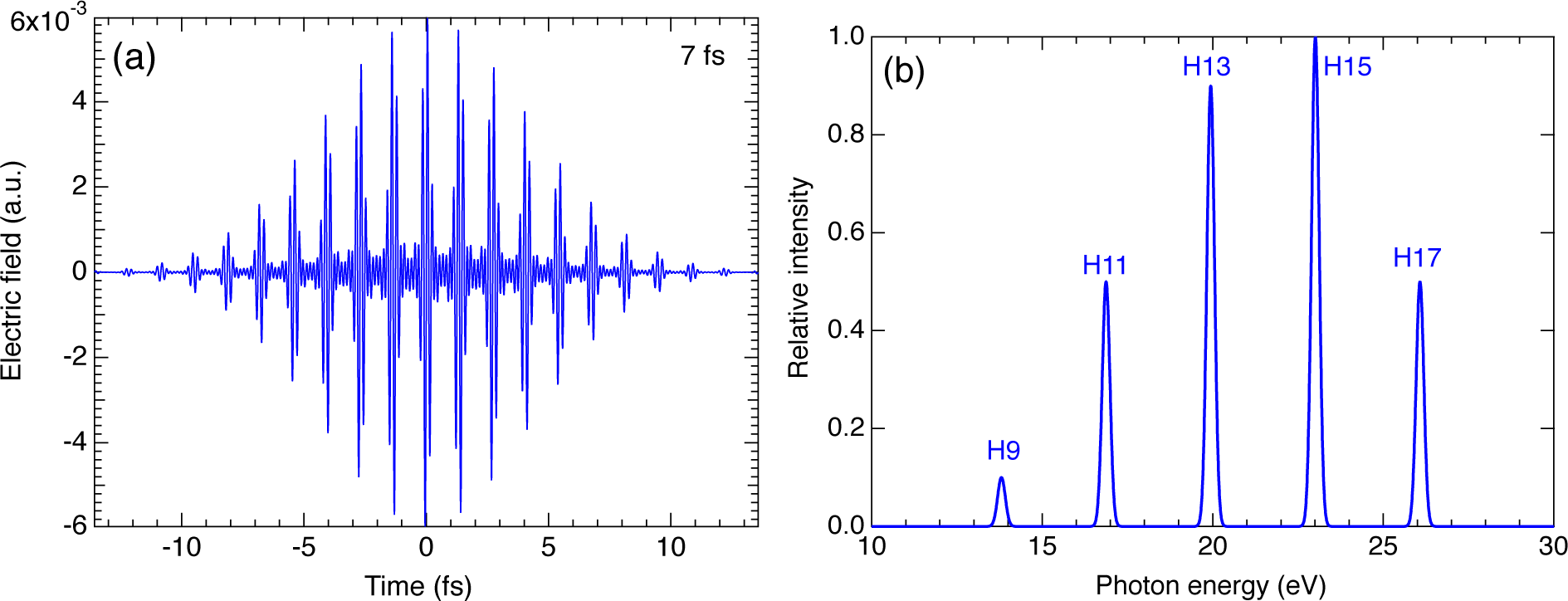
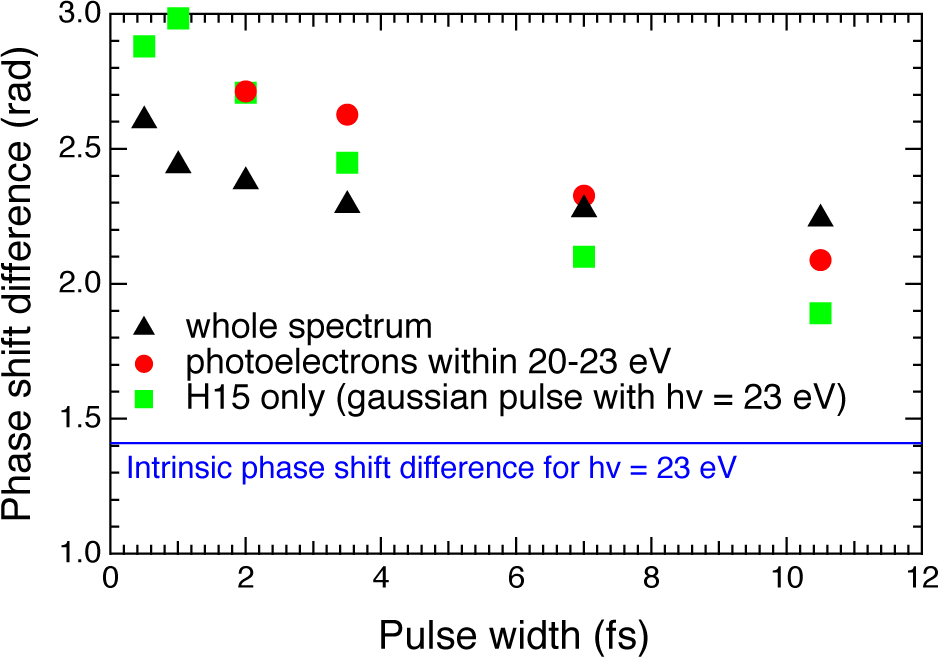
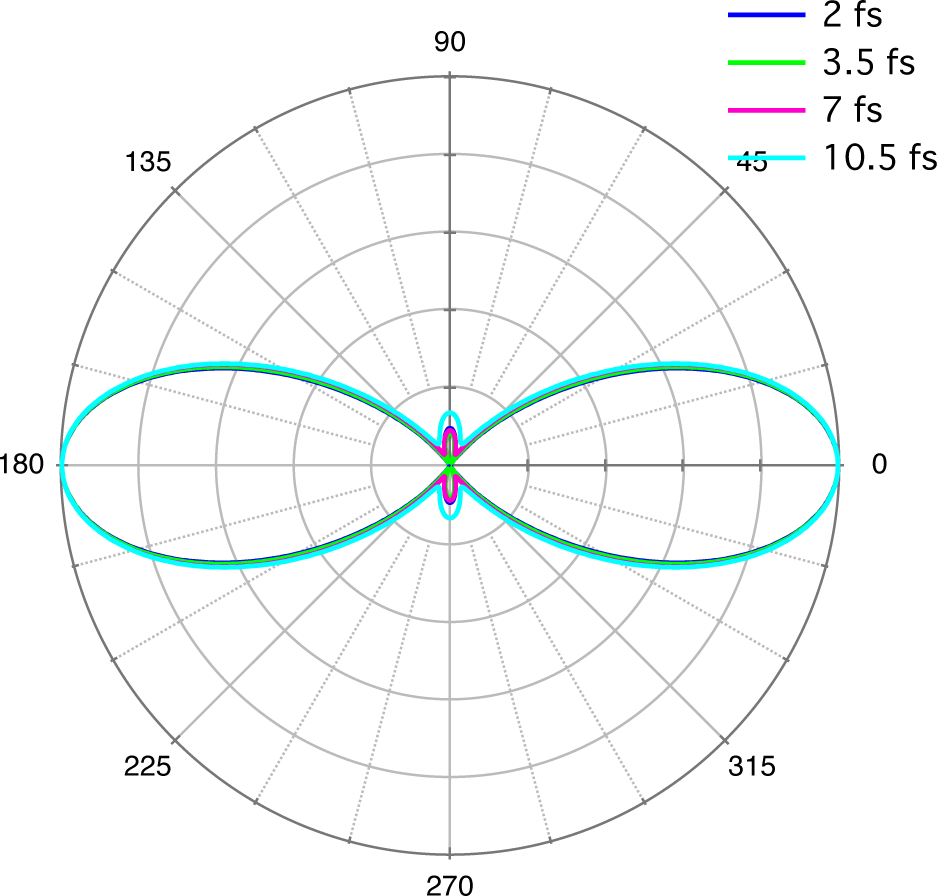
3.2. High-Harmonic Pulses Containing Multiple Harmonic Orders
4. Conclusions
Acknowledgments
References and Note
- Gontier, Y.; Trahin, M. Multiphoton processes in a hydrogen atom. Phys. Rev. A 1971, 4, 1896–1906. [Google Scholar]
- Beers, B.L.; Armstrong, L. Exact solution of a realistic model for two-photon ionization. Phys. Rev. A 1975, 12, 2447–2454. [Google Scholar]
- Crance, M.; Feneuiile, S. Resonant multiphoton ionization induced by pulsed excitation. Phys. Rev. A 1977, 16, 1587–1593. [Google Scholar]
- Andrews, D.L. Resonance two-photon ionisation with ultrashort laser pulses. J. Phys. B 1977, 10, L659–L662. [Google Scholar]
- McClean, W.A.; Swain, S. Theory of resonant two-photon ionisation. J. Phys. B 1978, 11, 1717–1732. [Google Scholar]
- McClean, W.A.; Swain, S. The time dependence of two-photon ionisation. J. Phys. B 1979, 12, 2291–2308. [Google Scholar]
- Jackson, R.I.; O’Brien, D.P.; Swain, S. The photoelectron energy distribution in two-photon ionisation. J. Phys. B 1982, 15, 3385–3403. [Google Scholar]
- Dixit, S.N.; Lambropoulos, P. Theory of photoelectron angular distributions in resonant multiphoton ionization. Phys. Rev. A 1983, 27, 861–874. [Google Scholar]
- Ishikawa, K.; Midorikawa, K. Two-photon ionization of He+ as a nonlinear optical effect in the soft-x-ray region. Phys. Rev. A 2002, 65. [Google Scholar] [CrossRef]
- Selstø, S.; Palacios, A.; Fernandez, J.; Martin, F. Electron angular distribution in resonance-enhanced two-photon ionization of H2+ by ultrashort laser pulses. Phys. Rev. A 2007, 75. [Google Scholar] [CrossRef]
- Varma, H.R.; Ciappina, M.F.; Rohringer, N.; Santra, R. Above-threshold ionization in the X-ray regime. Phys. Rev. A 2009, 80. [Google Scholar] [CrossRef]
- Smith, S.J.; Leuchs, G. Angular correlation in multiphoton ionization of atoms. Adv. At. Mol. Phys 1988, 24, 157–221. [Google Scholar]
- Lambropoulos, P.; xMaragikis, G.; Zhang, J. Two-electron atoms in strong fields. Phys. Rep 1998, 305, 203–293. [Google Scholar]
- Wang, Z.-M.; Elliott, D.S. Determination of the phase difference between even and odd continuum wave functions in atoms through quantum interference measurements. Phys. Rev. Lett 2001, 87. [Google Scholar] [CrossRef]
- Reid, K.L. Photoelectron angular distributions. Annu. Rev. Chem 2003, 54, 397–424. [Google Scholar]
- Kabachnik, N.M.; Fritzsche, S.; Grum-Grzhimailo, A.N.; Meyer, M.; Ueda, K. Coherence and correlations in photoinduced Auger and fluorescence cascades in atoms. Phys. Rep 2007, 451, 155–233. [Google Scholar]
- Ackermann, W.; Asova, G.; Ayvazyan, V.; Azima, A.; Baboi, N.; Bhr, J.; Balandin, V.; Beutner, B.; Brandt, A.; Bolzmann, A.; et al. Operation of a free-electron laser from the extreme ultraviolet to the water window. Nat. Photon 2007, 1, 336–342. [Google Scholar]
- Shintake, T.; Tanaka, H.; Hara, T.; Tanaka, T.; Togawa, K.; Yabashi, M.; Otake, Y.; Asano, Y.; Bizen, T.; Fukui, T.; et al. A compact free-electron laser for generating coherent radiation in the extreme ultraviolet region. Nat. Photon 2008, 2, 555–559. [Google Scholar]
- Emma, P.; Akre, R.; Arthur, J.; Bionta, R.; Bostedt, C.; Bozek, J.; Brachmann, A.; Bucksbaum, P.; Coffee, R.; Decker, F.-J.; et al. First lasing and operation of an ångstrom-wavelength free-electron laser. Nat. Photon 2010, 4, 641–647. [Google Scholar]
- Ishikawa, T.; Aoyagi, H.; Asaka, T.; Asano, Y.; Azumi, N.; Bizen, T.; Ego, H.; Fukami, K.; Fukui, T.; Furukawa, Y.; et al. A compact X-ray free-electron laser emitting in the sub-ångström region. Nat. Photon 2012, 6, 540–544. [Google Scholar]
- Nagasono, M.; Suljoti, E.; Pietzsch, A.; Hennies, F.; Wellhöfer, M.; Hoeft, J.-T.; Martins, M.; Wurth, W.; Treusch, R.; Feldhaus, J.; Schneider, J.R.; Föhlisch, A. Resonant two-photon absorption of extreme-ultraviolet free-electron-laser radiation in helium. Phys. Rev. A 2007, 75, 051406(R)-1–051406(R)-4. [Google Scholar]
- Santra, R.; Kryzhevoi, N. V.; Cederbaum, L. S. X-ray two-photon photoelectron spectroscopy: A theoretical study of inner-shell spectra of the organic para-aminophenol molecule. Phys. Rev. Lett 2009, 103, 013002-1–013002-4. [Google Scholar]
- Young, L.; Kanter, E. P.; Krässig, B.; Li, Y.; March, A. M.; Pratt, S. T.; Santra, R.; Southworth, S. H.; Rohringer, N.; DiMauro, L. F.; et al. Femtosecond electronic response of atoms to ultra-intense X-rays. Nat. (Lond.) 2010, 466, 56–61. [Google Scholar]
- Cryan, J.P.; Glownia, J. M.; Andreasson, J.; Belkacem, A.; Berrah, N.; Blaga, C. I.; Bostedt, C.; Bozek, J.; Buth, C.; DiMauro, L. F.; et al. Auger electron angular distribution of double core-hole states in the molecular reference frame. Phys. Rev. Lett 2010, 105, 083004-1–083004-5. [Google Scholar]
- Fang, L.; Hoener, M.; Gessner, O.; Tarantelli, F.; Pratt, S.T.; Kornilov, O.; Buth, C.; Gühr, M.; Kanter, E.P.; Bostedt, C.; et al. Double core-hole production in N2: Beating the Auger clock. Phys. Rev. Lett 2010, 105, 083005-1–083005-5. [Google Scholar]
- Berrah, N.; Fang, L.; Murphy, B.; Osipov, T.; Ueda, K.; Kukk, E.; Feifel, R.; van der Meulen, P.; Salen, P.; Schmidt, H.T.; et al. Double-core-hole spectroscopy for chemical analysis with an intense X-ray femtosecond laser. Proc. Nat. Acad. Sci. USA 2011, 108, 16912–16915. [Google Scholar]
- Doumy, G.; Roedig, C.; Son, S.-K.; Blaga, C. I.; DiChiara, A. D.; Santra, R.; Berrah, N.; Bostedt, C.; Bozek, J. D.; Bucksbaum, P. H.; et al. Nonlinear atomic response to intense ultrashort X rays. Phys. Rev. Lett 2011, 106, 083002-1–083002-4. [Google Scholar]
- Salén, P.; van der Meulen, P.; Schmidt, H. T.; Thomas, R. D.; Larsson, M.; Feifel, R.; Piancastelli, M. N.; Fang, L.; Murphy, B.; Osipov, T.; et al. Experimental verification of the chemical sensitivity of two-site double core-hole states formed by an X-ray free-electron laser. Phys. Rev. Lett 2012, 108, 153003-1–153003-5. [Google Scholar]
- Haber, L.H.; Doughty, B.; Leone, S.R. Continuum phase shifts and partial cross sections for photoionization from excited states of atomic helium measured by high-order harmonic optical pump-probe velocity map imaging. Phys. Rev. A 2009, 79, 031401(R)-1–031401(R)-4. [Google Scholar]
- Rouzée, A.; Johnsson, P.; Gryzlova, E.V.; Fukuzawa, H.; Yamada, A.; Siu, W.; Huismans, Y.; Louis, E.; Bijkerk, F.; Holland, D.M.P.; et al. Angle-resolved photoelectron spectroscopy of sequential three-photon triple ionization of neon at 90.5 eV photon energy. Phys. Rev. A 2011, 83, 031401(R)-1–031401(R)-4. [Google Scholar]
- Hasegawa, H.; Takahashi, E.J.; Nabekawa, Y.; Ishikawa, K.L.; Midorikawa, K. Multiphoton ionization of He by using intense high-order harmonics in the soft-X-ray region. Phys. Rev. A 2005, 71, 023407-1–023407-5. [Google Scholar]
- Kobayashi, Y.; Sekikawa, T.; Nabekawa, Y.; Watanabe, S. 27-fs extreme ultraviolet pulse generation by high-order harmonics. Opt. Lett 1998, 23, 64–66. [Google Scholar]
- Mitzner, R.; Sorokin, A.A.; Siemer, B.; Roling, S.; Rutkowski, M.; Zacharias, H.; Neeb, M.; Noll, T.; Siewert, F.; Eberhardt, W.; et al. Direct autocorrelation of soft-X-ray free-electron-laser pulses by time-resolved two-photon double ionization of He. Phys. Rev. A 2009, 80, 025402-1–025402-4. [Google Scholar]
- Sorgenfrei, F.; Schlotter, W.F.; Beeck, T.; Nagasono, M.; Gieschen, S.; Meyer, H.; Föhlisch, A.; Beye, M.; Wurth, W. The extreme ultraviolet split and femtosecond delay unit at the plane grating monochromator beamline PG2 at FLASH. Rev. Sci. Instrum 2010, 81, 043107-1–043107-7. [Google Scholar]
- Moshammer, R.; Pfeifer, Th.; Rudenko, A.; Jiang, Y.H.; Foucar, L.; Kurka, M.; Kühnel, K.U.; Schröter, C.D.; Ullrich, J.; Herrwerth, O.; et al. Second-order autocorrelation of XUV FEL pulses via time resolved two-photon single ionization of He. Opt. Exp 2011, 19, 21698–21706. [Google Scholar]
- Nabekawa, Y.; Shimizu, T.; Okino, T.; Furusawa, K.; Hasegawa, H.; Yamanouchi, K.; Midorikawa, K. Interferometric autocorrelation of an attosecond pulse train in the single-cycle regime. Phys. Rev. Lett 2006, 97, 153904-1–153904-4. [Google Scholar]
- Nikolopoulos, L.A.A.; Lambropoulos, P. Multichannel theory of two-photon single and double ionization of helium. J. Phys. B 2001, 34, 545–564. [Google Scholar]
- Van der Hart, H.W.; Bingham, P. Two- and three-photon ionization of He between 1013 and 1014 Wcm−2. J. Phys. B 2005, 38, 207–221. [Google Scholar]
- Sato, T.; Iwasaki, A.; Ishibashi, K.; Okino, T.; Yamanouchi, K.; Adachi, J.; Yagishita, A.; Yazawa, H.; Kannari, F.; Aoyama, M.; et al. Determination of the absolute two-photon ionization cross section of He by an XUV free electron laser. J. Phys. B 2011, 44, 161001-1–161001-5. [Google Scholar]
- Hishikawa, A.; Hishikawa, A.; Fushitani, M.; Hikosaka, Y.; Matsuda, A.; Liu, C.-N.; Morishita, T.; Shigemasa, E.; Nagasono, M.; Tono, K.; et al. Enhanced nonlinear double excitation of He in intense extreme ultraviolet laser fields. Phys. Rev. Lett 2011, 107, 243003-1–243003-5. [Google Scholar]
- Guyétand, O.; Gisselbrecht, M.; Huetz, A.; Agostini, P.; Taïeb, R.; Véniard, V.; Maquet, A.; Antonucci, L.; Boyko, O.; Valentin, C.; Douillet, D. Multicolour above-threshold ionization of helium: quantum interference effects in angular distributions. J. Phys. B 2005, 35, L357–L363. [Google Scholar]
- Haber, L.H.; Doughty, B.; Leone, S.R. Energy-dependent photoelectron angular distributions of two-color two-photon above threshold ionization of atomic helium. Phys. Rev. A 2011, 84, 013416-1–013416-9. [Google Scholar]
- O’Keeffe, P.; Bolognesi, P.; Richter, R.; Moise, A.; Ovcharenko, E.; Pravica, L.; Sergo, R.; Stebel, L.; Cautero, G.; Avaldi, L. Photoelectron imaging in pump-probe experiments combining synchrotron and laser radiation. J. Phys 2010, 235, 012006-1–012006-8. [Google Scholar]
- Ishikawa, K.L.; Ueda, K. Competition of resonant and nonresonant paths in resonance-enhanced two-photon single ionization of He by an ultrashort extreme-ultraviolet pulse. Phys. Rev. Lett 2012, 108. [Google Scholar] [CrossRef]
- Dudovich, N.; Dayan, B.; Gallagher Faeder, S.M.; Silberberg, Y. Transform-limited pulses are not optimal for resonant multiphoton transitions. Phys. Rev. Lett 2001, 86, 47–50. [Google Scholar]
- Wollenhapt, M.; Klug, M.; Köhler, J.; Bayer, T.; Sarpe-Tudoran, C.; Baumert, T. Photoelectron angular distributions from strong-field coherent electronic excitation. Appl. Phys. B 2009, 95, 245–259. [Google Scholar]
- Reid, K.L. Photoelectron angular distributions: Developments in applications to isolated molecular systems. Mol. Phys 2012, 110, 131–147. [Google Scholar]
- NIST: Atomic Spectra Database—Energy Levels Form. Available online: http://physics.nist.gov/PhysRefData/ASD/levels_form.html accessed on 28 February 2013.
- FERMI HomePage. Available online: http://www.elettra.trieste.it/FERMI/ accessed on 28 February 2013.
- Pindzola, M.S.; Robicheaux, F. Time-dependent close-coupling calculations of correlated photoionization processes in helium. Phys. Rev. A 1998, 57, 318–324. [Google Scholar]
- Pindzola, M.S.; Robicheaux, F. Two-photon double ionization of He and H−. J. Phys. B 1998, 31, L823–L831. [Google Scholar]
- Colgan, J.; Pindzola, M.S.; Robicheaux, F. Fully quantal (γ, 2e) calculations for absolute differential cross sections of helium. J. Phys. B 2001, 34, L457–L466. [Google Scholar]
- Parker, J.S.; Moore, L. R.; Meharg, K.J.; Dundas, D.; Taylor, K. T. Double-electron above threshold ionization of helium. J. Phys. B 2001, 34, L69–L78. [Google Scholar]
- Ishikawa, K.L.; Midorikawa, K. Above-threshold double ionization of helium with attosecond intense soft x-ray pulses. Phys. Rev. A 2005, 72, 013407-1–013407-8. [Google Scholar]
- Mitzner, R.; Siemer, B.; Neeb, M.; Noll, T.; Siewert, F.; Roling, S.; Rutkowski, M.; Sorokin, A. A.; Richter, M.; Juranic, P.; et al. Spatio-temporal coherence of free electron laser pulses in the soft X-ray regime. Opt. Exp 2008, 16, 19909–19919. [Google Scholar]
- Schlotter, W.F.; Sorgenfrei, F.; Beeck, T.; Beye, M.; Gieschen, S.; Meyer, H.; Nagasono, M.; Fohlisch, A.; Wurth, W. Longitudinal coherence measurements of an extreme-ultraviolet free-electron laser. Opt. Lett 2010, 35, 372–374. [Google Scholar]
- Bonifacio, R.; de Salvo, L.; Pierini, P.; Piovella, N.; Pellegrini, C. Longitudinal coherence measurements of an extreme-ultraviolet free-electron laser. Phys. Rev. Lett 1994, 73, 70–73. [Google Scholar]
- Saldin, E.L.; Schneidmiller, E.A.; Yurkov, M.V. Statistical properties of radiation from VUV and X-ray free electron laser. Opt. Commun 1998, 148, 383–403. [Google Scholar]
- Krinsky, S.; Gluckstern, R.L. Analysis of statistical correlations and intensity spiking in the self-amplified spontaneous-emission free-electron laser. Phys. Rev. ST Accel. Beams 2003, 6, 050701-1–050701-10. [Google Scholar]
- NIST Handbook of Mathematical Functions; Olver, F.W.J.; Lozier, D.W.; Boisvert, R.F.; Clark, C.W. (Eds.) Cambridge: New York, NY, USA, 2010.
- Oza, D.H. Phase shifts and resonances for electron scattering by He+ below the N = 2 threshold. Phys. Rev. A 1986, 33, 824–838. [Google Scholar]
- Chang, T.N.; Fang,, T.K. Effect of positive-energy orbitals on the photoionization cross-sections and oscillator-strengths of He and divalent atoms. Phys. Rev. A 1995, 52, 2638–2644. [Google Scholar]
- Gien, T.T. Accurate calculation of phase shifts for electron-He+ collisions. J. Phys. B 2002, 35, 4475–4490. [Google Scholar]
- W and δ are related to X and ∆ in [29] as and δ = ∆.
- Kulander, K.C.; Schafer, K.J.; Krause, J.L. Time-Dependent Studies of Multiphoton Processes. In Atoms in Intense Laser Fields; Gavrila, M., Ed.; Academic: New York, NY, USA, 1992; Volume 247. [Google Scholar]
- Ishikawa, K.; Midorikawa, K. Coherent control of extreme uv absorption and photoemission by the simultaneous irradiation of ultrashort extreme uv and laser pulses. Phys. Rev. A 2002, 65, 031403(R)-1–031403(R)-4. [Google Scholar]
- Ishikawa, K. Photoemission and ionization of He+ under simultaneous irradiation of fundamental laser and high-order harmonic pulses. Phys. Rev. Lett 2003, 91, 043002-1–043002-4. [Google Scholar]
- Ishikawa, K.L. Temporal Young’s interference experiment by attosecond double and triple soft-x-ray pulses. Phys. Rev. A 2006, 74, 023806-1–023806-4. [Google Scholar]
- Ishikawa, K.L.; Takahashi, E.J.; Midorikawa, K. Single-attosecond pulse generation using a seed harmonic pulse train. Phys. Rev. A 2007, 75, 021801(R)-1–021801(R)-4. [Google Scholar]
- Schiessl, K.; Ishikawa, K.L.; Persson, E.; Burgdörfer, J. Quantum path interference in the wavelength dependence of high-harmonic generation. Phys. Rev. Lett 2007, 99, 253903-1–253903-4. [Google Scholar]
- Ishikawa, K.L.; Takahashi, E.J.; Midorikawa, K. Wavelength dependence of high-order harmonic generation with independently controlled ionization and ponderomotive energy. Phys. Rev. A 2009, 80, 011807(R)-1–011807(R)-4. [Google Scholar]
- Arbo, D.G.; Ishikawa, K.L.; Schiessl, K.; Persson, E.; Burgdörfer, J. Intracycle and intercycle interferences in above-threshold ionization: The time grating. Phys. Rev. A 2010, 81, 021403(R)-1–021403(R)-4. [Google Scholar]
- Ishikawa, K.L.; Kawazura, Y.; Ueda, K. Two-photon ionization of atoms by ultrashort laser pulses. J. Mod. Opt 2010, 57, 999–1007. [Google Scholar]
- Fano, U.; Cooper, J.W. Spectral distribution of atomic oscillator strengths. Rev. Mod. Phys 1968, 40, 441–507. [Google Scholar]
- Schultze, M.; Fieß, M.; Karpowicz, N.; Gagnon, J.; Korbman, M.; Hofstetter, M.; Neppl, S.; Cavalieri, A.L.; Komninos, Y.; Mercouris, Th.; et al. Delay in photoemission. Science 2010, 328, 1658–1662. [Google Scholar]
- Klünder, K.; Dahlström, J.M.; Gisselbrecht, M.; Fordell, T.; Swoboda, M.; Guénot, D.; Johnsson, P.; Caillat, J.; Mauritsson, J.; Maquet, A.; et al. Probing single-photon ionization on the attosecond time scale. Phys. Rev. Lett 2011, 106, 143002-1–143002-4. [Google Scholar]
- Swoboda, M.; Fordell, T.; Klünder, K.; Dahlström, J.M.; Miranda, M.; Buth, C.; Schafer, K.J.; Mauritsson, J.; L’Huillier, A.; Gisselbrecht, M. Phase measurement of resonant two-photon ionization in helium. Phys. Rev. Lett 2010, 104, 103003-1–103003-4. [Google Scholar]
- Dahlström, J.M.; L’Huillier, A.; Maquet, A. Introduction to attosecond delays in photoionization. J. Phys. B 2012, 45, 183001-1–183001-32. [Google Scholar]
- Dahlström, J.M.; Guénot, D.; Klünder, K.; Gisselbrecht, M.; Mauritsson, J.; L’Huillier, A.; Maquet, A.; Taïeb, R. Theory of attosecond delays in laser-assisted photoionization. Chem. Phys 2012, in press. [Google Scholar]
- Togashi, T.; Takahashi, E.J.; Midorikawa, K.; Aoyama, M.; Yamakawa, K.; Sato, T.; Iwasaki, A.; Owada, S.; Okino, T.; Yamanouchi, K.; et al. Extreme ultraviolet free electron laser seeded with high-order harmonic of Ti:sapphire laser. Opt. Exp 2010, 19, 317–324. [Google Scholar]
- Pfeifer, T.; Jiang, Y.; Düsterer, S.; Moshammer, R.; Ullrich, J. Partial-coherence method to model experimental free-electron laser pulse statistics. Opt. Lett 2010, 35, 3441–3443. [Google Scholar]
- Okino, T.; Yamanouchi, K.; Shimizu, T.; Ma, R.; Nabekawa, Y.; Midorikawa, K. Attosecond nonlinear Fourier transformation spectroscopy of CO2 in extreme ultraviolet wavelength region. J. Chem. Phys 2008, 129, 161103-1–161103-4. [Google Scholar]
- Varjú, K.; Mairesse, Y.; Carré, B.; Gaarde, B.; Johnsson, P.; Kazamias, S.; López-Martens, R.; Mauritsson, J.; Schafer, K.J.; Balcou, Ph.; L’huillier, A.; Salières, P. Frequency chirp of harmonic and attosecond pulses. J. Mod. Opt 2005, 52, 379–394. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
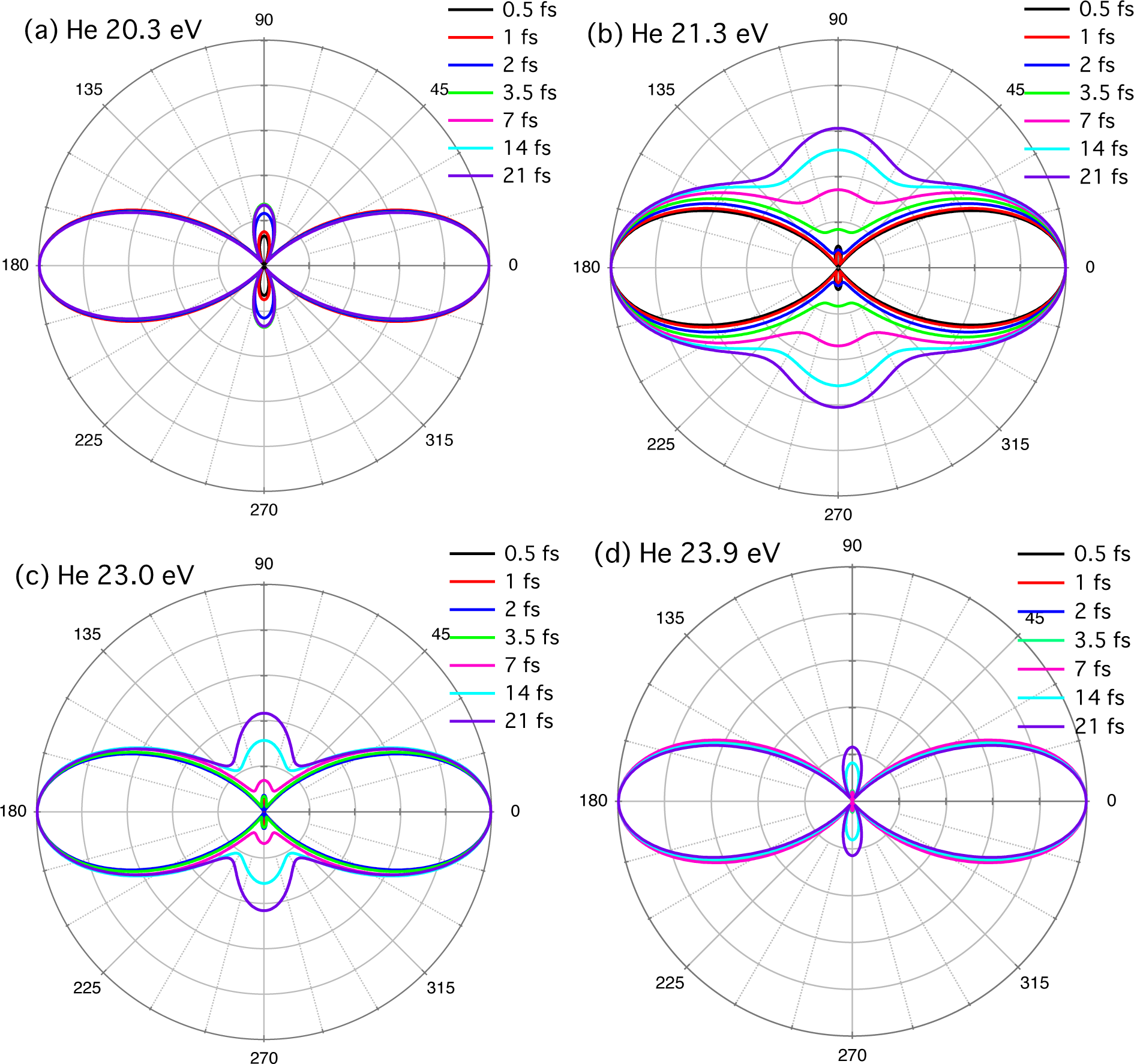{kind=link}
{kind=link}
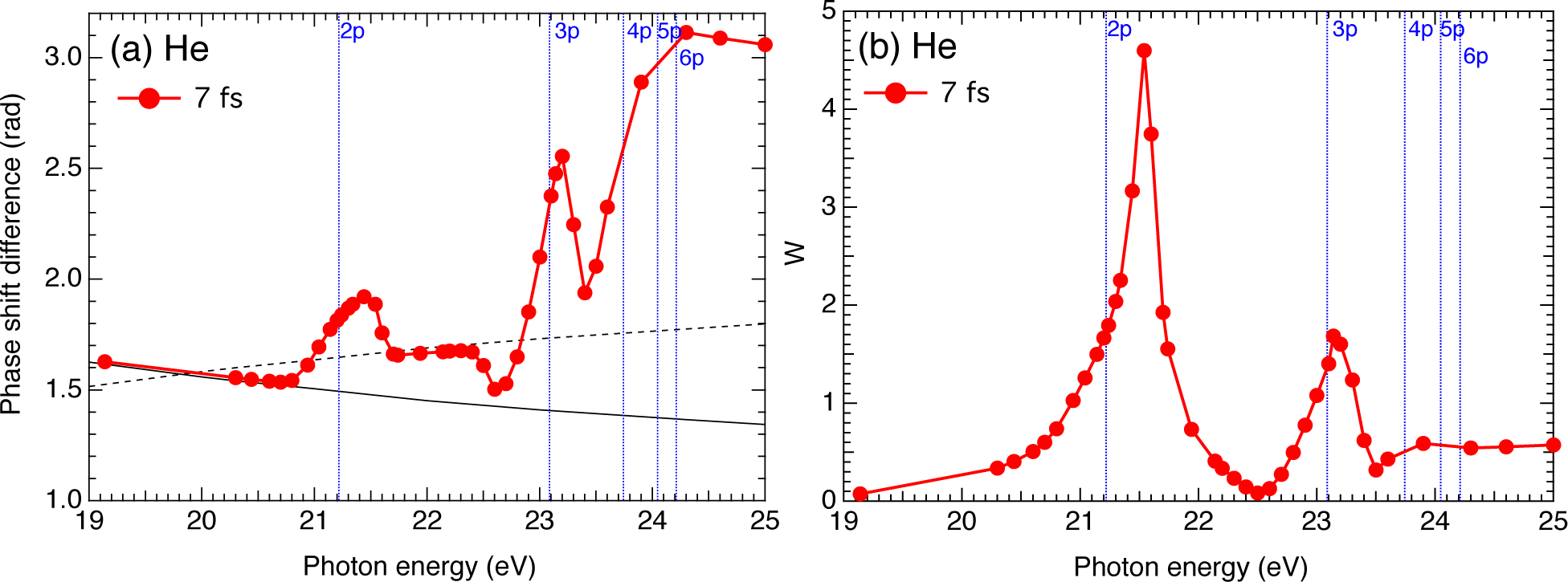{kind=link}
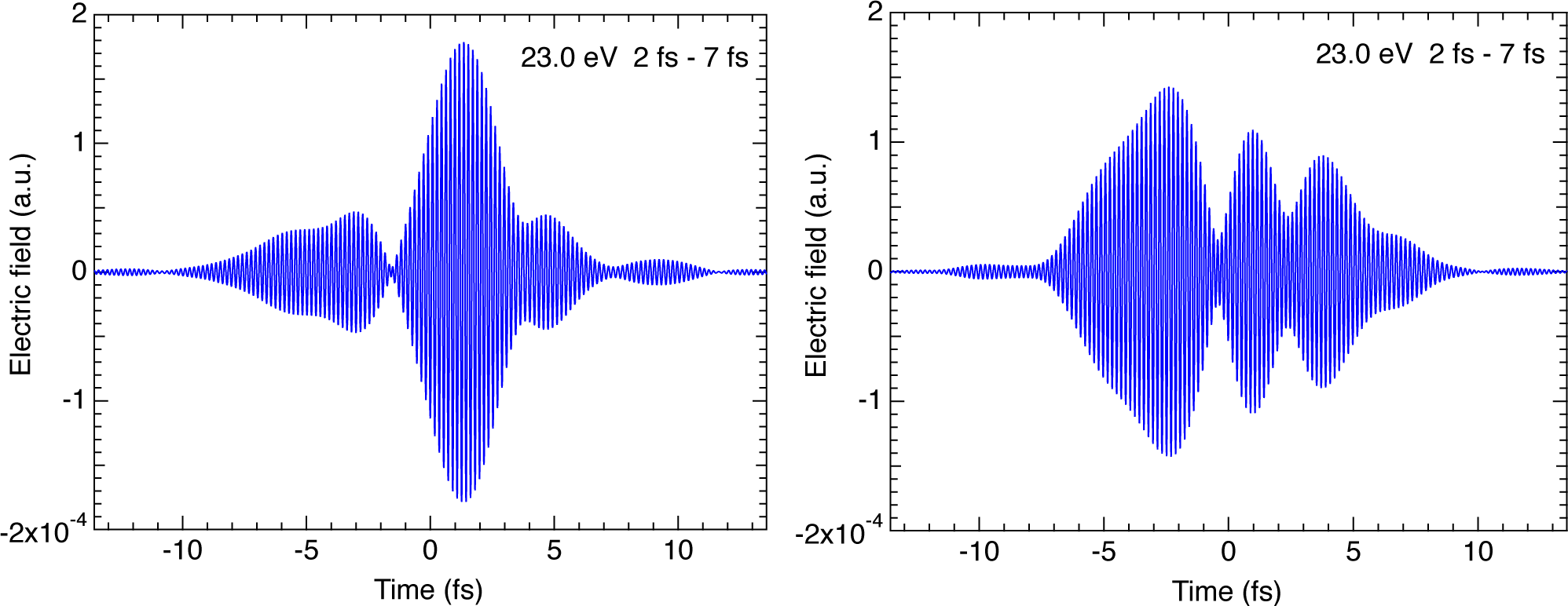{kind=link}
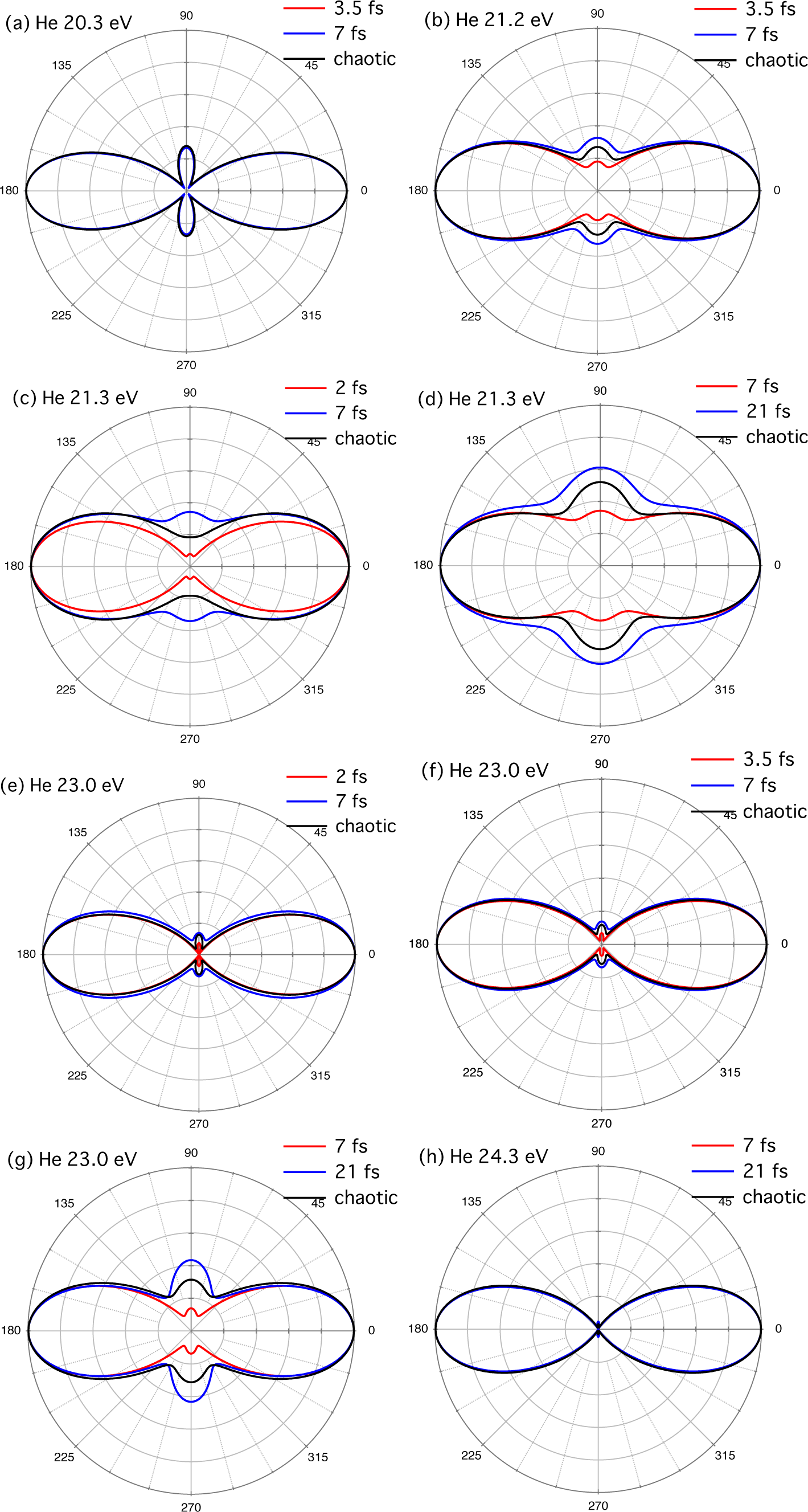{kind=link}
| ħω (eV) | CT (fs) | MPW (fs) | W | δ |
|---|---|---|---|---|
| 21.2 | 2 | 5 | 1.31 ± 0.08 | 2.03 ± 0.04 |
| 2 | 7 | 1.42 ± 0.09 | 1.91 ± 0.04 | |
| 3.5 | 7 | 1.49 ± 0.09 | 1.87 ± 0.03 | |
| 2 | 2 | 1.05 | 2.26 | |
| 3.5 | 3.5 | 1.41 | 2.05 | |
| 5 | 5 | 1.57 | 1.92 | |
| 7 | 7 | 1.67 | 1.81 | |
| 21.3 | 2 | 7 | 2.21 ± 0.17 | 2.26 ± 0.06 |
| 7 | 21 | 1.92 ± 0.08 | 1.62 ± 0.009 | |
| 2 | 2 | 1.13 | 2.34 | |
| 7 | 7 | 2.04 | 1.87 | |
| 21 | 21 | 2.31 | 1.59 | |
| 23.0 | 2 | 7 | 0.645 ± 0.061 | 2.14 ± 0.08 |
| 3.5 | 7 | 0.890 ± 0.058 | 2.15 ± 0.06 | |
| 7 | 21 | 1.50 ± 0.154 | 1.79 ± 0.033 | |
| 2 | 2 | 0.464 | 2.71 | |
| 3.5 | 3.5 | 0.679 | 2.45 | |
| 7 | 7 | 1.08 | 2.10 | |
| 21 | 21 | 1.31 | 1.58 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ishikawa, K.L.; Ueda, K. Photoelectron Angular Distribution and Phase in Two-Photon Single Ionization of H and He by a Femtosecond and Attosecond Extreme-Ultraviolet Pulse. Appl. Sci. 2013, 3, 189-213. https://doi.org/10.3390/app3010189
Ishikawa KL, Ueda K. Photoelectron Angular Distribution and Phase in Two-Photon Single Ionization of H and He by a Femtosecond and Attosecond Extreme-Ultraviolet Pulse. Applied Sciences. 2013; 3(1):189-213. https://doi.org/10.3390/app3010189
Chicago/Turabian StyleIshikawa, Kenichi L., and Kiyoshi Ueda. 2013. "Photoelectron Angular Distribution and Phase in Two-Photon Single Ionization of H and He by a Femtosecond and Attosecond Extreme-Ultraviolet Pulse" Applied Sciences 3, no. 1: 189-213. https://doi.org/10.3390/app3010189
APA StyleIshikawa, K. L., & Ueda, K. (2013). Photoelectron Angular Distribution and Phase in Two-Photon Single Ionization of H and He by a Femtosecond and Attosecond Extreme-Ultraviolet Pulse. Applied Sciences, 3(1), 189-213. https://doi.org/10.3390/app3010189