
HG-LGBM: A Hybrid Model for Microbiome-Disease Prediction Based on Heterogeneous Networks and Gradient Boosting

Abstract
1. Introduction
2. Materials and Methods
2.1. Data Preprocessing and Feature Extraction
2.1.1. Microbiome Similarity
- If there is functional similarity between a pair of microorganisms (i.e., ), the functional similarity and Gaussian kernel similarity are integrated using the following formula:
- If there is no functional similarity between a pair of microorganisms (i.e., ), only the GIP similarity is considered, as expressed in the following equation:
2.1.2. Disease Similarity
- If there is functional similarity between a pair of diseases (i.e., ), the functional similarity and Gaussian kernel similarity are integrated using the following formula:
- If there is no functional similarity between a pair of diseases (i.e., ), only the GIP similarity is considered, as expressed in the following equation:
2.2. Heterogeneous Graph Neural Network Model
2.2.1. Multi-Head Attention Mechanism Calculation
2.2.2. Message Passing and Aggregation
3. LightGBM Method
- Prediction Initialization: For each microbiome–disease pair, LightGBM initializes the predicted value using the mean of the training data labels.
- Gradient Calculation: For each training sample i, the gradient of the loss function is calculated as , where is the loss function, is the true label, and is the predicted value.
- Decision Tree Construction: The GOSS sampling method is used to select samples with larger gradients, thereby accelerating the tree construction.
- Prediction Update: The output of the new tree is added to the existing predicted values, updating the predictions for the microbiome–disease pairs.
- Iterative Training: The model is iteratively optimized through gradient descent and multiple tree constructions until the preset number of trees is reached or error convergence occurs.
4. Results
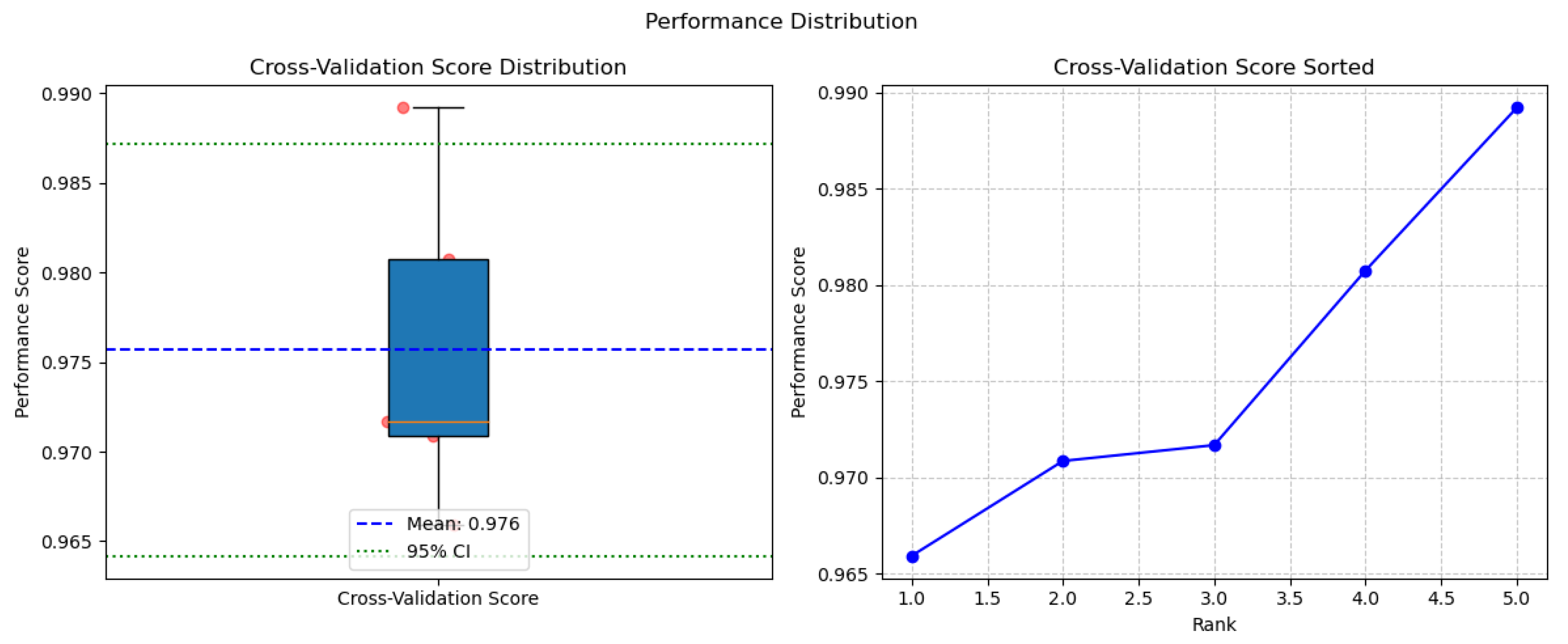
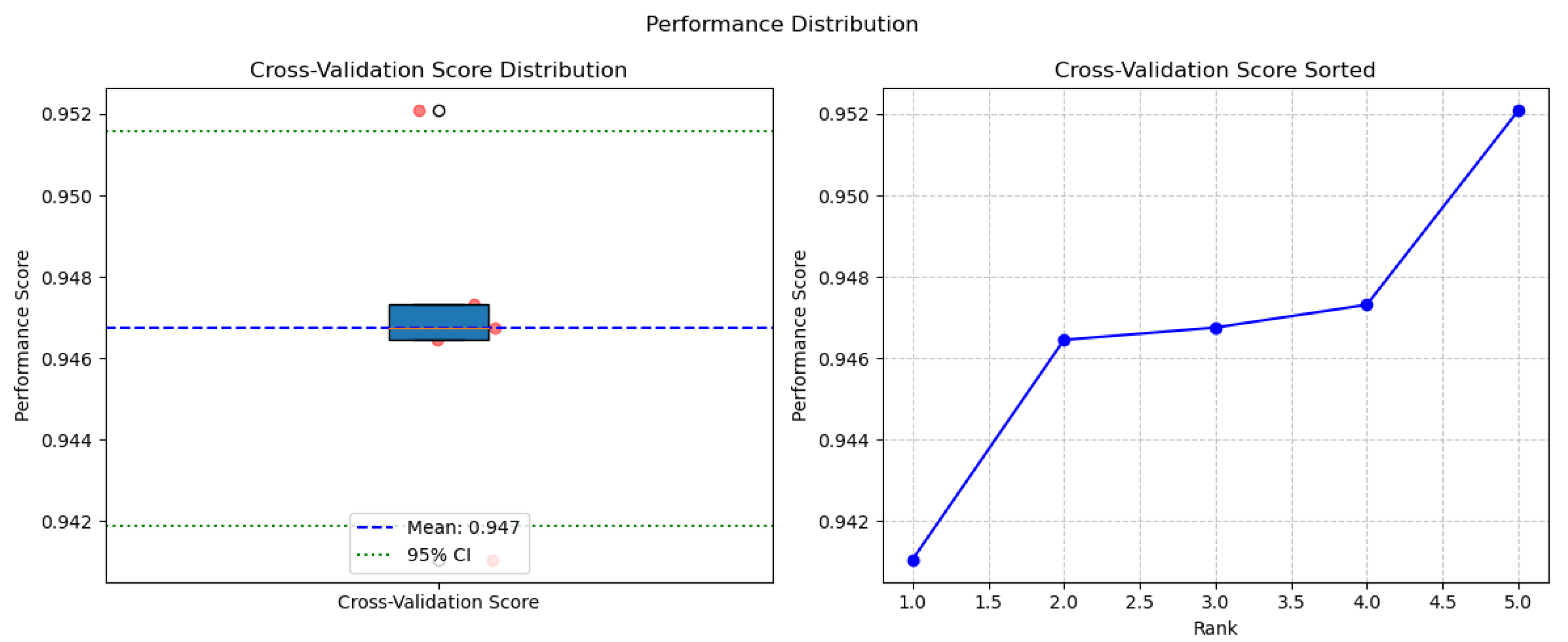
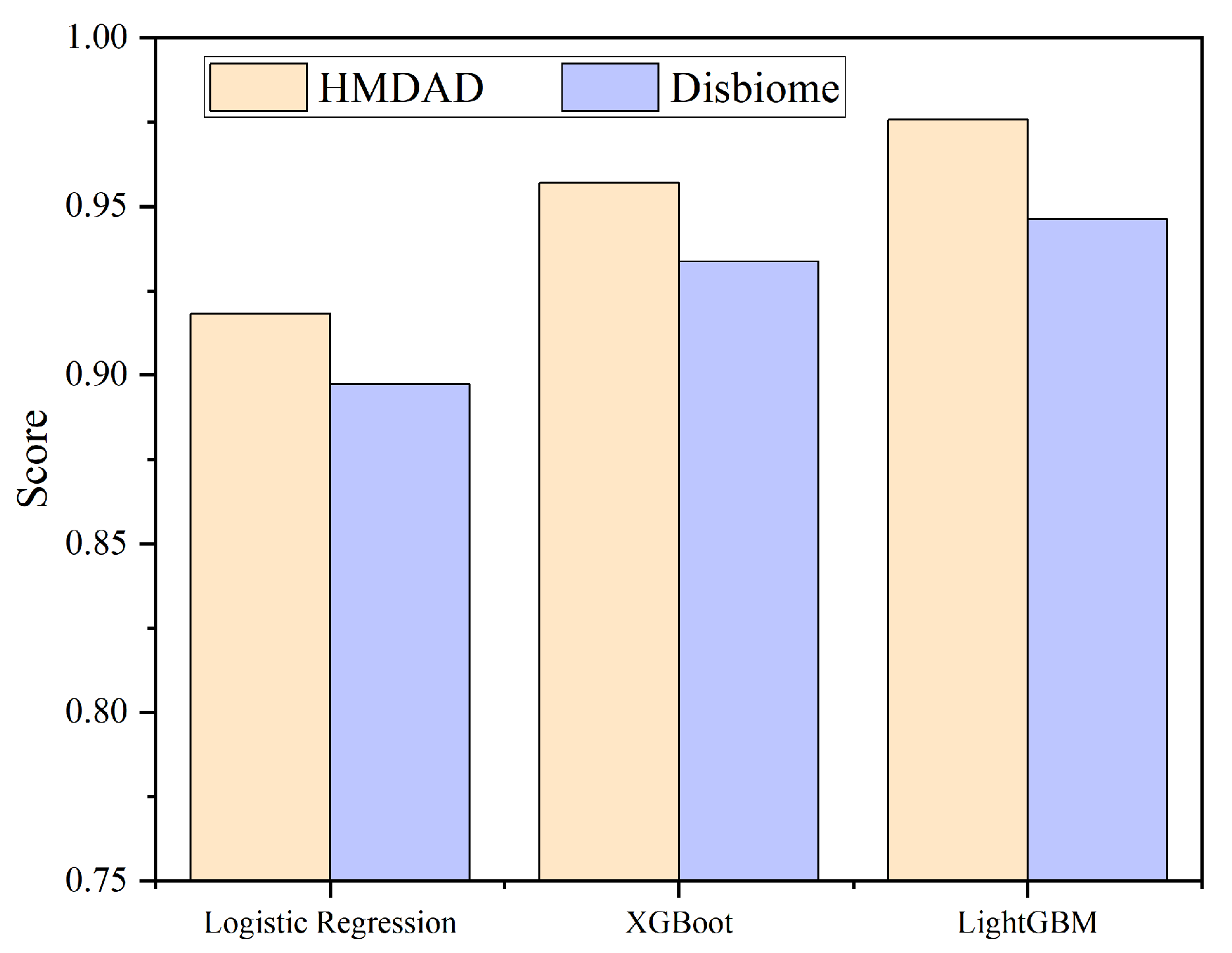
4.1. Performance Analysis
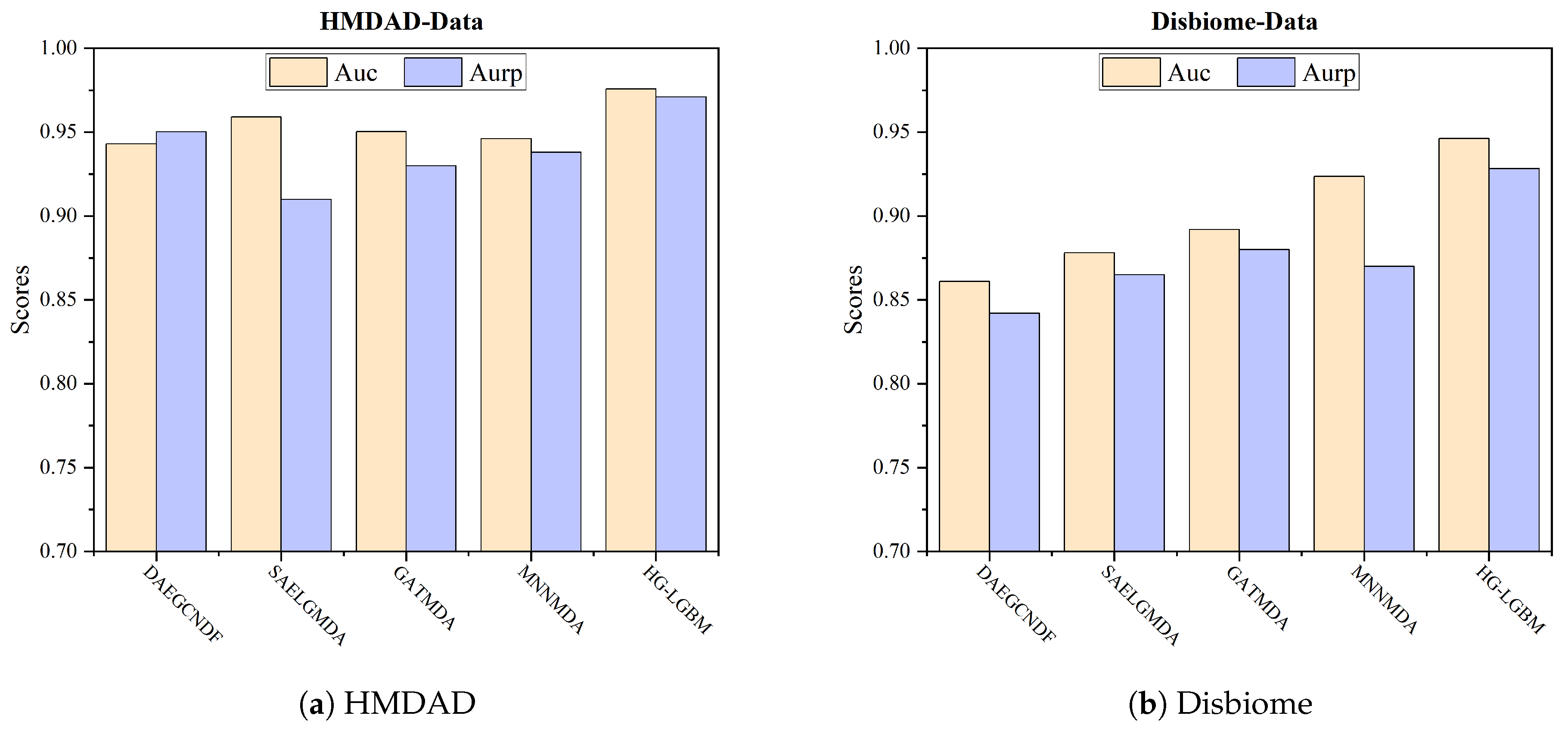
4.2. Model Comparison
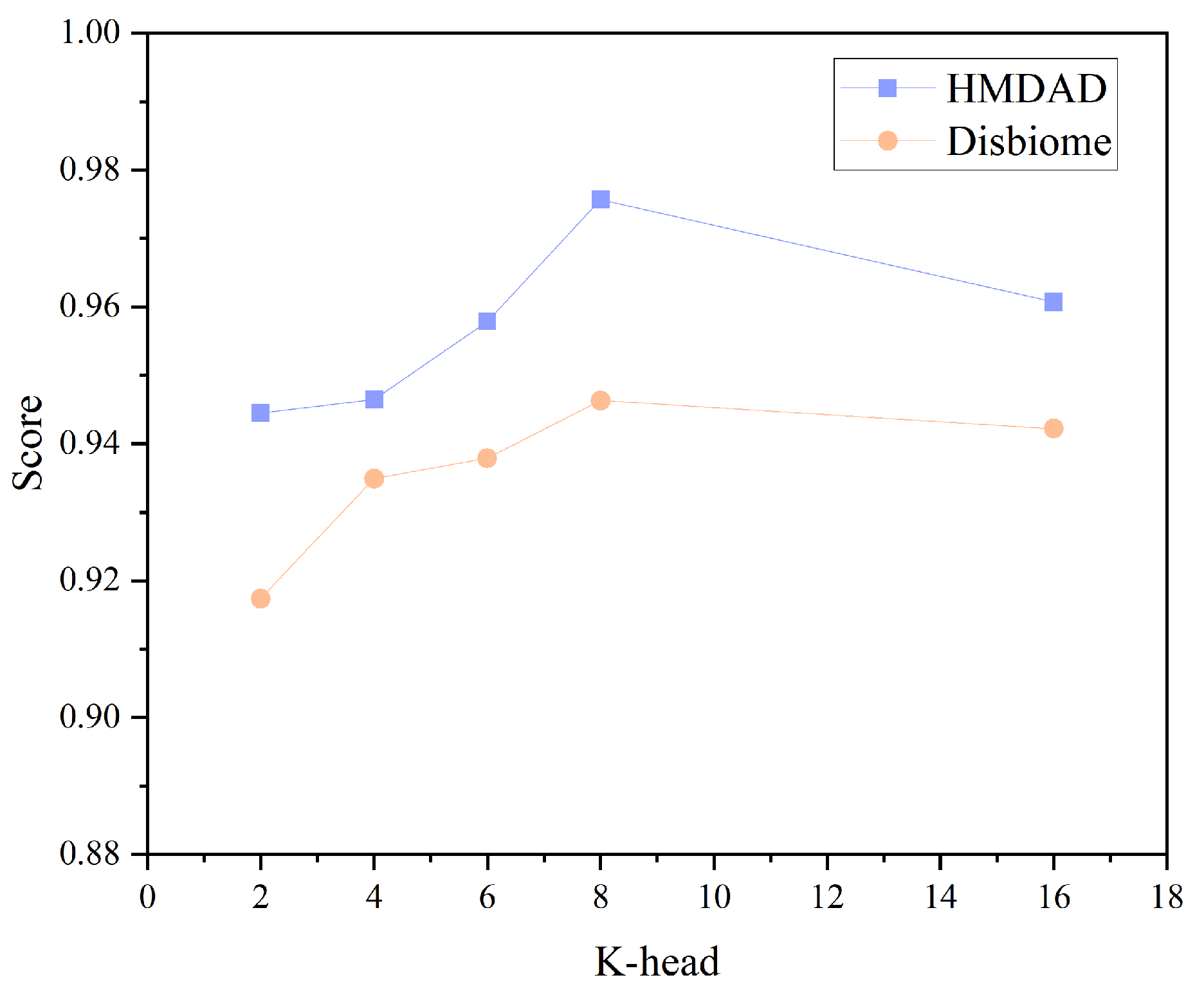
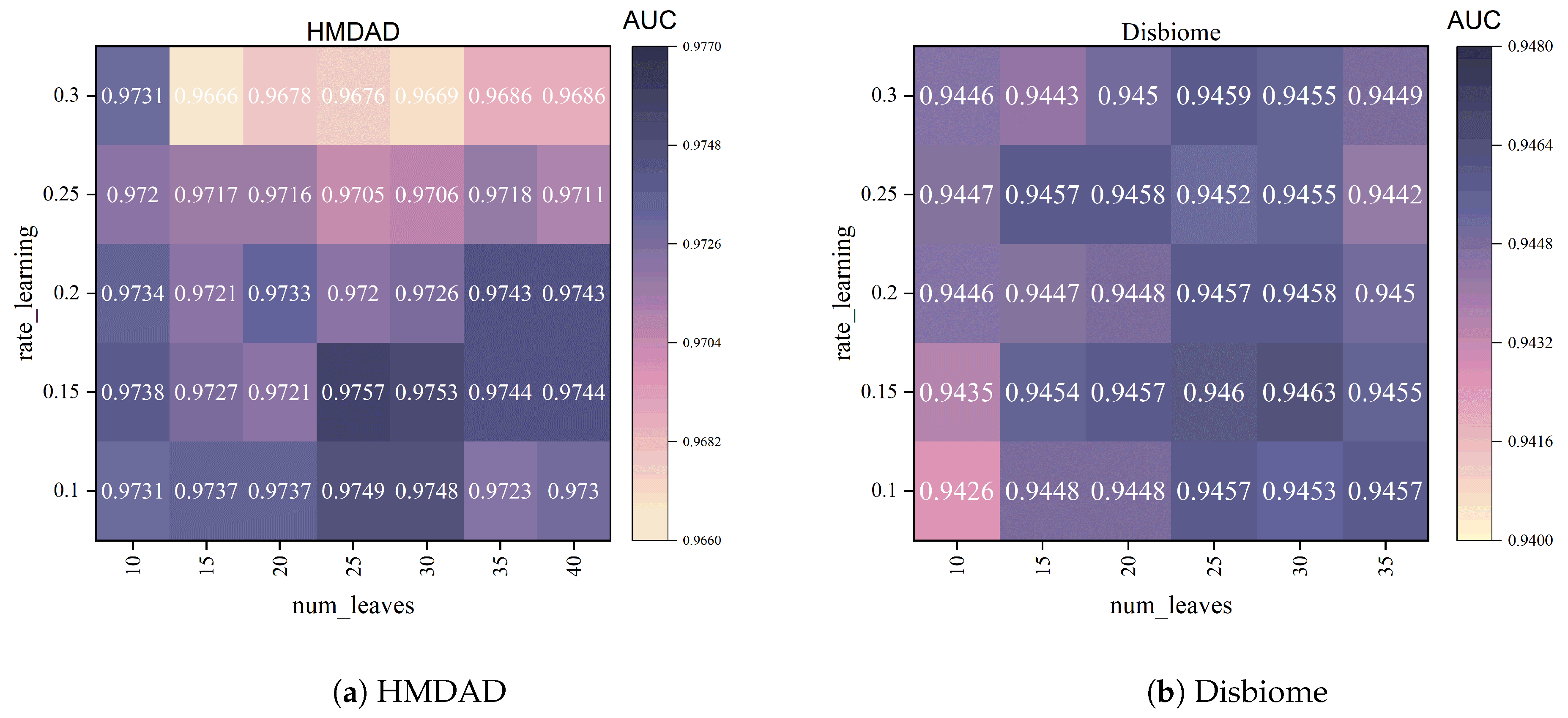
4.3. Hyperparameter Analysis
- num_leaves: This parameter controls the number of leaves in a tree, affecting the model’s complexity. A larger num_leaves allows the model to be more flexible and learn more complex relationships, but it may lead to overfitting. A smaller num_leaves helps reduce overfitting, but it may limit the model’s expressiveness. We tested several different num_leaves values, including [5, 10, 15, 20, 25, 30, 35, 40], and ultimately selected num_leaves = 25, which showed the best balance on the validation set.
- learning_rate: This parameter represents the learning rate for model iteration. A higher learning rate can speed up convergence, but it may lead to instability and overfitting. A lower learning rate provides more stable training, but it may require more iterations to converge. We tested several learning_rate values (0.1, 0.15, 0.2, 0.25, 0.3) and ultimately selected learning_rate = 0.15, which provided the best balance between learning speed and prediction performance during training.
- max_depth: This parameter controls the maximum depth of each tree, which determines the tree’s complexity. If max_depth is set too deep, it may cause overfitting; if it is set too shallow, it may cause underfitting. In our experiments, we set max_depth = −1 to allow the model to adaptively determine the depth based on the data, better capturing complex patterns in the data.
5. Case Study
Microbial Association Analysis of Inflammatory Bowel Disease (IBD)
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aggarwal, N.; Kitano, S.; Puah, G.R.Y.; Kittelmann, S.; Hwang, I.Y.; Chang, M.W. Microbiome and Human Health: Current Understanding, Engineering, and Enabling Technologies. Chem. Rev. 2023, 123, 31–72. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2021, 19, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Fischbach, M.; Belkaid, Y. Skin microbiota–host interactions. Nature 2018, 553, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Hou, K.; Wu, Z.X.; Chen, X.Y.; Wang, J.Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J.; et al. Microbiota in health and diseases. Signal Transduct. Target. Ther. 2022, 7, 135. [Google Scholar] [CrossRef]
- Brown, J.; Hazen, S. Microbial modulation of cardiovascular disease. Nat. Rev. Microbiol. 2018, 16, 171–181. [Google Scholar] [CrossRef]
- Sepich-Poore, G.D.; Zitvogel, L.; Straussman, R.; Hasty, J.; Wargo, J.A.; Knight, R. The microbiome and human cancer. Science 2021, 371, eabc4552. [Google Scholar] [CrossRef]
- Wu, J.; Wang, K.; Wang, X.; Pang, Y.; Jiang, C. The role of the gut microbiome and its metabolites in metabolic diseases. Protein Cell 2021, 12, 360–373. [Google Scholar] [CrossRef]
- Schirmer, M.; Garner, A.; Vlamakis, H.; Xavier, R.J. Microbial genes and pathways in inflammatory bowel disease. Nat. Rev. Microbiol. 2019, 17, 497–511. [Google Scholar] [CrossRef]
- Stripling, J.; Rodriguez, M. Current evidence in delivery and therapeutic uses of fecal microbiota transplantation in human diseases-clostridium difficile disease and beyond. Am. J. Med. Sci. 2018, 356, 424–432. [Google Scholar] [CrossRef]
- Benech, N.; Sokol, H. Fecal microbiota transplantation in gastrointestinal disorders: Time for precision medicine. Genome Med. 2020, 12, 58. [Google Scholar] [CrossRef]
- Cunningham, M.; Vinderola, G.; Charalampopoulos, D.; Lebeer, S.; Sanders, M.E.; Grimaldi, R. Applying probiotics and prebiotics in new delivery formats—Is the clinical evidence transferable? Trends Food Sci. Technol. 2021, 112, 495–506. [Google Scholar] [CrossRef]
- Kahouli, I.; Malhotra, M.; Westfall, S.; Alaoui-Jamali, M.A.; Prakash, S. Design and validation of an orally administrated active L. fermentum-L. acidophilus probiotic formulation using colorectal cancer Apc Min/+ mouse model. Appl. Microbiol. Biotechnol. 2017, 101, 1999–2019. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Ogasawara, N.; Funaki, Y.; Mizuno, M.; Iida, A.; Goto, C.; Koikeda, S.; Kasugai, K.; Joh, T. Transglucosidase improves the gut microbiota profile of type 2 diabetes mellitus patients: A randomized double-blind, placebo-controlled study. BMC Gastroenterol. 2013, 13, 81. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Zitvogel, L.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef]
- Qi, X.; Li, X.; Zhao, Y.E.; Wu, X.; Chen, F.; Ma, X.; Zhang, F.; Wu, D. Treating steroid refractory intestinal acute graft-vs.-host disease with fecal microbiota transplantation: A pilot study. Front. Immunol. 2018, 9, 2195. [Google Scholar] [CrossRef]
- Morton, J.T.; Aksenov, A.A.; Nothias, L.F.; Foulds, J.R.; Quinn, R.A.; Badri, M.H.; Swenson, T.L.; Goethem, M.W.V.; Northen, T.R.; Knight, R.; et al. Learning representations of microbe–metabolite interactions. Nat. Methods 2019, 16, 1306–1314. [Google Scholar] [CrossRef]
- Jiang, C.; Feng, J.; Shan, B.; Chen, Q.; Yang, J.; Wang, G.; Peng, X.; Li, X. Predicting microbe-disease associations via graph neural network and contrastive learning. Front. Microbiol. 2024, 15, 1483983. [Google Scholar] [CrossRef]
- Chen, X.; Huang, Y.-A.; You, Z.-H.; Yan, G.-Y.; Wang, X.-S. A novel approach based on KATZ measure to predict associations of human microbiota with non-infectious diseases. Bioinformatics 2017, 33, 733–739. [Google Scholar] [CrossRef]
- Huang, Z.-A.; Chen, X.; Zhu, Z.; Liu, H.; Yan, G.-Y.; You, Z.-H.; Wen, Z. PBHMDA: Path-Based Human Microbe-Disease Association Prediction. Front. Microbiol. 2017, 8, 233. [Google Scholar] [CrossRef]
- Chen, Y.; Lei, X. Metapath Aggregated Graph Neural Network and Tripartite Heterogeneous Networks for Microbe-Disease Prediction. Front. Microbiol. 2022, 13, 919380. [Google Scholar] [CrossRef]
- Zou, S.; Zhang, J.; Zhang, Z. A novel approach for predicting microbe-disease associations by bi-random walk on the heterogeneous network. PLoS ONE 2017, 12, e0184394. [Google Scholar] [CrossRef] [PubMed]
- He, B.-S.; Peng, L.-H.; Li, Z. Human Microbe-Disease Association Prediction With Graph Regularized Non-Negative Matrix Factorization. Front. Microbiol. 2018, 9, 2560. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Bing, P.; Zhang, M.; Tian, G.; Ma, J.; Li, H.; Bao, M.; He, K.; He, J.; He, B.; et al. MNNMDA: Predicting human microbe-disease association via a method to minimize matrix nuclear norm. Comput. Struct. Biotechnol. J. 2023, 21, 1414–1423. [Google Scholar] [CrossRef] [PubMed]
- Zitnik, M.; Nguyen, F.; Wang, B.; Leskovec, J.; Goldenberg, A.; Hoffman, M.M. Machine learning for integrating data in biology and medicine: Principles, practice, and opportunities. Inf. Fusion 2019, 50, 71–91. [Google Scholar] [CrossRef]
- Wang, F.; Yang, H.; Wu, Y.; Peng, L.; Li, X. SAELGMDA: Identifying human microbe–disease associations based on sparse autoencoder and LightGBM. Front. Microbiol. 2023, 14, 1207209. [Google Scholar] [CrossRef]
- Long, Y.; Luo, J.; Zhang, Y.; Xia, Y. Predicting human microbe–disease associations via graph attention networks with inductive matrix completion. Brief. Bioinform. 2021, 22, bbaa146. [Google Scholar] [CrossRef]
- Lu, S.; Liang, Y.; Li, L.; Miao, R.; Liao, S.; Zou, Y.; Yang, C.; Ouyang, D. Predicting potential microbe-disease associations based on auto-encoder and graph convolution network. BMC Bioinform. 2023, 24, 476. [Google Scholar] [CrossRef]
- Gong, H.; You, X.; Jin, M.; Meng, Y.; Zhang, H.; Yang, S.; Xu, J. Graph neural network and multi-data heterogeneous networks for microbe-disease prediction. Front. Microbiol. 2022, 13, 1077111. [Google Scholar] [CrossRef]
- Wen, S.; Liu, Y.; Yang, G.; Chen, W.; Wu, H.; Zhu, X.; Wang, Y. A method for miRNA-disease association prediction using machine learning decoding of multi-layer heterogeneous graph Transformer encoded representations. Sci. Rep. 2024, 14, 20490. [Google Scholar] [CrossRef]
- Kamneva, O.K. Genome composition and phylogeny of microbes predict their co-occurrence in the environment. PLoS Comput. Biol. 2017, 13, e1005366. [Google Scholar] [CrossRef]
- Van Laarhoven, T.; Nabuurs, S.B.; Marchiori, E. Gaussian interaction profile kernels for predicting drug–target interaction. Bioinformatics 2011, 27, 3036–3043. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Zhang, Q.; Ma, Q.; Yu, B. LightGBM-PPI: Predicting protein-protein interactions through LightGBM with multi-information fusion. Chemom. Intell. Lab. Syst. 2019, 191, 54–64. [Google Scholar] [CrossRef]
- Shaker, B.; Yu, M.S.; Song, J.S.; Ahn, S.; Ryu, J.Y.; Oh, K.S.; Na, D. LightBBB: Computational prediction model of blood–brain-barrier penetration based on LightGBM. Bioinformatics 2020, 37, 1135–1139. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Datasets | Microbes | Diseases | Associations |
|---|---|---|---|
| HMDAD | 292 | 39 | 450 |
| Disbiome | 1052 | 218 | 4351 |
| Metric | HMDAD (Mean) | Disbiome (Mean) |
|---|---|---|
| AUC | 0.9757 | 0.9463 |
| AUPR | 0.9711 | 0.9283 |
| F1-score | 0.9246 | 0.8868 |
| Accuracy | 0.9233 | 0.8836 |
| Recall | 0.9336 | 0.9126 |
| Specificity | 0.9123 | 0.8549 |
| Precision | 0.9162 | 0.8620 |
| Rank | Microbe | Evidence (PMID) |
|---|---|---|
| 1 | Veillonella | 38980940 |
| 2 | Prevotella | 38053528 |
| 3 | Bifidobacterium | 38368394 |
| 4 | Clostridium coccoides | 26994772 |
| 5 | Bacteroidetes | 31071294 |
| 6 | Klebsiella | 38545880 |
| 7 | Haemophilus | 34334167 |
| 8 | Firmicutes | 35951774 |
| 9 | Lactobacillus | 37773196 |
| 10 | Enterococcus | 38788722 |
| Rank | Microbe | Evidence (PMID) |
|---|---|---|
| 1 | Clostridium coccoides | 26084032 |
| 2 | Proteobacteria | 37263983 |
| 3 | Actinobacteria | 38648753 |
| 4 | Staphylococcus | 36037202 |
| 5 | Lachnospiraceae | 36893736 |
| 6 | Haemophilus | 35663463 |
| 7 | Lactobacillus | 37192617 |
| 8 | Collinsella aerofaciens | 37704113 |
| 9 | Enterococcus | 35090978 |
| 10 | Desulfovibrio | 38484555 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, J.; Xu, C.; Liu, Y. HG-LGBM: A Hybrid Model for Microbiome-Disease Prediction Based on Heterogeneous Networks and Gradient Boosting. Appl. Sci. 2025, 15, 4452. https://doi.org/10.3390/app15084452
Guo J, Xu C, Liu Y. HG-LGBM: A Hybrid Model for Microbiome-Disease Prediction Based on Heterogeneous Networks and Gradient Boosting. Applied Sciences. 2025; 15(8):4452. https://doi.org/10.3390/app15084452
Chicago/Turabian StyleGuo, Jun, Chunyan Xu, and Ying Liu. 2025. "HG-LGBM: A Hybrid Model for Microbiome-Disease Prediction Based on Heterogeneous Networks and Gradient Boosting" Applied Sciences 15, no. 8: 4452. https://doi.org/10.3390/app15084452
APA StyleGuo, J., Xu, C., & Liu, Y. (2025). HG-LGBM: A Hybrid Model for Microbiome-Disease Prediction Based on Heterogeneous Networks and Gradient Boosting. Applied Sciences, 15(8), 4452. https://doi.org/10.3390/app15084452