1. Introduction
The biological activity of free amino acids and peptides is a matter of particular interest for the food industry and scientists [
1,
2]. Significant efforts have been made by researchers to find and characterize natural peptides that have displayed different bioactivities [
1,
2]. Amongst the biological activities reported, the capacity of peptides to inhibit the reactions initiated by radicals involved in the deterioration of foods or biological matrices has been extensively explored [
3]. In this context, classical methodologies originally employed for quantifying the antioxidant capacity (AC) of enriched polyphenolic samples have been applied to free amino acids, peptides, and proteins [
2,
4,
5,
6]. Reported data have shown the bleaching of stable radicals, such as 2,2-Diphenyl-1-picrylhydrazyl (DPPH) and 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) radical anions (ABTS), elicited by free amino acids, peptides, and their complex mixtures [
3,
7,
8]. The reducing activity of these samples has also been studied with regard to ferric ions (ferric reducing antioxidant power, FRAP) [
3,
9]. Particular importance has been given to the methodologies based on competitive reactions, where a target molecule (probe) competes with additives (antioxidants, XH) for peroxyl radicals (ROO
•), with the latter species commonly generated by the thermolysis of 2,2′-azo-bis(2-amidinopropane) dihydrochloride (AAPH), according to Reactions (1) to (4) [
10,
11].
In these assays, the AC of a particular sample is frequently assessed by determinations of the area under the curve (AUC) or the initial consumption rate of the kinetic profiles of the consumption of the probe (usually followed by UV–visible or fluorescence spectroscopy). These data are commonly normalized to the response given by 6-hydroxy-2,5,8-tetramethyl chroman-2-carboxylic acid (Trolox), a hydrosoluble vitamin E analog. While the determination of the AUC is employed in the oxygen radical absorbance (ORAC) assay, analysis of the initial consumption rates has been lesser used to asses AC. Fluorescein (FL) is the most accepted probe in the ORAC assay (ORAC-FL); however, other molecules, such as pyrogallol red (PGR) and pyranine (PYR), have also been employed as target molecules in this methodology [
12,
13,
14]. The reactivity of FL, and the low concentration employed in the assay (usually 70 nM), explain why this probe is easily protected by XH [
12]. The latter is inferred from the presence of lag times in the kinetic profiles which suggest that the ORAC-FL index is mainly governed by the stoichiometry of the reactions. In addition to this, the use of FL as probe would imply that alkoxyl radicals (RO
•) generated from self-reactions of ROO
• (Reactions (5) and (6)) are the principal oxidants of the system [
15].
In contrast to FL, the high reactivity of PGR, and its use at a higher concentration (5 μM), would explain why this probe is usually protected by XH without generating lag times in the kinetics [
12]. As the consumption of PGR follows a zero-order kinetic limit, all ROO
• generated by the thermolysis of AAPH are neutralized, minimizing the formation of RO
• [
12]. Thus, the ORAC assay using PGR as a probe (ORAC-PGR) gives values mainly associated with the reactivity of the sample towards ROO
• [
12]. Similarly to PGR, PYR is highly reactive towards ROO
• with its consumption, in the micromolar concentration range, also following a zero-order kinetic limit [
13]. Nevertheless, despite its high reactivity, PYR protection is usually characterized by the presence of clear lag times in the kinetics [
13,
14]. Interestingly, such behavior has been explained by repairing reactions where XH reacts with the secondary radical of PYR (PYR
•), in accordance with Reactions (7) and (8) [
13,
14,
16,
17,
18].
These reactions would imply that the use of PYR as a probe gives values influenced by Reaction (8) and not necessarily by competitive reactions towards ROO
•. However, it should be noted that the protection of PYR by low reactive XH involves processes without producing lag times in the kinetics, a pattern explained by the presence of dual mechanisms in which competition by ROO
• (Reaction (4)), and PYR repair (Reaction (8)), would be present [
13]. Remarkably, the presence of lag times in the kinetics of the ORAC assay due to Reaction (8) has not been linked exclusively to the use of PYR as a probe. Repairing FL by the reactions of its secondary radical with XH has also been carried out to explain the lag times usually registered in the measurements of the ORAC-FL index. The lag times generated by XH in the kinetics of FL consumption have been shown to be dependent on the oxidation potential of XH [
19].
Despite the reported evidence showing that some peptides display AC, the role that this bioactivity plays in complex systems is still under investigation. It is necessary to obtain new insights aimed at understanding the mechanisms of reactions, as well as the meaning of each methodology employed when the AC of bioactive peptides is investigated. In this context, despite the use of PYR as probe to assess the AC of a variety of samples [
20,
21], there is currently limited information regarding the AC of free amino acids and peptides evaluated by PYR-based procedures. Consequently, we have developed a series of competitive experiments using PYR as a target molecule, aimed at investigating the AC of free and peptide tryptophan (Trp) and tyrosine (Tyr) residues, two reactive amino acids towards ROO
•.
3. Results and Discussion
Consumption of PYR by ROO
• followed a zero-order kinetic limit in PYR over a wide concentration range, corroborating its high reactivity towards these radicals [
13]. In the presence of 10 mM AAPH, a progressive decrease in the fluorescence intensity of PYR over the incubation time was observed (
Figure 1), giving initial consumption rates (R
0) between 0.3 ± 0.04 and 0.5 ± 0.06 µM/min for PYR concentrations between 1 and 100 µM. At 5 µM PYR, a R
0 of 0.32 µM/min was calculated, implying that 2.5 moles of ROO
• were trapped per mole of PYR (a flux of 0.8 µM/min of ROO
• is generated from 10 mM AAPH at 37 °C [
10]). In agreement with previous reports [
13,
18], this value would imply a total trap of ROO
• by PYR, suggesting a minimal yield of RO
• production (Reaction (6)).
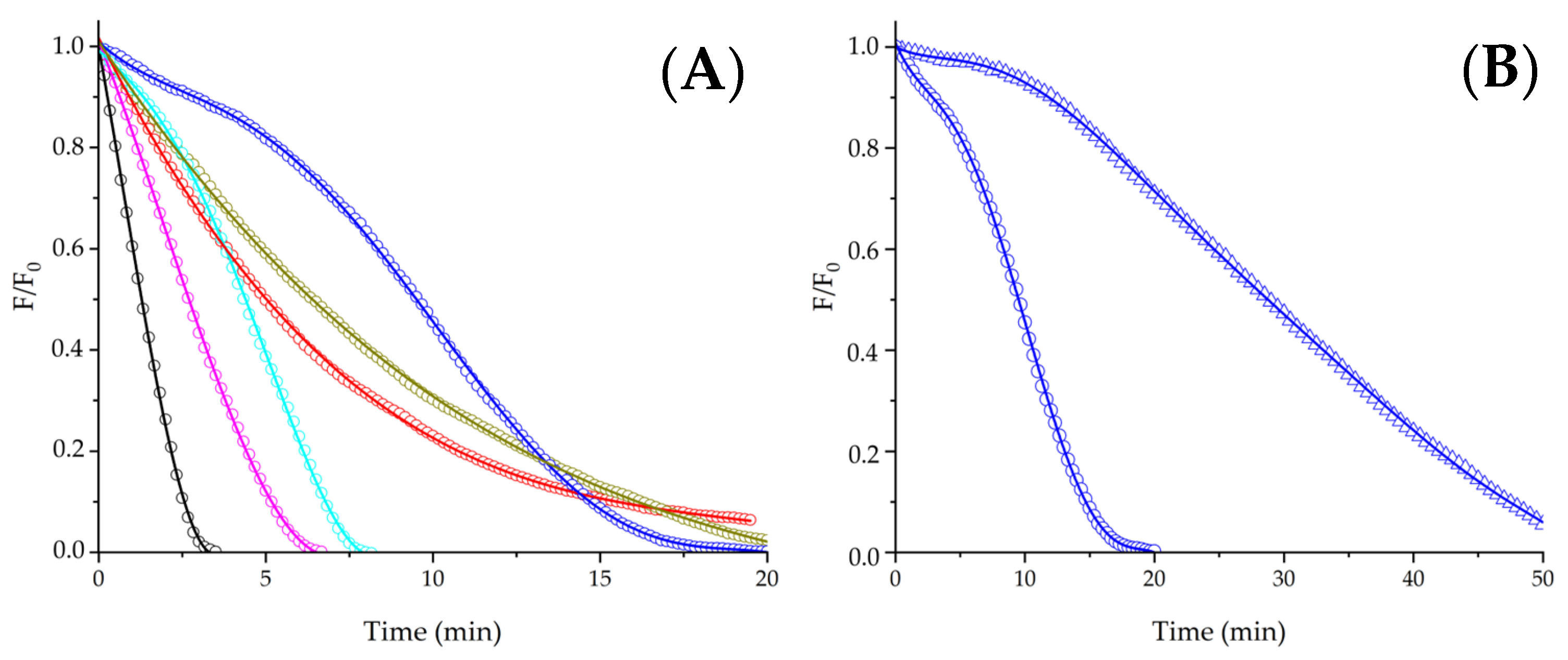
As depicted in Panel A of
Figure 2, free Trp protected PYR without generating lag times in the kinetics, reflecting only changes in R
0. At 1 mM Trp, a R
0 = 0.12 ± 0.01 µM/min was determined, indicating a 2.5-fold-times slower consumption of PYR than control experiments. The absence of lag times suggests that competition between PYR and Trp for ROO
• is the main process, with a minimal occurrence of Reaction (8). Aiming to obtain new insights regarding the protection of PYR elicited by peptides containing a single Trp residue, we carried out experiments using two dipeptides (Gly-Trp and Trp-Gly), and Gly-Trp-Gly. Due to these peptides having similar reactivities towards ROO
• (inferred from their direct consumption mediated by ROO
• [
22]), we speculated that simple competitions with PYR for ROO
• should be reflected in similar values of R
0. In contrast to such hypothesis, as depicted in
Figure 2, Panel A, the chemical structure of the peptides affected R
0, with values (expressed as µM/min) following the order as shown:
This order is not in consonance with the bond dissociation energy (BDE) of the hydrogen–nitrogen bond at position 1 of the indole ring of the Trp residues, which follows the order as shown [
22]:
Thus, despite the apparent simplicity of the protection of PYR afforded by Trp, Gly-Trp, Trp-Gly, and Gly-Trp-Gly, the observed behavior should not be interpreted as a simple competitive scheme where the changes in R
0 are only explained by the ability of peptides to neutralize ROO
•. The addition of high concentrations of free Gly (1–3 mM) to solutions containing 5 µM PYR and 10 mM AAPH did not affect the rate of PYR consumption (
Supplementary Figure S1), discarding some effect of Gly in the obtained results. In such a context, it seems reasonable to postulate that intermediates and oxidation products generated during the oxidation of Trp could have some participation in the protection of PYR. The latter could involve reactions involving the oxidation products of Trp, or Trp-derived radicals with PYR or PYR
•. These reactions could be affected by the chemical structure of the peptides, probably modulating the PYR-based AC index. Interestingly, the results obtained using PYR as a probe contrast with the ORAC-FL index of Trp, Gly-Trp, Trp-Gly, and Gly-Trp-Gly, which did not show differences (2.5 ± 0.3, 2.4 ± 0.2, 2.6 ± 0.2, and 2.4 ± 0.3, respectively), reflecting a similar protection of FL mediated by the additives [
22]. Thus, comparing the AC of these peptides using PYR and FL as probes clearly demonstrates the dependence of competitive assays with the probe employed, with the differences explained by complex mechanisms involving, as mentioned, reactions towards ROO
•, and RO
•, as well as reactions of secondary radicals of the probes and Trp. For example, reactions involving hydroperoxides could be present, since these species have shown to extend the original damage on proteins inflicted by oxidants [
23]. Similarly, comparison of results with systems using PGR as a probe should also denote important differences, since this target molecule has shown to be protected only for highly reactive derivatives. Consumption of 5 µM PGR mediated by 10 mM AAPH was not affected for PYR, reflecting the different reactivity of both probes (data not shown).
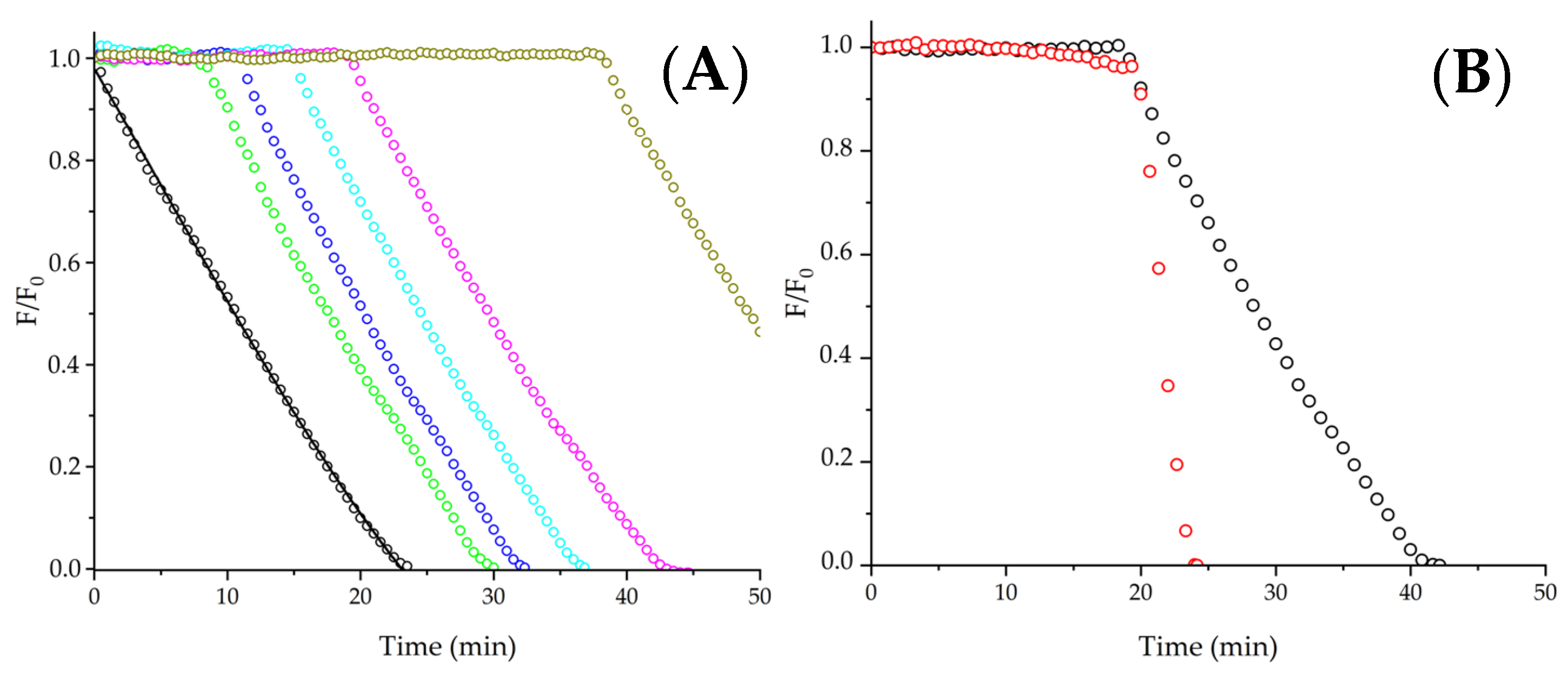
To explore if the protective pattern presented in
Figure 2, Panel A, can be extended to larger peptides, we assessed the consumption of PYR in the presence of three peptides: Asn-Trp-Asp-Asp-Met, Glu-Val-Trp-Lys-Ser-Ala-Glu, and Ser-Val-Trp-Ile-Gly-Gly-Ser-Ile. As expected, under our experimental conditions, these peptides lack secondary structures, displaying a random conformation, according to circular dichroism measurements (
Supplementary Figure S2). Similarly to Gly-Trp, a low protection of PYR, without lag times in the kinetics, was observed in the presence of 1 mM Asn-Trp-Asp-Asp-Met (
Figure 2, Panel B). As Met is also susceptible to oxidation by ROO
• [
24], the consumption of 5 µM PYR elicited by ROO
• was assessed in the presence of 5 mM free Met. Similar values of R
0 that control experiments were determined, showing a lack of PYR protection given by Met (
Supplementary Figure S3). In contrast to the kinetics registered in the presence of the pentapeptide (Asn-Trp-Asp-Asp-Met), Glu-Val-Trp-Lys-Ser-Ala-Glu and Ser-Val-Trp-Ile-Gly-Gly-Ser-Ile (hepta- and octa- peptides, respectively) inhibited the consumption of PYR, generating lag times in the kinetic profiles. These results demonstrated an effect of the chemical structure of peptides on the mechanisms involved in their AC. Analogously to shorter peptides, reactions involving intermediates or oxidation products were probably involved; however, since lag times were detected in the presence of the larger species (hepta- and octa-peptides), it is reasonable to infer that their AC was Influenced by a PYR-repairing mechanism. Remarkably, the length of the lag times was dependent on the peptide studied; while the lag time produced by 1 mM Glu-Val-Trp-Lys-Ser-Ala-Glu was ~5 min, the lag time generated by the same concentration of Ser-Val-Trp-Ile-Gly-Gly-Ser-Ile was close to 11 min. If the lag times are explained by the direct reaction of peptides with ROO
•, these values would imply that 4 and 8.8 µM of ROO
• were neutralized by the hepta- and octa-peptides, respectively. By contrast, if Reaction (8) explains the observed lag times, these values would reflect the total amount of PYR
• generated within the system (assuming a low extent of termination reactions of PYR
•). Nonetheless, independently of the processes explaining the presence of lag times in the kinetics, their lengths indicate different stoichiometry in the processes, reflecting the complexity of the reactions involved. Since a high concentration of peptides (1 mM) was necessary to protect PYR at the micromolar order (5 µM), the lengths of the lag times would suggest a very low yield of PYR repair by peptides (or reactions with ROO
•), explaining the low AC determined for the peptides investigated. Therefore, despite the complexity of the mechanisms, it is evident that the AC of Trp residues is considerably lower than that of classical antioxidants, such as a wide variety of phenols which have shown significant protection of PYR on a micromolar level [
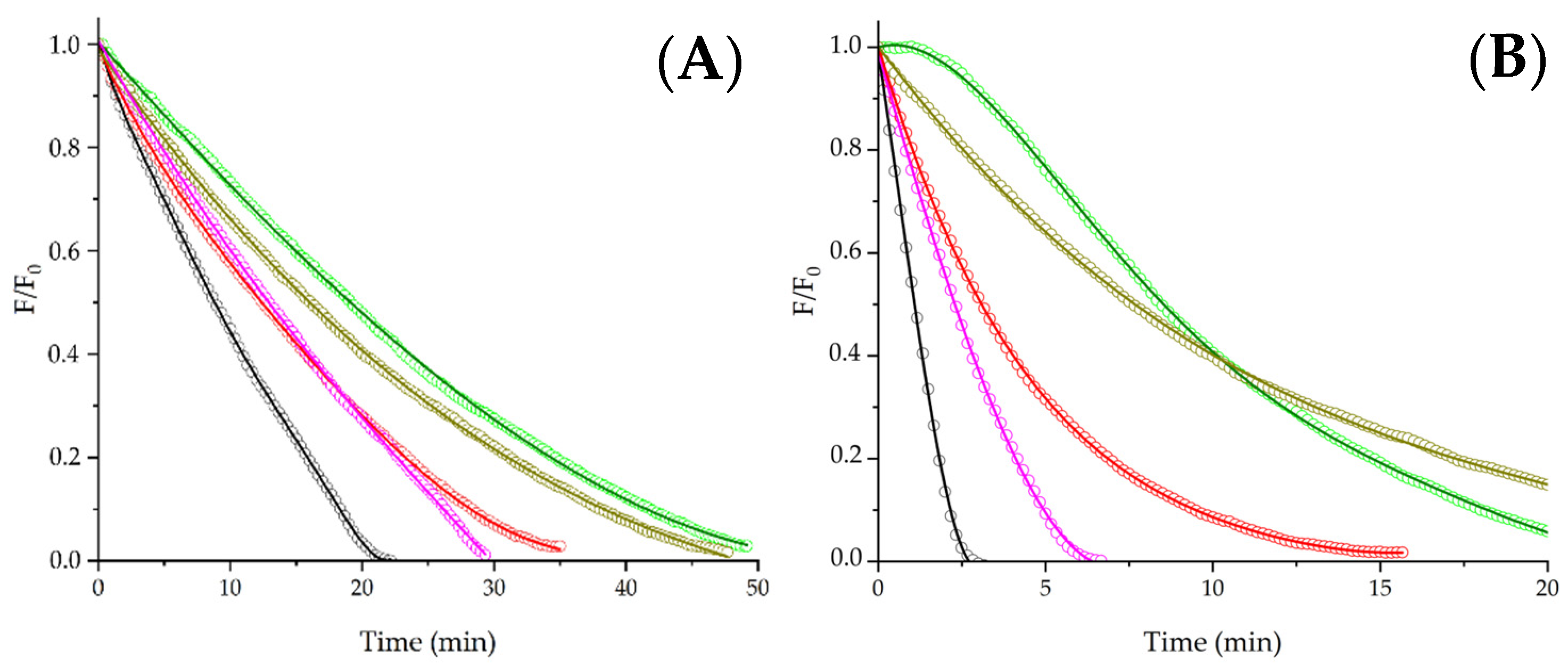
14]. Aiming to explore if the kinetics of PYR protection afforded by free Trp and the studied peptides depend on the PYR concentration, we developed experiments employing 1 μM PYR. If repairing PYR is the main process of the protective effect, the kinetics should be independent of the initial concentration of PYR, giving similar lag times or R
0 values. In contrast to the latter, if competition by ROO
• is the main reaction pathway, the pattern of kinetics should be affected by changes in the PYR concentration. In the absence of additives, the consumption of 1 µM PYR showed a similar R
0 to 5 µM PYR (~0.2–0.3 µM/min). As presented in
Figure 3, Panel A, the protection of 1 µM PYR elicited by free Trp, Gly-Trp-Gly, and Asn-Trp-Asp-Asp-Met was not affected by the change in PYR concentration, i.e., similar R
0 values were determined at 1 and 5 µM PYR. This result would indicate that even in the absence of lag times, the reparation of PYR could be involved in the protection given by free Trp, Gly-Trp-Gly, and Asn-Trp-Asp-Asp-Met. By contrast, as presented in
Figure 3, Panel A, the lag times generated by Glu-Val-Trp-Lys-Ser-Ala-Glu and Ser-Val-Trp-Ile-Gly-Gly-Ser-Ile were strongly affected by the change of PYR concentration. As presented in
Figure 3, Panel B, 1 mM Ser-Val-Trp-Ile-Gly-Gly-Ser-Ile generated a 3-times shorter lag time at 1 µM PYR than those registered at 5 µM PYR. This behavior would imply a complex mechanism where the presence of lag times in the kinetics would not be exclusively associated with Reaction (8); however, the lower protection observed at 1 µM PYR is not easily explained by simple competitive reactions between PYR and the peptide for ROO
•. As mentioned above, the pattern of the kinetics would represent a complex set of reactions with the participation of intermediates (particularly radicals) or oxidation products of Trp. In this context, it should be considered that the chemistry of Trp oxidation involves the formation of tryptophanyl radicals (Trp
•), which can react with O
2, (to form oxygenated products such as alcohols and hydroperoxides), dismutate, or generate Trp dimers by radical–radical reactions [
25,
26]. Therefore, our results suggest that the presence of lag times only in the kinetics of the PYR protection elicited by Trp containing peptides does not necessarily imply PYR repair as unique mechanism. By contrast, the absence of lag times in the kinetics does not necessarily involve competitive reactions towards ROO
• as exclusive processes.
Aiming to corroborate whether the obtained results agree with the PYR repair demonstrated in the presence of phenolic derivatives, we carried out experiments employing Trolox as an additive. In line with previous reports [
13,
14], clear induction times were generated by the protection given by Trolox for PYR consumption (
Figure 4, Panel A). The values of the lag times showed a linear dependence, with the initial concentration of Trolox indicating that ~3.0 moles of ROO
• were trapped per each mole of Trolox. These lag times were not affected by changes in the PYR concentrations, evidencing that repairing PYR is the main reaction in the presence of this additive.
Trying to explore if the above discussed patterns of PYR protection can be extended to other amino acids and peptides, experiments using free tyrosine (Tyr), as well as peptides containing a single Tyr residue, were developed. The oxidation of Tyr by ROO
• involves the abstraction of an electron (as hydrogen atom at pH 7.4) to produce tyrosyl radicals (Tyr
•) [
27]. Subsequently, Tyr
• react with O
2 (this is a slow reaction with k = 1 × 10
3 M
−1s
−1, [
27]) to form a Tyr-OO
•, or self-react producing di-tyrosine dimers (di-Tyr), amongst other reaction pathways [
27]. As presented in
Figure 5, Panel A, the consumption of 5 μM PYR was inhibited by 1 mM free Tyr and peptides following the order of R
0:
Similarly to the peptides containing Trp, the chemical structure of peptides including a single Tyr residue also affected the protection of PYR. Interestingly, the tripeptide Tyr-Tyr-Tyr showed a higher protection of PYR than free Tyr; however, this effect was not the expected to confer 3-fold-times higher protection, implying that each Tyr residue of Tyr-Tyr-Tyr did not react independently. Several reasons can be postulated to explain such an effect; however, it seems plausible to propose that the protective effect of Tyr-Tyr-Tyr is not easily explained by competitive reactions.
The latter is in line with the pattern of the kinetic profiles obtained for the consumption of 1 μM PYR in the presence of the Tyr residues. In the case of free Tyr and Gln-Arg-Tyr-Arg-Val-Leu-Asn-Ala-Ser, R
0 values of 0.20 ± 0.06 and 0.23 ± 0.04 µM/min were determined, respectively. These values are similar to those assessed at 5 µM PYR, suggesting some influence of PYR repair in the mechanism. Nonetheless, at 1 µM PYR, Tyr-Tyr-Tyr showed a R
0 value significantly lower (0.09 ± 0.02) than the value determined at 5 µM PYR, indicating the relevance of competitive reactions for ROO
•. In the case of Gly-Tyr, important changes were induced by changing the PYR concentration. At 5 µM PYR, kinetics did not show lag times; however, a clear lag time was detected when 1 µM PYR was employed (
Figure 4, Panel B), demonstrating that even when lag times are present in the protection of PYR, competition by ROO
• is an important process.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

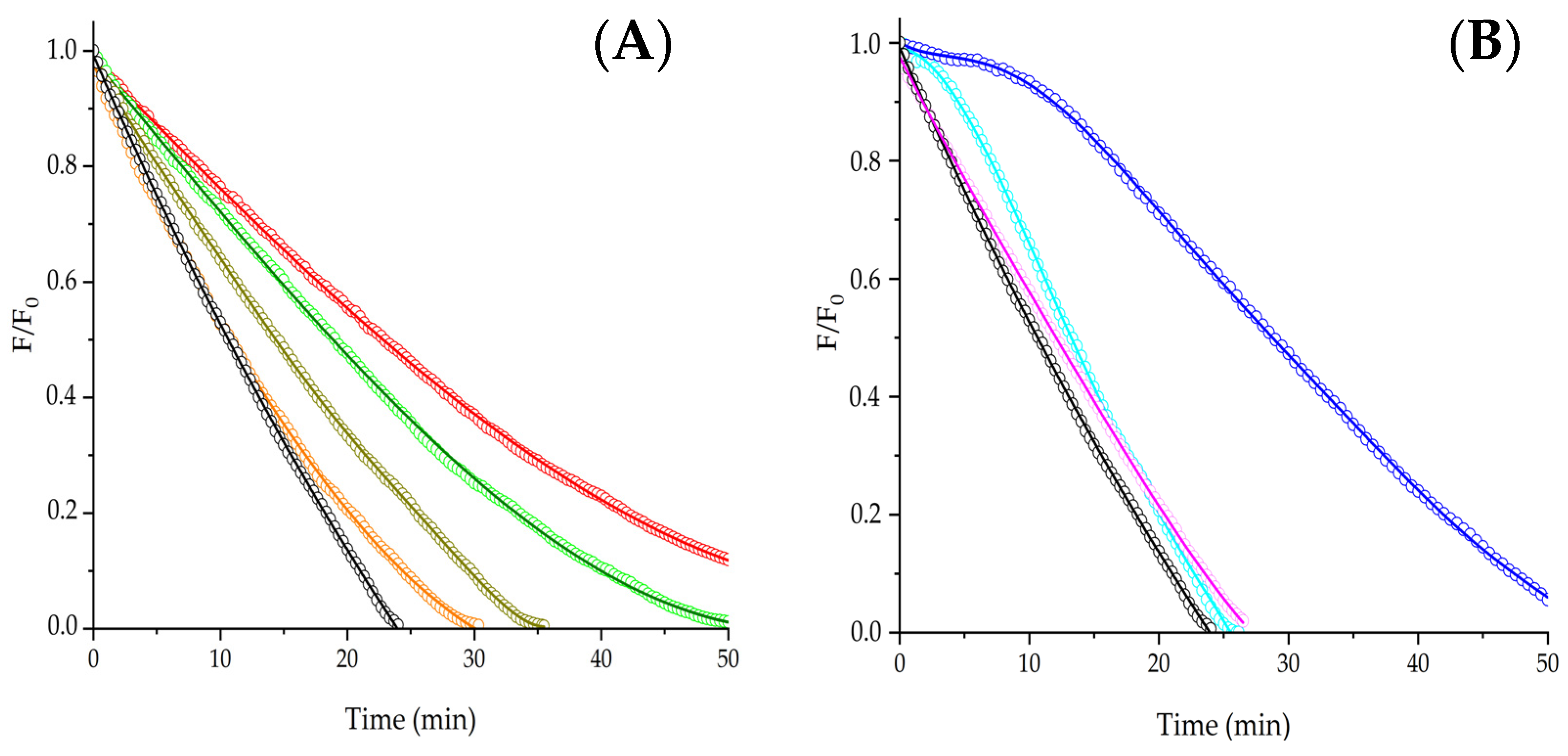
 ) and 5 (
) and 5 ( ) μM PYR elicited by 1 mM Ser-Val-Trp-Ile-Gly-Gly-Ser-Ile. F/F0 = normalized fluorescence intensity.
) μM PYR elicited by 1 mM Ser-Val-Trp-Ile-Gly-Gly-Ser-Ile. F/F0 = normalized fluorescence intensity.