Deep Learning-Driven Molecular Generation and Electrochemical Property Prediction for Optimal Electrolyte Additive Design


Abstract
1. Introduction
2. Materials and Methods
2.1. Electrolyte Additives Dataset
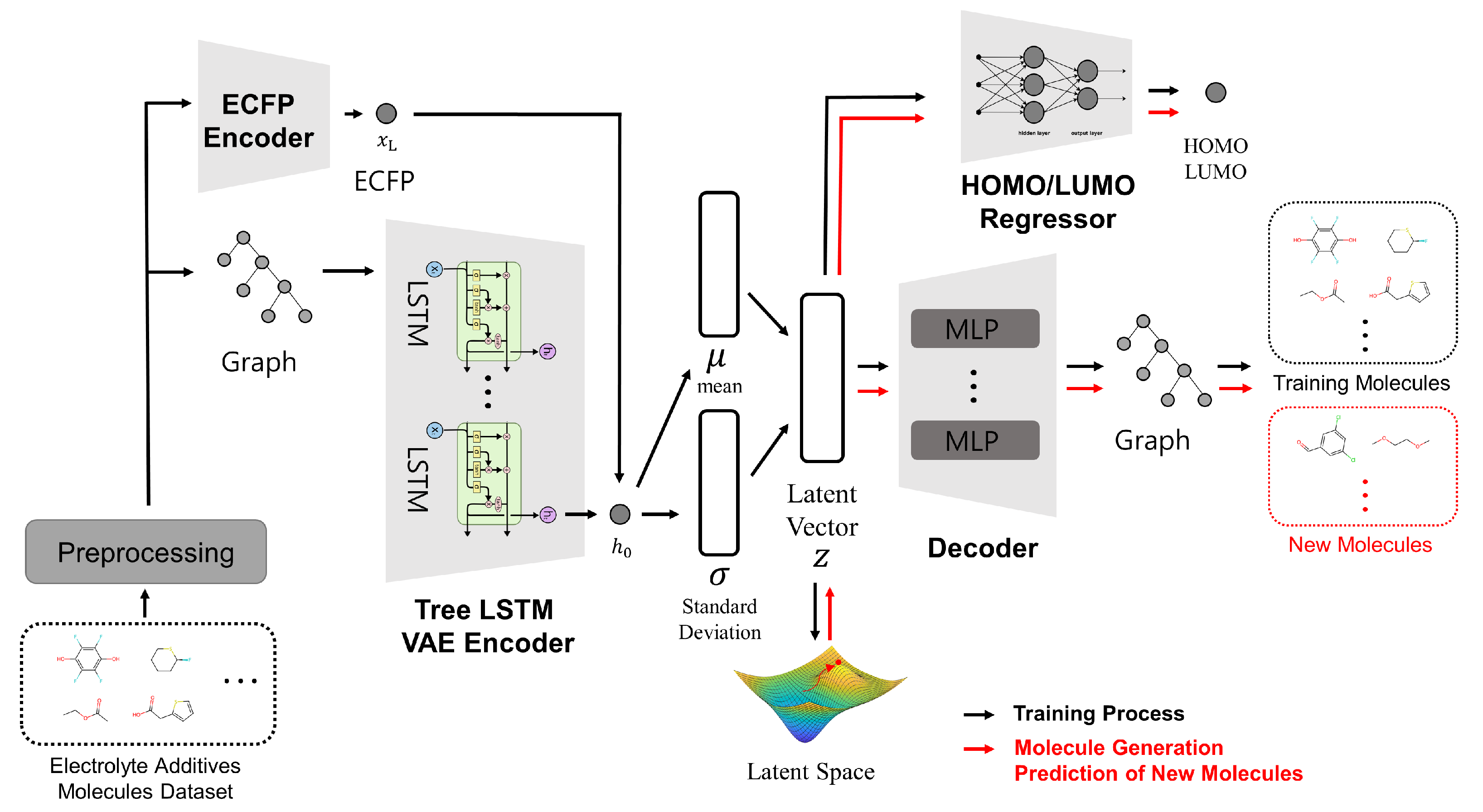
2.2. Model’s Architecture
- Architecture aligned with our requirements and objectives. The dataset described in the following Section 2.1 includes significant candidate molecules used in lithium-ion battery electrolytes. We aim to extract meaningful and valuable information from this high-quality dataset to explore new molecules that could serve as potential electrolyte additives. Additionally, we require accurate predictions of electrochemical parameters such as HOMO/LUMO, which are critical for electrolyte additives. If new materials can be discovered and their HOMO/LUMO values predicted without direct experimentation, lithium-ion battery researchers would be able to anticipate experimental results quickly and efficiently. The VAE-based model with its continuous latent space representation fits these requirements and objectives, enabling us to explore the desired chemical space effectively.
- Performance. Among the VAE-based 3D molecule generation models NPVAE demonstrates the best performance outperforming models such as ChemicalVAE, CG-VAE, JT-VAE, and HierVAE, with a 2D reconstruction accuracy of 0.813 [22,26,27,28]. Based on experimental results from the literature NPVAE was deemed the most suitable model for achieving our performance goals.
- Utilization of structural information. Deep learning models that handle large sets of molecules typically employ various molecular representation methods. While SMILES is a simple and widely used format, it has limitations in capturing full structural information [29]. The NPVAE model overcomes this by converting SMILES inputs into graph-based representations, incorporating structural information through the use of a Tree-LSTM model.
- Chirality handling. Chirality refers to the geometric property of a molecule where its mirror image cannot be superimposed on the original structure, which is crucial in many chemical applications including pharmaceuticals. The NPVAE model by utilizing 3D molecular structures can effectively incorporate chirality enabling more accurate modeling of stereochemical properties. In NPVAE, the model determines whether a molecule is chiral or not by incorporating chirality information from its ECFP.
3. Results
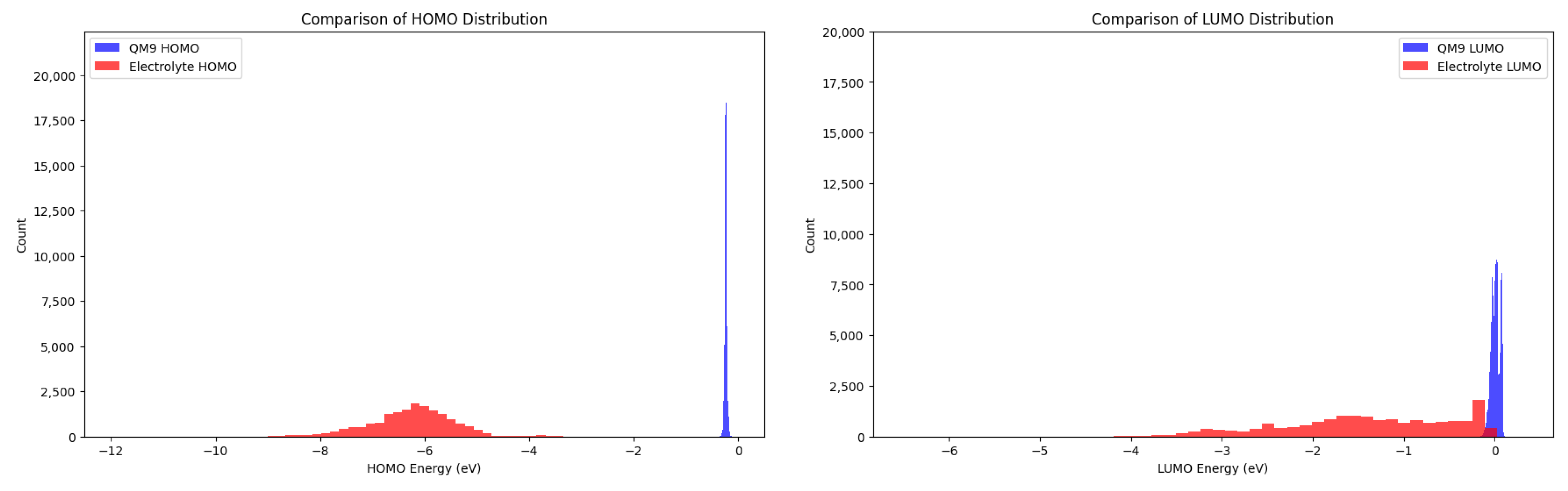
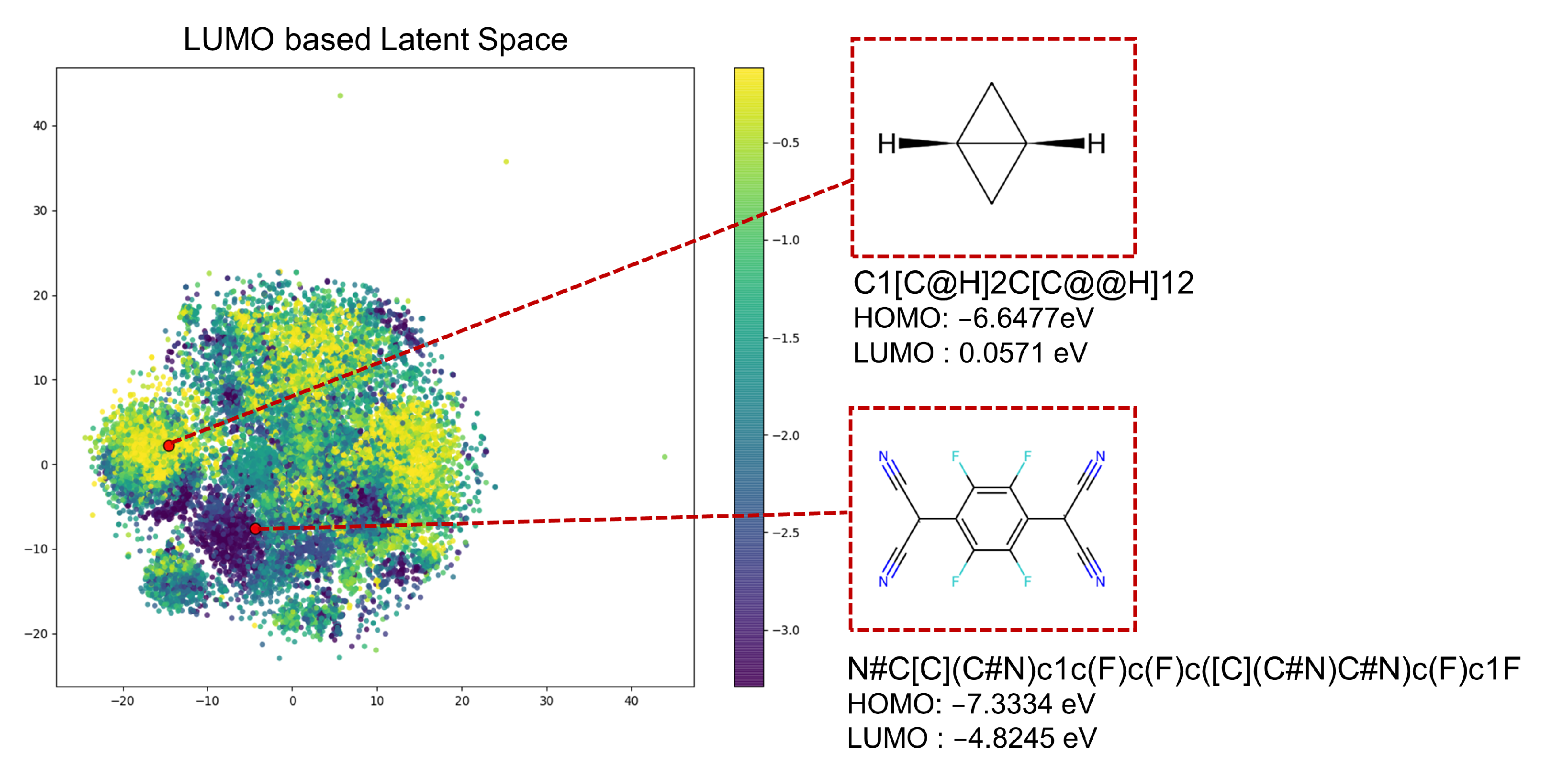
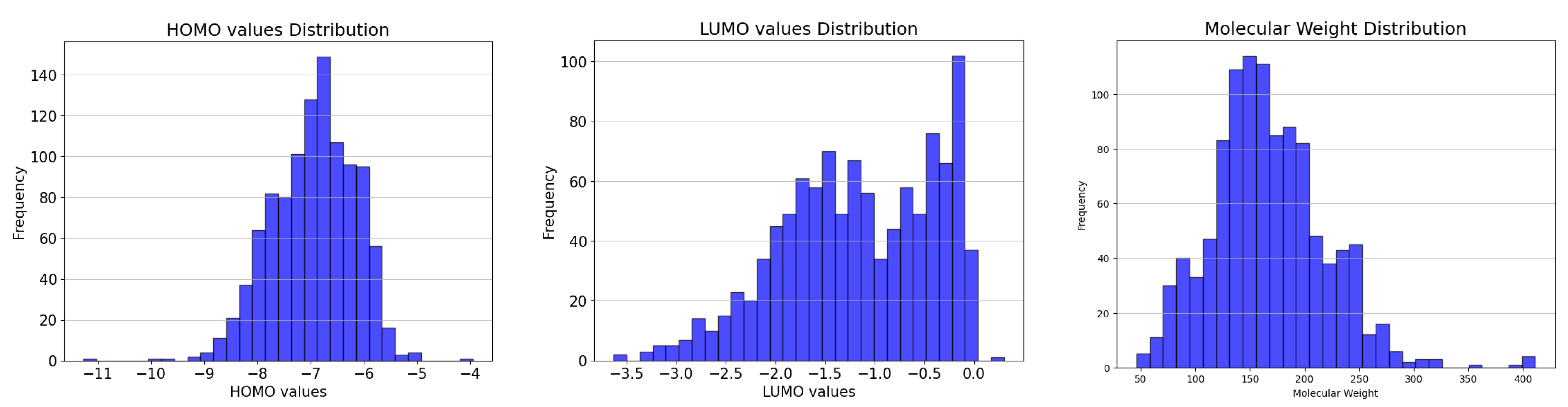
3.1. Dataset Analysis
3.2. Results and Experimental Setup
3.2.1. Reconstruction
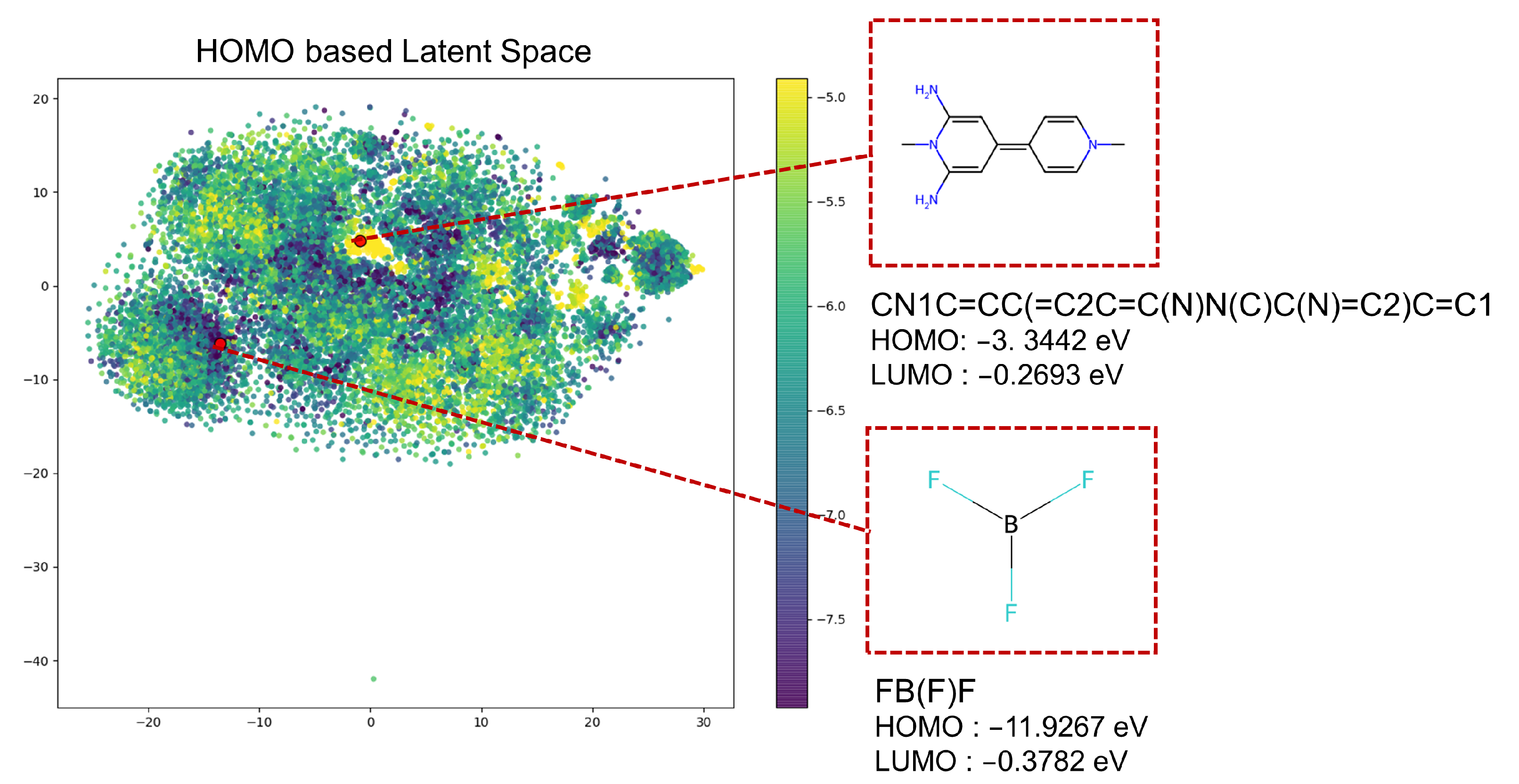
3.2.2. Generation
3.2.3. Molecular Property Prediction
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| HOMO | Highest occupied molecular orbital |
| LUMO | Lowest unoccupied molecular orbital |
| DFT | Density Functional Theory |
| SEI layer | Solid electrolyte interphase |
| GNN | Graph neural network |
| VAE | Variational Autoencoder |
Appendix A
Appendix A.1. Preprocessing
- Initial fragmentation: The molecular structure is represented as a graph , where V denotes atoms and E denotes bonds. Bonds that are not part of ring structures but connect ring atoms are identified and removed, breaking the structure into subgraphs .
- Frequency-based filtering: Among the fragmented substructures, those that do not contain ring structures and appear infrequently (below a set frequency threshold, ) are selected. These substructures become unique labels in the vocabulary, denoted as .
- Functional group decomposition: Further decomposition focuses on specific functional groups. For instance:
- -
- Amide groups: Substructures containing amide groups are identified, and bonds involving the amide C(=O)N are broken to create individual labels.
- -
- Carboxyl and ester bonds: Substructures with carboxyl or ester groups are similarly decomposed of separating bonds involving C(=O)O.
- -
- Aldehyde and ketone groups: Bonds within aldehyde or ketone groups C(=O) are also separated to generate labeled substructures.
- -
- Hydroxyl and ether bonds: Finally, bonds between oxygen and carbon in hydroxyl or ether groups are broken, adding further meaningful labels to the vocabulary.
Appendix A.2. NP-VAE Encoder Process
Appendix A.3. NP-VAE Decoder Process
- Root label prediction: Predicts a substructure label for the root node by applying fully connected layers to the latent variable z. This multi-class classification selects a substructure label from those generated during preprocessing. Specifically, transformations are applied to z through fully connected layers, with a softmax operation at the final layer to determine the most likely root label.
- Topological prediction: Determines whether a child node should be generated under the current node. Binary classification is performed to decide on generating a child node. If a child node is created, bond and label prediction steps follow. Otherwise, the process either terminates (at the root) or backtracks to the parent node for further structure generation.
- Bond prediction: Predicts the type of bond (single, double, or triple) between the current node’s substructure and the child node’s substructure. A ternary classification is applied through -layer fully connected transformations to .
- Label prediction: Predicts the substructure label for the newly generated child node. -layer fully connected layers are applied to for multi-class classification. The predicted substructure label is validated for chemical plausibility. If invalid, bond prediction attempts are adjusted until a valid connection is achieved.
- Latent variable update (z): Updates the latent variable to after label prediction or backtracking. is updated using a fully connected layer that integrates the feature vector , derived from Child-Sum Tree-LSTM. Node-specific features propagate to enrich feature representation.
- Conversion to compound structure: Constructs the compound structure from the generated substructure labels. The tree structure is converted to the final compound structure by linking substructure labels, with bonding information uniquely defining the compound.
- Chirality assignment: Assigns stereochemistry to ensure correct 3D structural representation. -layer fully connected transformations produce an ECFP value, and stereoisomers are generated. The final structure is chosen based on the smallest Euclidean distance between predicted and calculated ECFP.
Appendix B

References
- Reiser, P.; Neubert, M.; Eberhard, A.; Torresi, L.; Zhou, C.; Shao, C.; Metni, H.; van Hoesel, C.; Schopmans, H.; Sommer, T.; et al. Graph neural networks for materials science and chemistry. Commun. Mater. 2022, 3, 93. [Google Scholar] [CrossRef] [PubMed]
- Khemani, B.; Patil, S.; Kotecha, K.; Tanwar, S. A review of graph neural networks: Concepts, architectures, techniques, challenges, datasets, applications, and future directions. J. Big Data 2024, 11, 18. [Google Scholar] [CrossRef]
- Liu, Z.; Wan, G.; Prakash, B.A.; Lau, M.S.; Jin, W. A Review of Graph Neural Networks in Epidemic Modeling. In Proceedings of the 30th ACM SIGKDD Conference on Knowledge Discovery and Data Mining (KDD ’24), Barcelona, Spain, 25–29 August 2024; pp. 6577–6587. [Google Scholar] [CrossRef]
- Vora, L.K.; Gholap, A.D.; Jetha, K.; Thakur, R.R.S.; Solanki, H.K.; Chavda, V.P. Artificial Intelligence in Pharmaceutical Technology and Drug Delivery Design. Pharmaceutics 2023, 15, 1916. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.; Gurnani, R.; Kim, C.; Pilania, G.; Kwon, H.K.; Lively, R.P.; Ramprasad, R. Design of functional and sustainable polymers assisted by artificial intelligence. Nat. Rev. Mater. 2024, 9, 866–886. [Google Scholar] [CrossRef]
- Xu, K. Silicon electro-optic micro-modulator fabricated in standard CMOS technology as components for all silicon monolithic integrated optoelectronic systems. J. Micromech. Microeng. 2021, 31, 054001. [Google Scholar] [CrossRef]
- Merchant, A.; Batzner, S.; Schoenholz, S.S.; Aykol, M.; Cheon, G.; Cubuk, E.D. Scaling deep learning for materials discovery. Nature 2023, 624, 80–85. [Google Scholar] [CrossRef]
- Wu, X.; Wang, H.; Gong, Y.; Fan, D.; Ding, P.; Li, Q.; Qian, Q. Graph neural networks for molecular and materials representation. J. Mater. Inform. 2023, 3, 12. [Google Scholar] [CrossRef]
- Zhou, J.; Cui, G.; Hu, S.; Zhang, Z.; Yang, C.; Liu, Z.; Wang, L.; Li, C.; Sun, M. Graph neural networks: A review of methods and applications. AI Open 2020, 1, 57–81. [Google Scholar] [CrossRef]
- Li, J.; Lim, K.; Yang, H.; Ren, Z.; Raghavan, S.; Chen, P.Y.; Buonassisi, T.; Wang, X. AI applications through the whole life cycle of material discovery. Matter 2020, 3, 393–432. [Google Scholar] [CrossRef]
- Liu, R.L.; Wang, J.; Shen, Z.H.; Shen, Y. AI for dielectric capacitors. Energy Storage Mater. 2024, 71, 103612. [Google Scholar] [CrossRef]
- Bilodeau, C.; Jin, W.; Jaakkola, T.; Barzilay, R.; Jensen, K.F. Generative models for molecular discovery: Recent advances and challenges. WIREs Comput. Mol. Sci. 2022, 12, e1608. [Google Scholar] [CrossRef]
- Pang, C.; Qiao, J.; Zeng, X.; Zou, Q.; Wei, L. Deep Generative Models in De Novo Drug Molecule Generation. J. Chem. Inf. Model. 2024, 64, 2174–2194. [Google Scholar] [CrossRef] [PubMed]
- Meyers, J.; Fabian, B.; Brown, N. De novo molecular design and generative models. Drug Discov. Today 2021, 26, 2707–2715. [Google Scholar] [CrossRef] [PubMed]
- Xue, D.; Gong, Y.; Yang, Z.; Chuai, G.; Qu, S.; Shen, A.; Yu, J.; Liu, Q. Advances and challenges in deep generative models for de novo molecule generation. WIREs Comput. Mol. Sci. 2019, 9, e1395. [Google Scholar] [CrossRef]
- Hu, W.; Liu, Y.; Chen, X.; Chai, W.; Chen, H.; Wang, H.; Wang, G. Deep Learning Methods for Small Molecule Drug Discovery: A Survey. IEEE Trans. Artif. Intell. 2024, 5, 459–479. [Google Scholar] [CrossRef]
- Walters, W.P.; Barzilay, R. Applications of Deep Learning in Molecule Generation and Molecular Property Prediction. Acc. Chem. Res. 2021, 54, 263–270. [Google Scholar] [CrossRef]
- Xu, Y.; Lin, K.; Wang, S.; Wang, L.; Cai, C.; Song, C.; Lai, L.; Pei, J. Deep Learning for Molecular Generation. Future Med. Chem. 2019, 11, 567–597. [Google Scholar] [CrossRef]
- Elton, D.C.; Boukouvalas, Z.; Fuge, M.D.; Chung, P.W. Deep learning for molecular design—A review of the state of the art. Mol. Syst. Des. Eng. 2019, 4, 828–849. [Google Scholar] [CrossRef]
- Sousa, T.; Correia, J.; Pereira, V.; Rocha, M. Generative Deep Learning for Targeted Compound Design. J. Chem. Inf. Model. 2021, 61, 5343–5361. [Google Scholar] [CrossRef]
- Lim, J.; Ryu, S.; Kim, J.W.; Kim, W.Y. Molecular generative model based on conditional variational autoencoder for de novo molecular design. J. Cheminform. 2018, 10, 31. [Google Scholar] [CrossRef]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Bombarelli, R.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Duvenaud, D.; Maclaurin, D.; Blood-Forsythe, M.A.; Chae, H.S.; Einzinger, M.; Ha, D.G.; Wu, T.; et al. Design of efficient molecular organic light-emitting diodes by a high-throughput virtual screening and experimental approach. Nat. Mater. 2016, 15, 1120–1127. [Google Scholar] [CrossRef] [PubMed]
- Kusner, M.J.; Paige, B.; Hernández-Lobato, J.M. Grammar variational autoencoder. In Proceedings of the 34th International Conference on Machine Learning, PMLR 70, Sydney, NSW, Australia, 6–11 August 2017; pp. 1945–1954. [Google Scholar]
- Dai, H.; Tian, Y.; Dai, B.; Skiena, S.S.; Song, L. Syntax-Directed Variational Autoencoder for Molecule Generation. In Proceedings of the 31st Conference on Neural Information Processing Systems (NIPS 2017), Long Beach, CA, USA, 4–9 December 2017. [Google Scholar]
- Liu, Q.; Allamanis, M.; Brockschmidt, M.; Gaunt, A.L. Constrained graph variational autoencoders for molecule design. In Proceedings of the 32nd International Conference on Neural Information Processing Systems (NIPS’18), Montreal, QC, Canada, 2–8 December 2018; pp. 7806–7815. [Google Scholar]
- Jin, W.; Barzilay, R.; Jaakkola, T. Junction Tree Variational Autoencoder for Molecular Graph Generation. arXiv 2019, arXiv:1802.04364. [Google Scholar]
- Jin, W.; Barzilay, R.; Jaakkola, T. Hierarchical generation of molecular graphs using structural motifs. In Proceedings of the 37th International Conference on Machine Learning (ICML’20), Virtual, 13–18 July 2020. [Google Scholar]
- Raghunathan, S.; Priyakumar, U.D. Molecular representations for machine learning applications in chemistry. Int. J. Quantum Chem. 2022, 122, e26870. [Google Scholar] [CrossRef]
- Wigh, D.S.; Goodman, J.M.; Lapkin, A.A. A review of molecular representation in the age of machine learning. WIREs Comput. Mol. Sci. 2022, 12, e1603. [Google Scholar] [CrossRef]
- Chang, J.; Ye, J.C. Bidirectional generation of structure and properties through a single molecular foundation model. Nat. Commun. 2024, 15, 2323. [Google Scholar] [CrossRef]
- Xu, Z.; Lei, X.; Ma, M.; Pan, Y. Molecular Generation and Optimization of Molecular Properties Using a Transformer Model. Big Data Min. Anal. 2023, 7, 142–155. [Google Scholar]
- Mswahili, M.E.; Jeong, Y.S. Transformer-based models for chemical SMILES representation: A comprehensive literature review. Heliyon 2024, 10, e39038. [Google Scholar] [CrossRef]
- Sadeghi, S.; Bui, A.; Forooghi, A.; Lu, J.; Ngom, A. Can large language models understand molecules? BMC Bioinform. 2024, 25, 225. [Google Scholar] [CrossRef]
- Liu, Y.; Guo, B.; Zou, X.; Li, Y.; Shi, S. Machine learning assisted materials design and discovery for rechargeable batteries. Energy Storage Mater. 2020, 31, 434–450. [Google Scholar] [CrossRef]
- Lombardo, T.; Duquesnoy, M.; El-Bouysidy, H.; Årén, F.; Gallo-Bueno, A.; Jørgensen, P.B.; Bhowmik, A.; Demortière, A.; Ayerbe, E.; Alcaide, F.; et al. Artificial Intelligence Applied to Battery Research: Hype or Reality? Chem. Rev. 2022, 122, 10899–10969. [Google Scholar] [CrossRef] [PubMed]
- Severson, K.A.; Attia, P.M.; Jin, N.; Perkins, N.; Jiang, B.; Yang, Z.; Chen, M.H.; Aykol, M.; Herring, P.K.; Fraggedakis, D.; et al. Data-driven prediction of battery cycle life before capacity degradation. Nat. Energy 2019, 4, 383–391. [Google Scholar] [CrossRef]
- Hu, X.; Li, S.E.; Yang, Y. Advanced Machine Learning Approach for Lithium-Ion Battery State Estimation in Electric Vehicles. IEEE Trans. Transp. Electrif. 2016, 2, 140–149. [Google Scholar] [CrossRef]
- Zahid, T.; Xu, K.; Li, W.; Li, C.; Li, H. State of charge estimation for electric vehicle power battery using advanced machine learning algorithm under diversified drive cycles. Energy 2018, 162, 871–882. [Google Scholar] [CrossRef]
- Chemali, E.; Kollmeyer, P.J.; Preindl, M.; Emadi, A. State-of-charge estimation of Li-ion batteries using deep neural networks: A machine learning approach. J. Power Sources 2018, 400, 242–255. [Google Scholar] [CrossRef]
- Lv, C.; Zhou, X.; Zhong, L.; Yan, C.; Srinivasan, M.; Seh, Z.W.; Liu, C.; Pan, H.; Li, S.; Wen, Y.; et al. Machine Learning: An Advanced Platform for Materials Development and State Prediction in Lithium-Ion Batteries. Adv. Mater. 2022, 34, 2101474. [Google Scholar] [CrossRef]
- Ling, C. A review of the recent progress in battery informatics. NPJ Comput. Mater. 2022, 8, 33. [Google Scholar] [CrossRef]
- Zheng, F.; Zhu, Z.; Lu, J.; Yan, Y.; Jiang, H.; Sun, Q. Predicting the HOMO-LUMO gap of benzenoid polycyclic hydrocarbons via interpretable machine learning. Chem. Phys. Lett. 2023, 814, 140358. [Google Scholar] [CrossRef]
- Amici, J.; Asinari, P.; Ayerbe, E.; Barboux, P.; Bayle-Guillemaud, P.; Behm, R.J.; Berecibar, M.; Berg, E.; Bhowmik, A.; Bodoardo, S.; et al. A Roadmap for Transforming Research to Invent the Batteries of the Future Designed within the European Large Scale Research Initiative BATTERY 2030+. Adv. Energy Mater. 2022, 12, 2102785. [Google Scholar] [CrossRef]
- Fichtner, M.; Edström, K.; Ayerbe, E.; Berecibar, M.; Bhowmik, A.; Castelli, I.E.; Clark, S.; Dominko, R.; Erakca, M.; Franco, A.A.; et al. Rechargeable Batteries of the Future—The State of the Art from a BATTERY 2030+ Perspective. Adv. Energy Mater. 2022, 12, 2102904. [Google Scholar] [CrossRef]
- Vegge, T.; Tarascon, J.M.; Edström, K. Toward Better and Smarter Batteries by Combining AI with Multisensory and Self-Healing Approaches. Adv. Energy Mater. 2021, 11, 2100362. [Google Scholar] [CrossRef]
- Haregewoin, A.M.; Wotango, A.S.; Hwang, B.J. Electrolyte additives for lithium ion battery electrodes: Progress and perspectives. Energy Environ. Sci. 2016, 9, 1955–1988. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, Q. Atomic Insights into the Fundamental Interactions in Lithium Battery Electrolytes. Acc. Chem. Res. 2020, 53, 1992–2002. [Google Scholar] [CrossRef]
- Jankowski, P.; Wieczorek, W.; Johansson, P. SEI-forming electrolyte additives for lithium-ion batteries: Development and benchmarking of computational approaches. J. Mol. Model. 2016, 23, 6. [Google Scholar] [CrossRef] [PubMed]
- Borodin, O. Challenges with prediction of battery electrolyte electrochemical stability window and guiding the electrode—Electrolyte stabilization. Curr. Opin. Electrochem. 2019, 13, 86–93. [Google Scholar] [CrossRef]
- Atkins, D.; Ayerbe, E.; Benayad, A.; Capone, F.G.; Capria, E.; Castelli, I.E.; Cekic-Laskovic, I.; Ciria, R.; Dudy, L.; Edström, K.; et al. Understanding Battery Interfaces by Combined Characterization and Simulation Approaches: Challenges and Perspectives. Adv. Energy Mater. 2022, 12, 2102687. [Google Scholar] [CrossRef]
- Bhowmik, A.; Castelli, I.E.; Garcia-Lastra, J.M.; Jørgensen, P.B.; Winther, O.; Vegge, T. A perspective on inverse design of battery interphases using multi-scale modelling, experiments and generative deep learning. Energy Storage Mater. 2019, 21, 446–456. [Google Scholar] [CrossRef]
- Sanchez-Lengeling, B.; Aspuru-Guzik, A. Inverse molecular design using machine learning: Generative models for matter engineering. Science 2018, 361, 360–365. [Google Scholar] [CrossRef]
- Ochiai, T.; Inukai, T.; Akiyama, M.; Furui, K.; Ohue, M.; Matsumori, N.; Inuki, S.; Uesugi, M.; Sunazuka, T.; Kikuchi, K.; et al. Variational autoencoder-based chemical latent space for large molecular structures with 3D complexity. Commun. Chem. 2023, 6, 249. [Google Scholar] [CrossRef]
- Qu, X.; Jain, A.; Rajput, N.N.; Cheng, L.; Zhang, Y.; Ong, S.P.; Brafman, M.; Maginn, E.; Curtiss, L.A.; Persson, K.A. The Electrolyte Genome project: A big data approach in battery materials discovery. Comput. Mater. Sci. 2015, 103, 56–67. [Google Scholar] [CrossRef]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. Commentary: The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef]
- Tai, K.S.; Socher, R.; Manning, C.D. Improved Semantic Representations From Tree-Structured Long Short-Term Memory Networks. arXiv 2015, arXiv:1503.00075. [Google Scholar]
- St. John, P.C.; Phillips, C.; Kemper, T.W.; Wilson, A.N.; Guan, Y.; Crowley, M.F.; Nimlos, M.R.; Larsen, R.E. Message-passing neural networks for high-throughput polymer screening. J. Chem. Phys. 2019, 150, 234111. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, R.; Dral, P.O.; Rupp, M.; von Lilienfeld, O.A. Quantum chemistry structures and properties of 134 kilo molecules. Sci. Data 2014, 1, 140022. [Google Scholar] [CrossRef]
- Polykovskiy, D.; Zhebrak, A.; Sanchez-Lengeling, B.; Golovanov, S.; Tatanov, O.; Belyaev, S.; Kurbanov, R.; Artamonov, A.; Aladinskiy, V.; Veselov, M.; et al. Molecular Sets (MOSES): A Benchmarking Platform for Molecular Generation Models. Front. Pharmacol. 2020, 11, 565644. [Google Scholar] [CrossRef]
- Buttenschoen, M.; Morris, G.M.; Deane, C.M. PoseBusters: AI-based docking methods fail to generate physically valid poses or generalise to novel sequences. Chem. Sci. 2024, 15, 3130–3139. [Google Scholar] [CrossRef]
- Ertl, P.; Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 2009, 1, 8. [Google Scholar] [CrossRef]
- Zhou, G.; Gao, Z.; Ding, Q.; Zheng, H.; Xu, H.; Wei, Z.; Zhang, L.; Ke, G. Uni-Mol: A Universal 3D Molecular Representation Learning Framework. In Proceedings of the Eleventh International Conference on Learning Representations (ICLR 2023), Kigali, Rwanda, 1–5 May 2023. [Google Scholar]
- Lu, S.; Gao, Z.; He, D.; Zhang, L.; Ke, G. Data-driven quantum chemical property prediction leveraging 3D conformations with Uni-Mol+. Nat. Commun. 2024, 15, 7104. [Google Scholar] [CrossRef]
- Rong, Y.; Bian, Y.; Xu, T.; Xie, W.; Wei, Y.; Huang, W.; Huang, J. Self-Supervised Graph Transformer on Large-Scale Molecular Data. arXiv 2020, arXiv:2007.02835. [Google Scholar]
- Pyzer-Knapp, E.O.; Suh, C.; Gómez-Bombarelli, R.; Aguilera-Iparraguirre, J.; Aspuru-Guzik, A. What Is High-Throughput Virtual Screening? A Perspective from Organic Materials Discovery. Annu. Rev. Mater. Res. 2015, 45, 195–216. [Google Scholar] [CrossRef]
- Bolloju, S.; Vangapally, N.; Elias, Y.; Luski, S.; Wu, N.L.; Aurbach, D. Electrolyte additives for Li-ion batteries: Classification by elements. Prog. Mater. Sci. 2025, 147, 101349. [Google Scholar] [CrossRef]
- Han, Y.K.; Lee, K.; Jung, S.C.; Huh, Y.S. Computational screening of solid electrolyte interphase forming additives in lithium-ion batteries. Comput. Theor. Chem. 2014, 1031, 64–68. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, S.; Fan, L.; Gao, L.; Kong, X.; Li, S.; Li, J.; Hong, X.; Lu, Y. Tuning the LUMO Energy of an Organic Interphase to Stabilize Lithium Metal Batteries. ACS Energy Lett. 2019, 4, 644–650. [Google Scholar] [CrossRef]
- Oliveira, A.F.; Da Silva, J.L.F.; Quiles, M.G. Molecular Property Prediction and Molecular Design Using a Supervised Grammar Variational Autoencoder. J. Chem. Inf. Model. 2022, 62, 817–828. [Google Scholar] [CrossRef]
- Cheng, L.; Assary, R.S.; Qu, X.; Jain, A.; Ong, S.P.; Rajput, N.N.; Persson, K.; Curtiss, L.A. Accelerating Electrolyte Discovery for Energy Storage with High-Throughput Screening. J. Phys. Chem. Lett. 2015, 6, 283–291. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Minimum (Min) | Maximum (Max) | Average (Avg) | Molecules |
|---|---|---|---|---|
| QM9 | 16 | 152 | 122 | 133,885 |
| Polymer | 83 | 1768 | 766 | 17,124 |
| Drug-and-Natural-Product | 3 | 8272 | 379 | 10,597 |
| Electrolyte | 9 | 705 | 183 | 17,271 |
| Metric | Value |
|---|---|
| PBValid | 0.995 |
| Validity | 1.000 |
| Novelty | 1.000 |
| Frag Similarity | 0.445 |
| Scaffold Similarity | 0.239 |
| IntDiv | 0.869 |
| SA Score | 0.4253 |
| Model | Dataset | Test Molecules | Property | MAE (eV) |
|---|---|---|---|---|
| Unimol | Electrolyte | 1000 | HOMO | 0.22385 |
| LUMO | 0.04176 | |||
| *Generated | 110 | HOMO | 0.10706 | |
| LUMO | 0.16052 | |||
| NPVAE | Electrolyte | 1000 | HOMO | 0.02099 |
| LUMO | 0.00158 | |||
| *Generated | 1060 | HOMO | 0.04996 | |
| LUMO | 0.06895 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoon, D.; Lee, J.; Lee, S. Deep Learning-Driven Molecular Generation and Electrochemical Property Prediction for Optimal Electrolyte Additive Design. Appl. Sci. 2025, 15, 3640. https://doi.org/10.3390/app15073640
Yoon D, Lee J, Lee S. Deep Learning-Driven Molecular Generation and Electrochemical Property Prediction for Optimal Electrolyte Additive Design. Applied Sciences. 2025; 15(7):3640. https://doi.org/10.3390/app15073640
Chicago/Turabian StyleYoon, Dongryun, Jaekyu Lee, and Sangyub Lee. 2025. "Deep Learning-Driven Molecular Generation and Electrochemical Property Prediction for Optimal Electrolyte Additive Design" Applied Sciences 15, no. 7: 3640. https://doi.org/10.3390/app15073640
APA StyleYoon, D., Lee, J., & Lee, S. (2025). Deep Learning-Driven Molecular Generation and Electrochemical Property Prediction for Optimal Electrolyte Additive Design. Applied Sciences, 15(7), 3640. https://doi.org/10.3390/app15073640