1. Introduction
The original simple fuel cell was invented by Wilhelm Ostwald in 1894, who thought that instead of typical combustion, certain fuels could undergo oxidation through controlled electromechanical mechanisms [
1]. It was not until the mid-1980s that fuel cells were given formal and practical consideration for generating on-demand electrical power in remote areas. Today, fuel cells are a renowned category with many industrial applications and substantial research in various types and operations.
A typical fuel cell contains two electrodes, a cathode and an anode, in two segments isolated by a proton conductive barrier material. The fuel flows through the anode compartment while the reactant flows through the cathode compartment. A redox reaction occurs facilitated by the passage of protons across the proton exchange barrier [
2]. The chemical reaction creates a net electrical potential difference at the cathode and anode sites. Hydrogen is a popular type of fuel cell where hydrogen gas is reacted with oxygen; however, the use of hydrogen peroxide as fuel has promising capabilities to improve upon hydrogen fuel cells. Hydrogen peroxide is stable in liquid form, making it easier to store and transport without the need for high-pressure methods like with hydrogen. Hydrogen peroxide, even at very high concentrations, can be reliably stored in containers made of passivated resistant stainless-steel, high-density polyethylene (HDPE), or glass. Pressurized hydrogen will almost always require the use of a reinforced metallic capsule, and despite all efforts, hydrogen has inherent material damaging risks. A concern with the use of hydrogen peroxide as both the fuel and oxidant pertains to how it can undergo a mixed potential reaction called hydrogen peroxide disproportionation. In this mixed reaction, oxidation and reduction can occur between the peroxide molecules without reacting with the electrode. This causes the degradation of the peroxide without the formation of any potential difference across the electrodes. The decomposition of Hydrogen peroxide in acidic solutions occurs through the redox reactions shown in Equations (
1) and (
2) [
3]. In an alkaline solution, the redox reaction differs as the peroxide enters the reaction in its dissociated form, as shown in Equations (
3) and (
4). In these reactions, the hydrogen peroxide reacts with the anode to produce hydrogen protons and electrons. The electrons travel from the anode to the cathode through a circuit to generate electricity. The electrons then react with the peroxide and hydrogen protons at the cathode electrode to produce water. The net cell electrochemical reaction of the hydrogen peroxide, as shown in Equation (
5), generates a predicted electrical potential of 1.09 V. This theoretical voltage is comparable to other common fuel cells such as hydrogen fuel cells (1.23 V) and methanol fuel cells (1.21 V).
Yamazaki et al. conducted the first academic investigation of a single-compartment hydrogen peroxide, published in 2008 [
4]. The study assessed the effectiveness of Gold (Au), Platinum (Pt), Palladium (Pd), Silver (Ag), and Nickel (Ni) as electrode materials using alkaline hydrogen peroxide as the common electrolyte. Since then, there have been various studies investigating hydrogen peroxide fuel cells as shown in
Table 1.
This paper uses different electrolyte pH values to investigate the use of seven pure elemental metals for the first time as potential electrocatalytic electrodes in a single-compartment hydrogen peroxide fuel cell. This study investigates the following rare earth metals: pure dysprosium (Dy), erbium (Er), gadolinium (Gd), holmium (Ho), terbium (Tb), antimony (Sb), and tantalum (Ta). These metals have not been investigated for use as catalytic electrodes in a peroxide fuel cell setting. However, they have been used as catalysts in related or close fields. A modified Dy catalyst was employed for low-temperature selective catalytic reduction denitrification, Tb mixed oxides were investigated as an alternative for three-way catalysts in engines, and Gd-based photocatalysts have demonstrated favourable properties for environmental remediation [
14,
15,
16,
17]. Dy, Tb, Er, Gd, and Ho were selected as representatives of rare metals that are non-hazardous and relatively inexpensive. Sb and Ta were selected as metals as they also have been used as catalysts in previous research papers. Ta has the added attribute of being resilient against corrosion in harsh chemical conditions such as extreme pH and highly oxidizing settings, like a fuel cell. This study compares the electrocatalytic performances and constructs plausible theories to explain the observed results and the catalytic mechanisms. Key findings from these investigations include that Dy, Er, Gd, Ho, and Tb can function as electrocatalytic electrode materials to produce a moderate cell output potential in the range of 0.14 V to 0.16 V in alkaline hydrogen peroxide electrolytes. (Sb) and (Ta) functioned as the highest-performing electrode in neutral (0.307 V and 0.2 V) and acidic (0.521 V and 0.457 V) electrolytes, respectively. Ta can also perform well in alkaline electrolytes with an output potential of 0.146 V. It was also observed that an acidic electrolyte leads to a more stable and greater energy output.
4. Discussion
This experiment is the first of its kind to explore several pure metals as electrode materials in a fuel cell using hydrogen peroxide as the oxidant and reductant. The experiments show that the metals combinations are capable of generating electrical energy at different levels when used in a single-compartment hydrogen fuel cell model. The results demonstrate consistent electrochemical activity when the novel metals are paired with themselves and other commonly used electrodes (like glassy carbon (GC), Ni, Pt, and Ag). The fabricated rare earth metal electrodes had the most stable activity in the alkaline peroxide electrolyte, as no visible oxidation or corrosion formed. In the Alkaline Electrolyte, any combination of electrodes produced a cell output potential of −0.2 V to 0.2 V. The most optimal combination was Dy with Ni, Ag, Ta, and Sb. This is followed by glassy carbon, as shown in
Table 5. Dy’s greater cell output potential may be due to it having a similar electrocatalytic activity to GC, as they both performed well with Er. However, this trend did not continue, as GC performed well with Gd, Ho, and Tb, while Dy had a lower output potential when paired with them.
When evaluating results, peak power density is useful for comparing results within similar test conditions, including electrolyte pH, fuel, electrolytic salt concentration, voltammetry sweep range, and sweep rate. As the conditions are kept similar in this experiment, the results can be compared, as shown in
Table 9. Ta had the highest electrical power density and alkaline peroxide electrolyte, and Ho had the highest among the rare earth metals. Ta and Sb performed well in the Acidic Electrolyte, delivering similar potential values, with Sb performing the best in an acidic solution with a pH of about 2. It should be noted the quality of the potential values does not correlate with the calculated power density values. Ho, with a larger power density in the alkaline peroxide electrolyte, generated a similar output potential to Er and Tb, despite those having a lower power density than Ho. One possibility for this could be that when Ho is paired with GC in the Alkaline Electrolyte, it produces a similar output potential to Er and Tb, except at a much higher output current. However, this would require further testing to understand.
Acidic peroxide electrolytes are more conducive to catalyzing reactions. Hydrogen peroxide is known to be more stable in acidic pH ranges. As mentioned in the introduction, Equations (
1) and (
2) are the redox reaction of hydrogen peroxide in acidic pH and Equations (
3) and (
4) is the reaction in alkaline pH. The alkaline pH enters the reaction in its dissociated form. This reduces the stability of the hydrogen peroxide in an alkaline pH. Therefore, the decrease in stability disproportionately increases the likelihood of hydrogen peroxide. It can be theorized that the decrease in the disproportionation rate of the peroxide in an Acidic Electrolyte allows it to be more efficient at generating electrocatalytic reactions in a fuel cell setup. This follows the trend of Acidic Electrolytes being more effective than Alkaline Electrolytes in the literature.
The cell output potential indicates how each electrode would act as an electrochemical catalyst (as an anode or cathode). This is determined by the sign of the output potential values when measuring the electrodes with a digital multimeter. Attaching the cathode to the positive lead and the negative lead to the anode will result in a positive electrical potential reading. If there is a negative sign, that means the leads were connected in reverse. In this experiment, the novel electrodes act as cathodes when paired with the glassy carbon electrode in the Alkaline Electrolyte. All the rare earth metal electrodes except for Dy displayed cathodic catalytic activity when paired with the Pt and Ni electrodes.
Other literature has predicted that the further the open circuit potential between two electrodes is, the more likely for the pair to generate a greater output potential in a fuel cell setup [
21]. This assumption was not entirely accurate in this experiment. Pairing Gd with Ta or Tb in an alkaline peroxide should have yielded the highest output potential; however, as
Table 5 shows, this is not the case. Therefore, there may be other mechanisms at play when predicting the output potential from a two-electrode fuel cell setup. This nuance is not commonly reported in other published papers for hydrogen peroxide fuel cells.
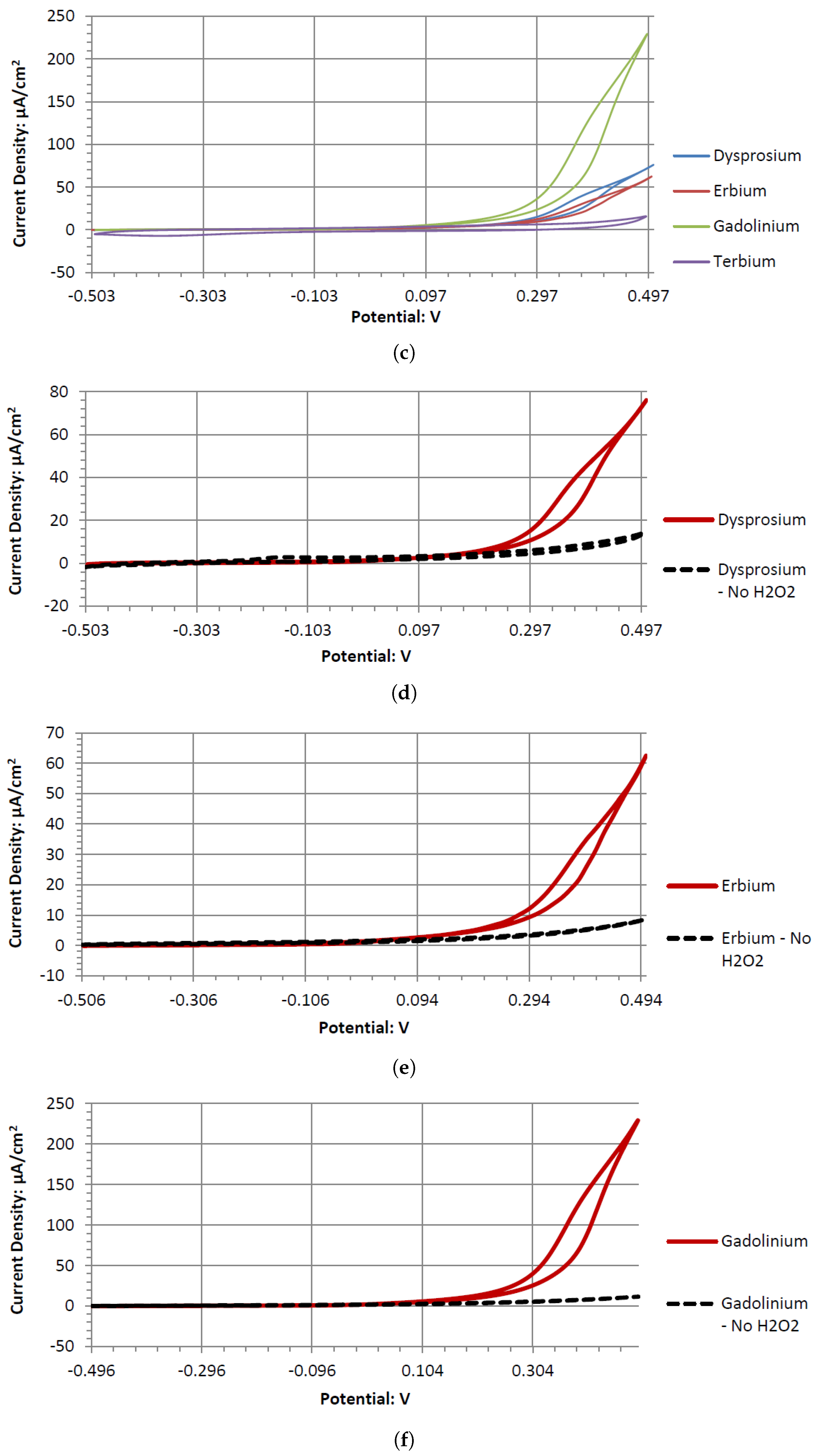
The Dy electrode operates similarly to Er, Gd, Ho, and Tb. However, it produces the least amount of output potential when paired with GC or Pt. It is consistently better than the other rare earth metals when paired with Ta, Ag, or Ni electrodes. Dy, Er, Gd, Ho, and Tb have similar atomic structures, empirically measured atomic radii ranging from 175 to 180 picometers, densities in the approximate range of 8 to 9 g/cm3, oxidation states of +1, +2, and +3, low electrical resistance values ranging roughly from 0.8 to 1.3 μΩ/m, and hexagonal close-packed crystal structures. They do have differences in electron configurations and atoms per electron shell, but no reasonable difference for Dy to behave catalytically differently from the other rare earth metals. Further studies are needed to determine the reasons for catalytic behavior.
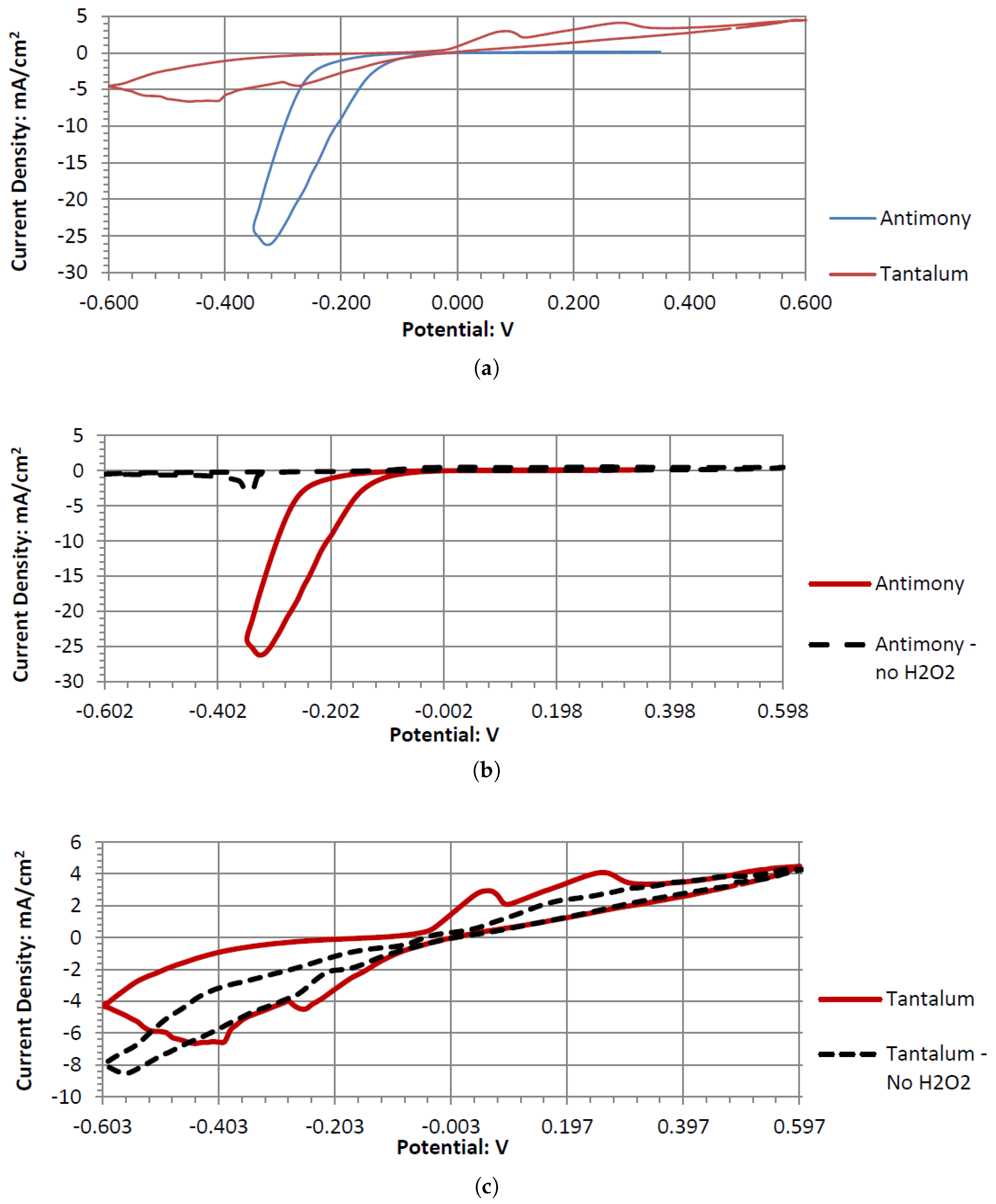
Sb and Ta were the only electrodes tested that were resistant to corrosion in the neutral and acidic hydrogen peroxide electrolytes. Both generated sizable cell output potentials when paired with the Pt electrode. Comparing the various pH peroxide electrolytes, acidic peroxide electrolytes produced a higher output potential. Hydrogen peroxide is also known to have better stability in acidic pH ranges. This leads to the conclusion that an acidic pH is more preferred for the fuel cell model. Sb performs better than Ta when paired with Pt in a neutral electrode. The performance difference decreases as the pH decreases. Also, as the pH decreases to 2, the differences in the output potential between Sb and Ta also decrease. As it decreases further to 1.08, Ta begins to outperform Sb, generating a higher output potential (when paired with Pt). The opposite trend is observed with the open circuit potentials. Sb has a higher OCP in acidic pH solutions and Ta has a higher OCP than Sb in a neutral pH. When paired with the Pt electrode, the Ta and Sb act as anodic electrocatalysts in the neutral and acidic peroxide electrolytes.
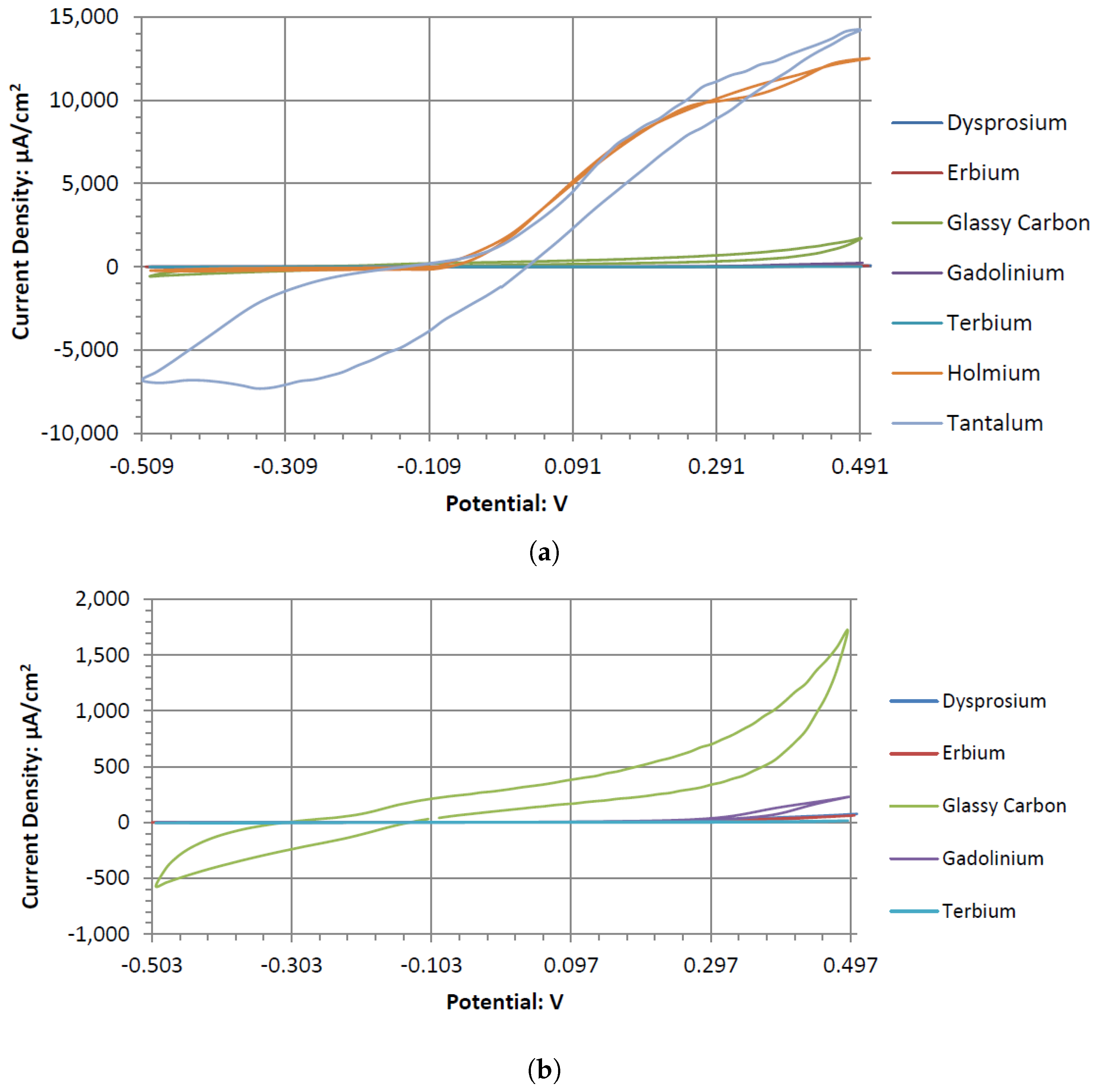
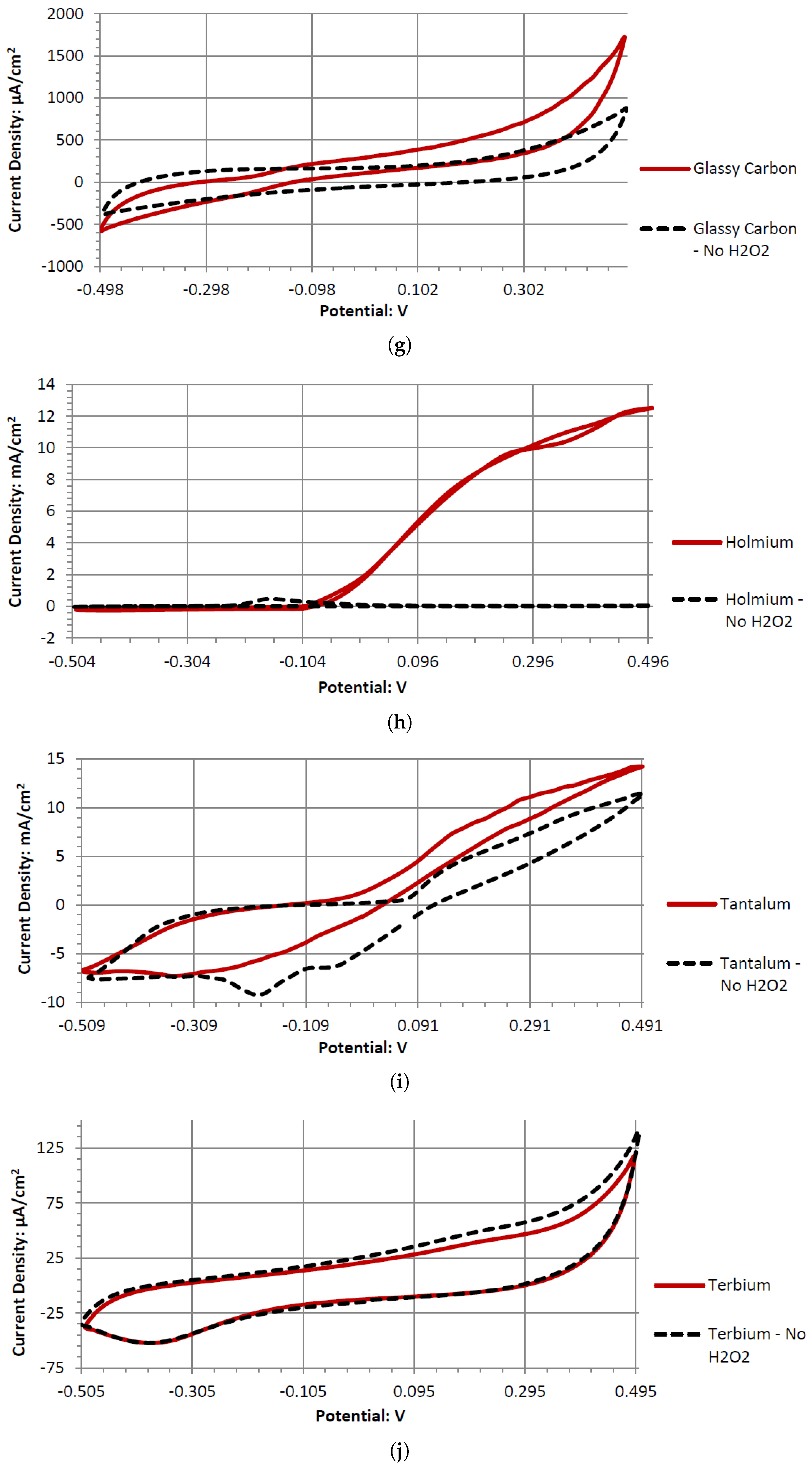
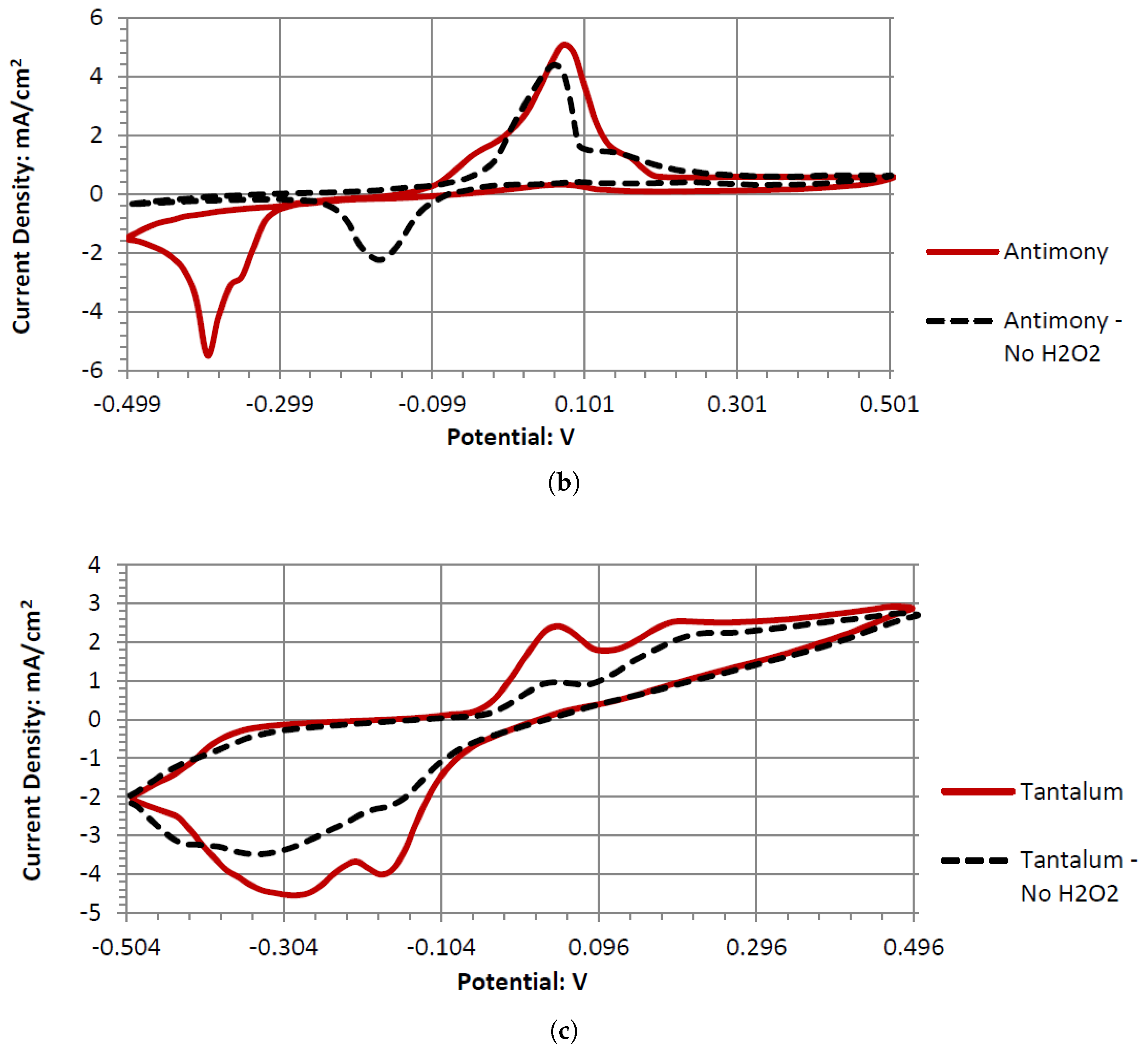
Dy, Er, and Gd produce typical catalytic cyclic voltammograms whether or not they are in peroxide or peroxide-free Alkaline Electrolytes. In the cyclic voltammograms, they have an anodic voltage sweep, indicating oxidative catalysts. Gd and Er voltammagrams have similar potentials around 0.1 V, whereas Dy has a higher potential of 0.2 V. The voltammograms of GC have similar shapes in the presence of hydrogen peroxide and without it. This could be due to GC’s natural defects, where the carbon oxidation can occur at microscopic fault areas through the electrochemical reactions with the dissolved oxygen in the water and hydrogen peroxide in both the anodic and cathodic sweeps. It is also possible the GC has low anodic catalytic activity with hydrogen peroxide, where Ta and Sb would be the main source of the output potentials shown in
Table 5.
Looking at Ho in the oxidative region at the early anodic sweep potentials, it had one of the strongest catalytic rises in its voltammogram. This rise lasted well beyond 0.5 V, after which it started to plateau. The plateau could be a sign of a need for higher mass transport of hydrogen peroxide to this electrode; however, a concentration of 0.3 M of hydrogen peroxide should be a sufficient amount of hydrogen peroxide molecules near the surface of the Ho electrode. Although Ho had a strong catalytic voltammogram, it cannot perform substantially better than the other novel electrodes in generating cell output potentials when paired with the glassy carbon electrode. The voltammogram of Ho had one of the highest current densities during the anodic voltage sweep, and therefore, it is possible that though it produces a similar output potential as Er, Gd, and Tb, a fuel cell of Ho and GC in alkaline peroxide could produce a higher electric current.
Tb’s voltammogram showed no catalytic activity in the anodic or cathodic directions when scanned in the range of −0.5 to 0.5 V. This is surprising, as Tb can generate a considerable cell output potential when paired with the GC electrode in an Alkaline Electrolyte. Given the inert catalytic activity, the measured potential may be a result of the paired electrodes catalytic activity. An electrode pairing that uses Tb would likely create a measurable amount of potential with low amounts of current.
Sb demonstrated a similar electrochemical activity to Tb in alkaline peroxide. The Sb electrode would generate a large output potential even though the electrode itself formed surface corrosion when used in a cyclic voltammetry experiment. This corrosion could be the result of a low current and a higher output potential. Therefore, forcefully sweeping the potentials in a cyclic voltammetry leads to the observed surface corrosion layer.
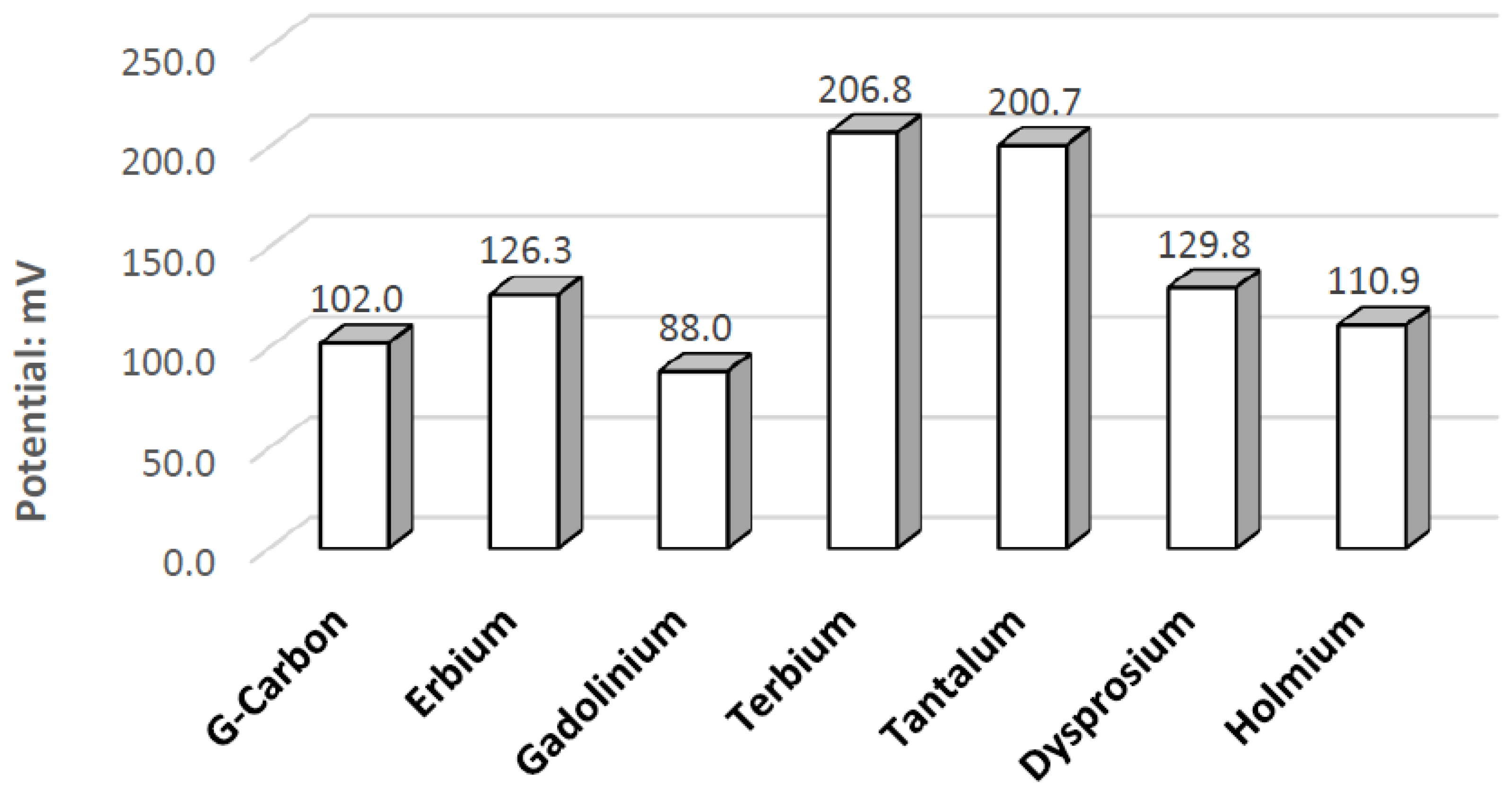
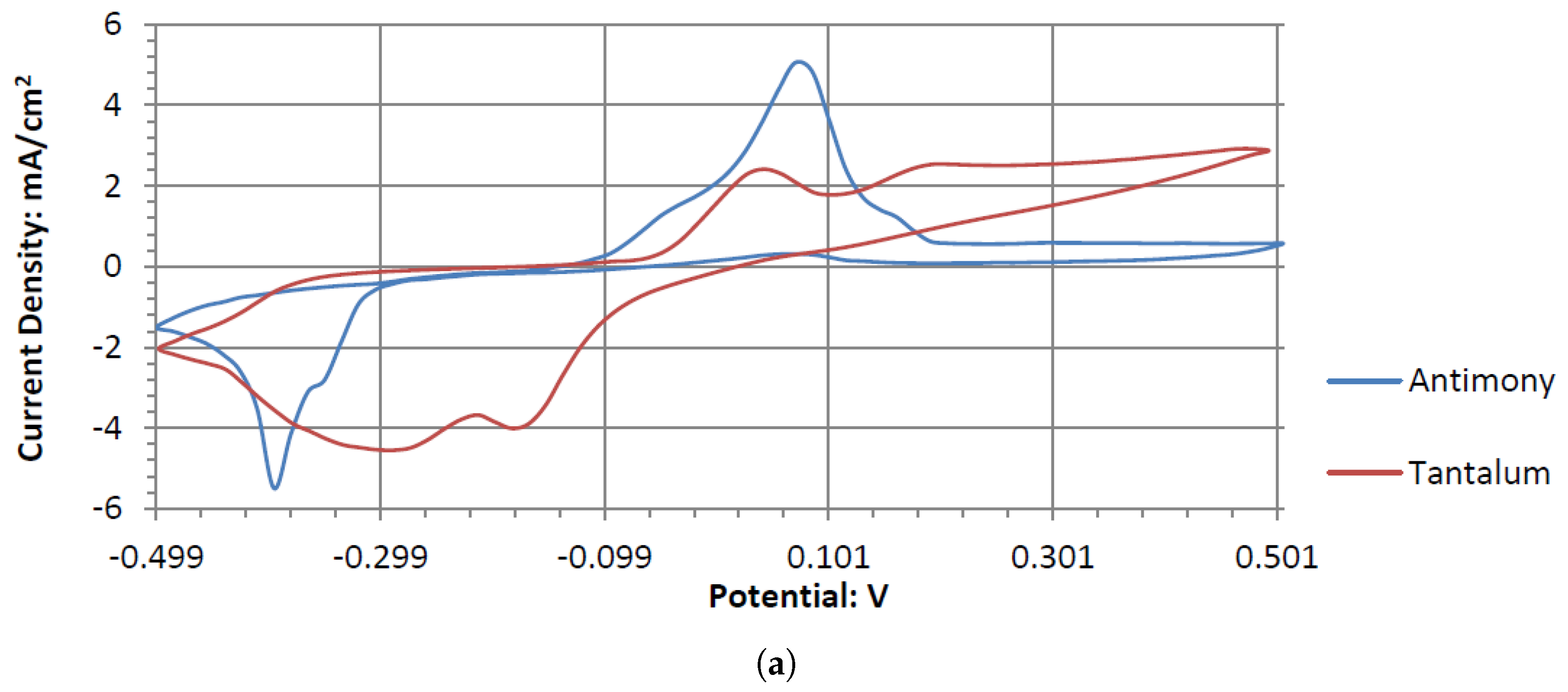
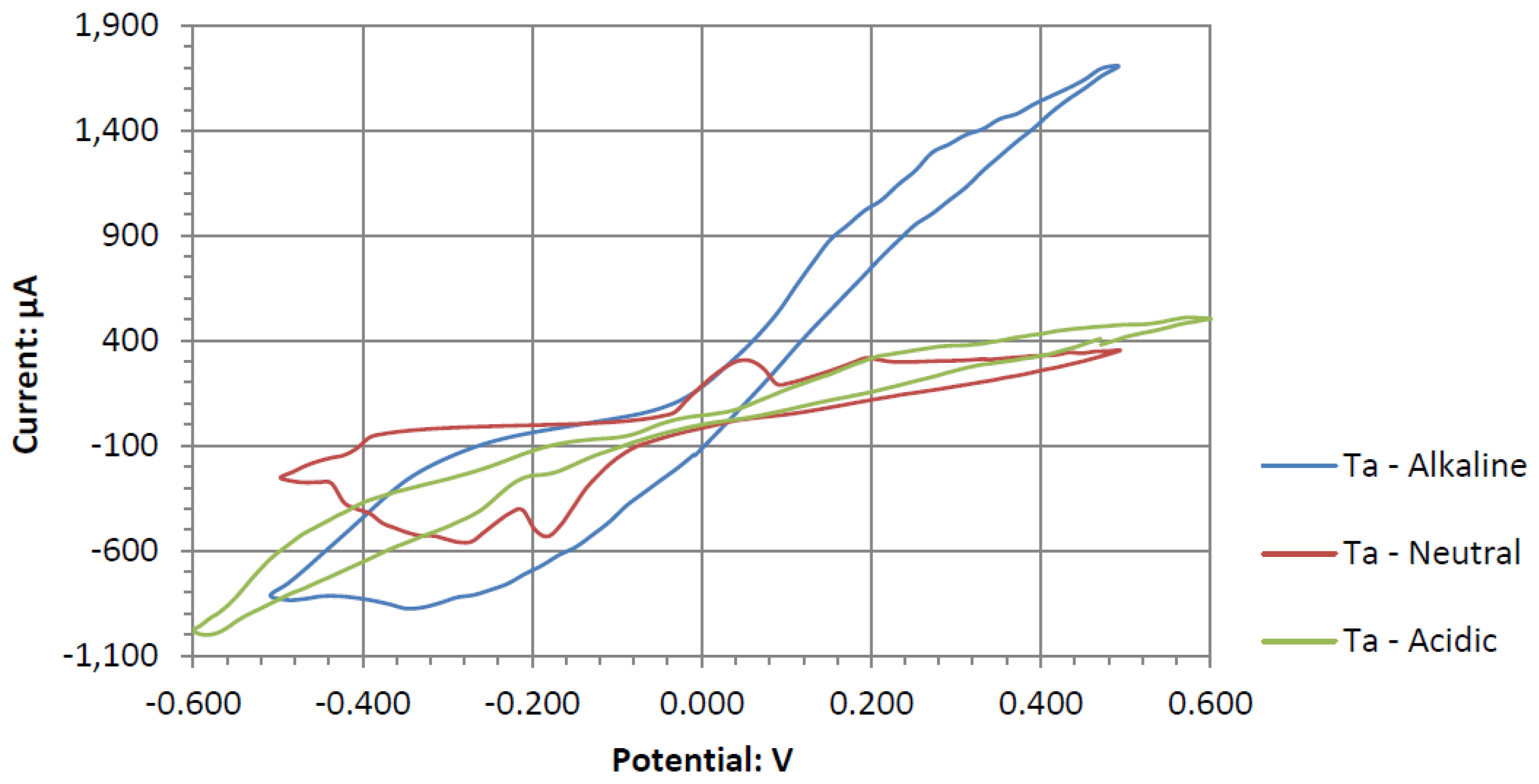
Ta is a strong electrode in alkaline peroxide. Compared to any other novel electrode tested, it has the greatest catalytic peak rise in the oxidative scan direction. The catalytic peak also increases by about 3 mA/cm2 at 0.5 V when hydrogen peroxide is included. In the electrolyte without hydrogen peroxide, it is composed of water and sodium hydroxide. It is unknown what is being oxidized during the Ta electrodes cyclic voltammetry, as it would require a much higher potential for the hydroxide ion to oxidize in alkaline pH to form water. It also takes a higher potential than 0.5 V to generate tantalum hydroxide or tantalum oxide. Ta was also one of the only electrodes that was compatible in the alkaline, neutral, and acidic pH ranges, keeping its polished surface after any electrochemical tests.
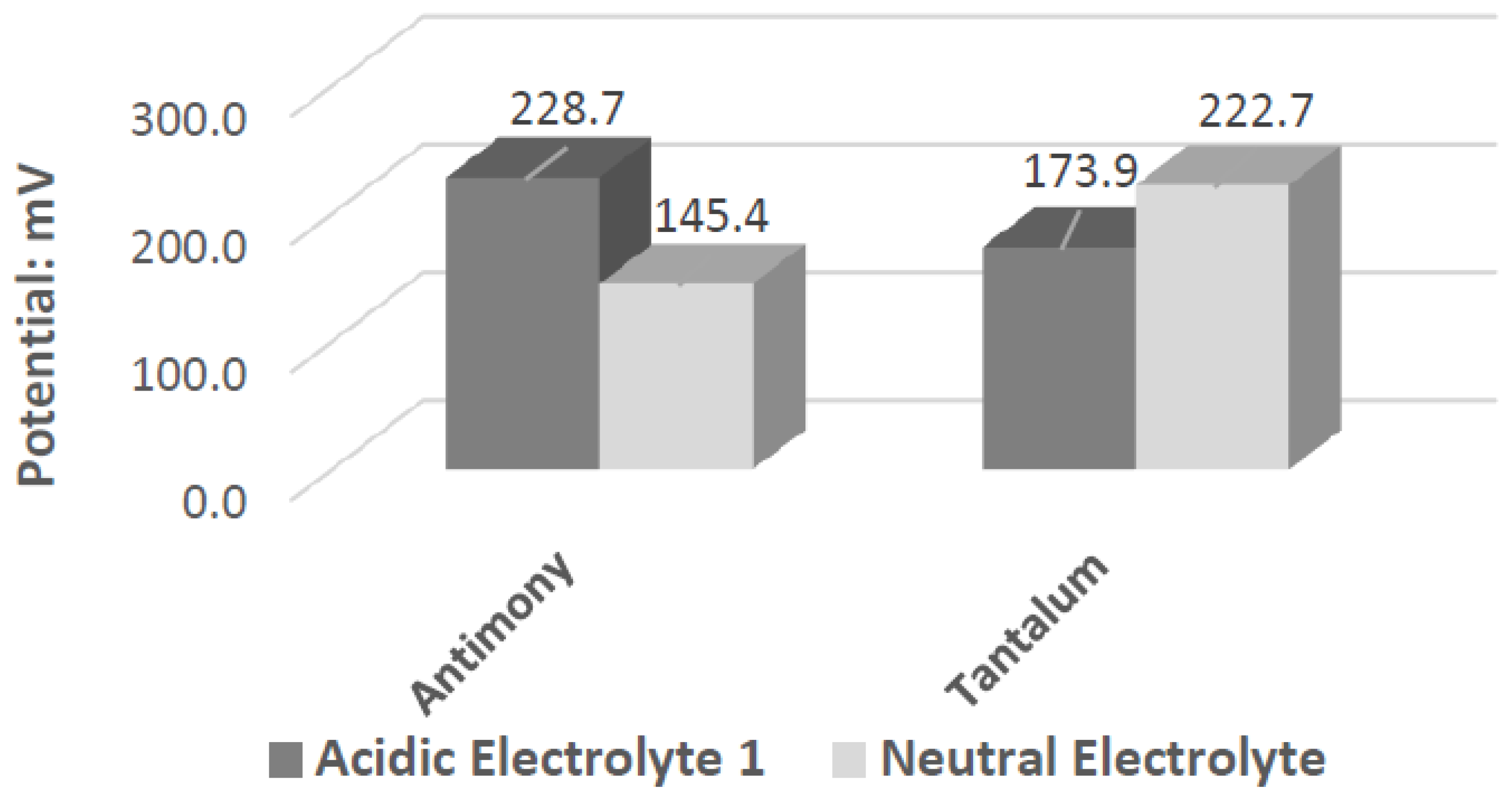
Ta’s different catalytic activity in the presence of hydrogen peroxide in acidic, alkaline, and neutral pH electrolytes can be observed by overlapping its voltammograms in
Figure 10. In the three pH electrolytes, Ta has similar onset potentials in the anodic potential sweep direction. The open circuit potentials in the various electrolytes decrease in order of neutral (222.7 mV), alkaline (200.7 mV), and acidic (173.9 mV). The slope of the catalytic voltammogram peaks in the anodic sweep decreases in the order of alkaline, acidic, and neutral. This demonstrates that a Ta electrode in an alkaline peroxide electrolyte will lead to a lower output potential and higher current compared to using an acidic peroxide electrolyte.
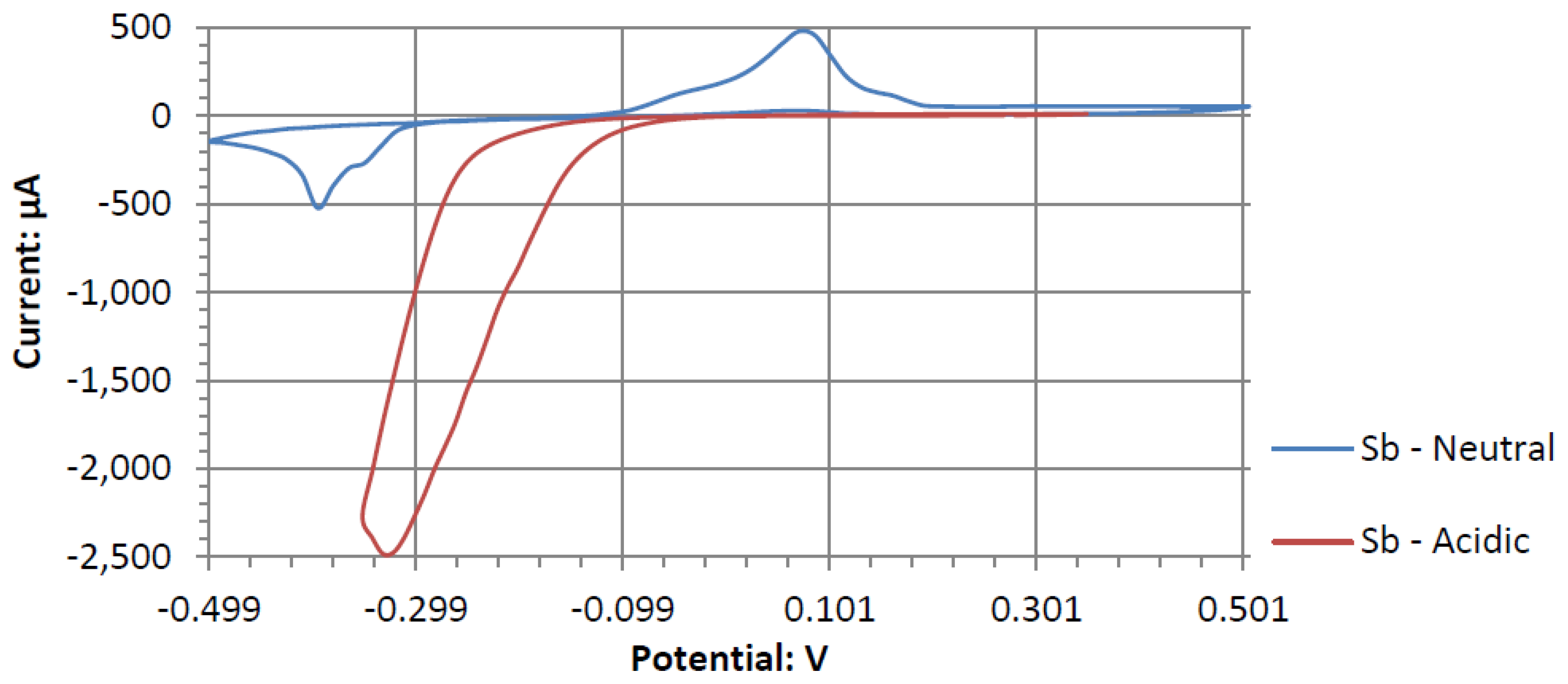
The voltammograms of ceSb in the different pH electrolytes is shown in
Figure 11. Sb has the most catalytic activity in the neutral pH. Given the shape of Antimony’s voltammogram in neutral peroxide resembles a possible reversible passivation reaction. The oxide layer formation and removal on the electrode surface are seen as the anodic and cathodic potential sweeps occur. The observed peaks can be the result of mass adsorption and desorption of salt ions to and from electrode surfaces. This behavior is not observed in the acidic peroxide electrolyte. The Acidic Electrolyte demonstrates a typical cathodic catalytical behavior with an onset potential of about −0.09 V.
The observed redox catalytic activities of Dy, Tb, Er, Dg, Gd, Ta, and Sb pure metal electrodes on hydrogen peroxide in their respective pH ranges is likely from molecular oxide layers forming on the electrode surface. Since all the metals can switch to higher and lower oxidation states, the oxide layer acts as an oxygen reservoir source. This oxygen reservoir introduces an excess of oxygen atoms, allowing the peroxide to undergo an electron-yielding decomposition reaction. The surface oxide formation occurs when the metal is submerged in the electrolyte and comes in contact with water, oxygen, and hydrogen peroxide. The switching activity causes the catalytic functions to accelerate the transfer of oxygen (and electrons) to and from electrode surfaces in the hydrogen peroxide redox reactions.
Figure 12 visually demonstrates this mechanism. Ta and Sb are observed to have a similar yet improved performance compared to the other pure metals. The reason for the difference in the catalytic mechanisms in the specific context of hydrogen peroxide fuel cells has not been investigated in the literature; however, it can be hypothesized that the formation of a thin metal oxide layer on the surface of the electrode may play a central role in the reaction and the electrical yield. Given that there is no investigation on this exact mechanism in the literature, there is an opportunity for a separate and chemically focused investigation in the field of inorganic chemistry catalysis to determine the exact reaction mechanism. One way to determine the involvement of the oxide layer in the catalytic reaction would be to treat the electrode surfaces with a known corrosion inhibitor agent, such as benzotriazole or tolyltriazole, right after they are fully polished. The corrosion-inhibiting molecules often help with preventing oxidation from occurring on the surface. If the corrosion-inhibitor-treated electrode has a drop in electrocatalytic output in the fuel cell, then there could be some evidence from this quick test. This testing would also have its shortcomings, as coverage of the electrode surface with a molecular layer of benzotriazole may also cause isolation of the metal surface from being able to effectively reach the electrolyte and its contents.
Explanations in Other Literature
As there is a lack of prior research on the use of these novel electrode materials in hydrogen peroxide fuel cells and the unavailability of advanced instrumentation, it is difficult to further investigate and precisely theorize the reasons for the newly investigated electrode behaviors. Some indirect references strengthen the hypothesis.
Rare earth metals have been indirectly investigated in their reaction with hydrogen peroxide. Cerium dioxide (CeO
2) and other oxides of lanthanum (La) have been used as the active catalysts in fuel cells to oxidize low molecular weight alcohol [
22]. In these electrochemical reactions, the rate of oxidation reactions to the molecules adsorbed onto their surface is enhanced by these rare metal oxides, providing a localized oxygen storage capacity. This publication describes CeO
2’s reaction with hydrogen peroxide using chemical Equations (
6) and (
7). The equations show that the oxide of Ce at lower oxidation states transform into an oxide with a high oxidation state, carrying more oxygen as an oxygen storage source. The higher oxidation state CeO
2 catalytically reacts with the hydrogen peroxide to return to its lower oxidation state, converting the hydrogen peroxide to water.
A different report demonstrated that rare earth metals such as Ce can act similarly to the infamous Fenton reaction, catalytically reacting and decomposing hydrogen peroxide [
23]. Another study examined the reaction mechanisms involved in using CeO
2 and gadolinium oxide to decompose hydrogen peroxide [
24]. The reactions involves the adsorption of hydrogen peroxide molecules onto the metal oxide surfaces, which proceeds to its catalytic decomposition to form water and oxygen. A thin layer of the respective oxides may be forming on the electrode surfaces when inserted into the alkaline peroxide electrolyte, which then acts as a catalytic surface for the peroxide oxidation to occur. Salts of La and lanthanum-doped zinc hydroxycarbonates also catalytically induce disproportionation of hydrogen peroxide [
25,
26]. Gadolinium-doped cerium oxide is reported to oxidize hydrogen peroxide to form hydroxyl radicals [
27]. Therefore, existing publications indicate that the electrocatalytic activities of some rare earth metals occur from their oxides instead of the metals in their pure unreacted forms.
Fewer publications investigate the electrocatalytic ability of Sb and Ta in hydrogen peroxide, as demonstrated by this experiment. One experiment observed that Sb has a higher order reaction order to form SB(OH)
4− as a hydroxide byproduct [
27], explaining Sb’s susceptibility to corrode in alkaline peroxide solution. However, this does not explain the same corrosion without the presence of hydrogen peroxide. Sb is commonly used in simple pH meter electrodes [
28,
29]. It provides a good degree of robustness to test the pH of highly alkaline and acidic solutions, where the pH function is related to the natural layer of antimony oxide formed on the electrode surface. In terms of catalytic activity, antimony oxide films have been used to produce hydrogen peroxide when an electric potential of 3.08 V is applied [
30]. Sb has demonstrated the capability to catalytically interact with hydrogen peroxide molecules as the addition of Sb to Pt has been shown to create a catalyst for oxidation of organic matters such as cinnamyl alcohol [
31]. It was also demonstrated that Sb alloys can decompose hydrogen peroxide through sufficient lowering of the needed activation energy [
32]. Combining the limited literature available, it is possible to theorize that the redox catalytic activity of Sb in hydrogen peroxide may involve the molecular oxide layer on the surface of the Sb electrode. The antimony oxide may provide a localized oxygen storage capacity to catalytically facilitate redox reactions. Since Sb
2O
3 leads to the formation of hydrogen peroxide, the reverse could occur if Sb
2O
3 is submerged with hydrogen peroxide and paired with another catalytically active electrode to consume the hydrogen peroxide to generate electric energy.
Similar uses and behaviors have been observed in the literature for tantalum oxides’ ability to interact with hydrogen peroxide. When tantalum oxide (Ta
2O
5) meets hydrogen peroxide, active oxygen generation is observed when forming hydroxyl radicals [
33]. Tantalum-based compounds show varied electrocatalytic activity with hydrogen peroxide. Tantalum carbide can decompose hydrogen peroxide, although at a slower rate than niobium carbide. In contrast, a chlorine salt of Ta catalyzes the oxidation of sulfides with high hydrogen peroxide concentrations, while Ta
2O
5 does not [
34]. Tantalum oxides are also used for oxygen reduction to produce hydrogen peroxide. The electrocatalytic behavior of Ta is thought to be due to its natural oxide layer, which protects against corrosion and enables peroxide redox reactions across various pH levels.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}