The Potential Therapeutic Role of Bruton Tyrosine Kinase Inhibition in Neurodegenerative Diseases

 ,
,  ,
,  ,
,  , and
, and 
{kind=link}
{kind=link}
Abstract
1. Introduction
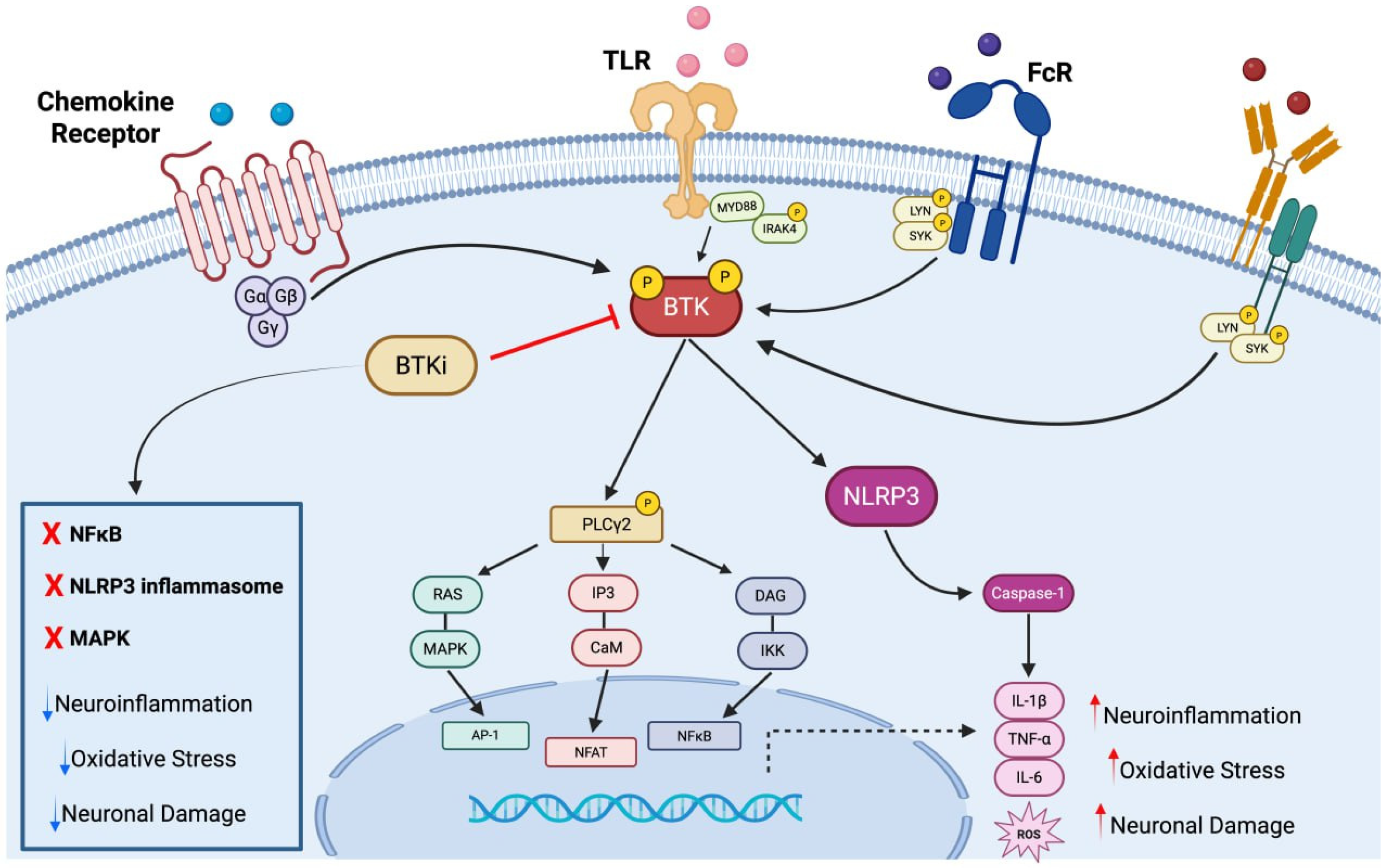
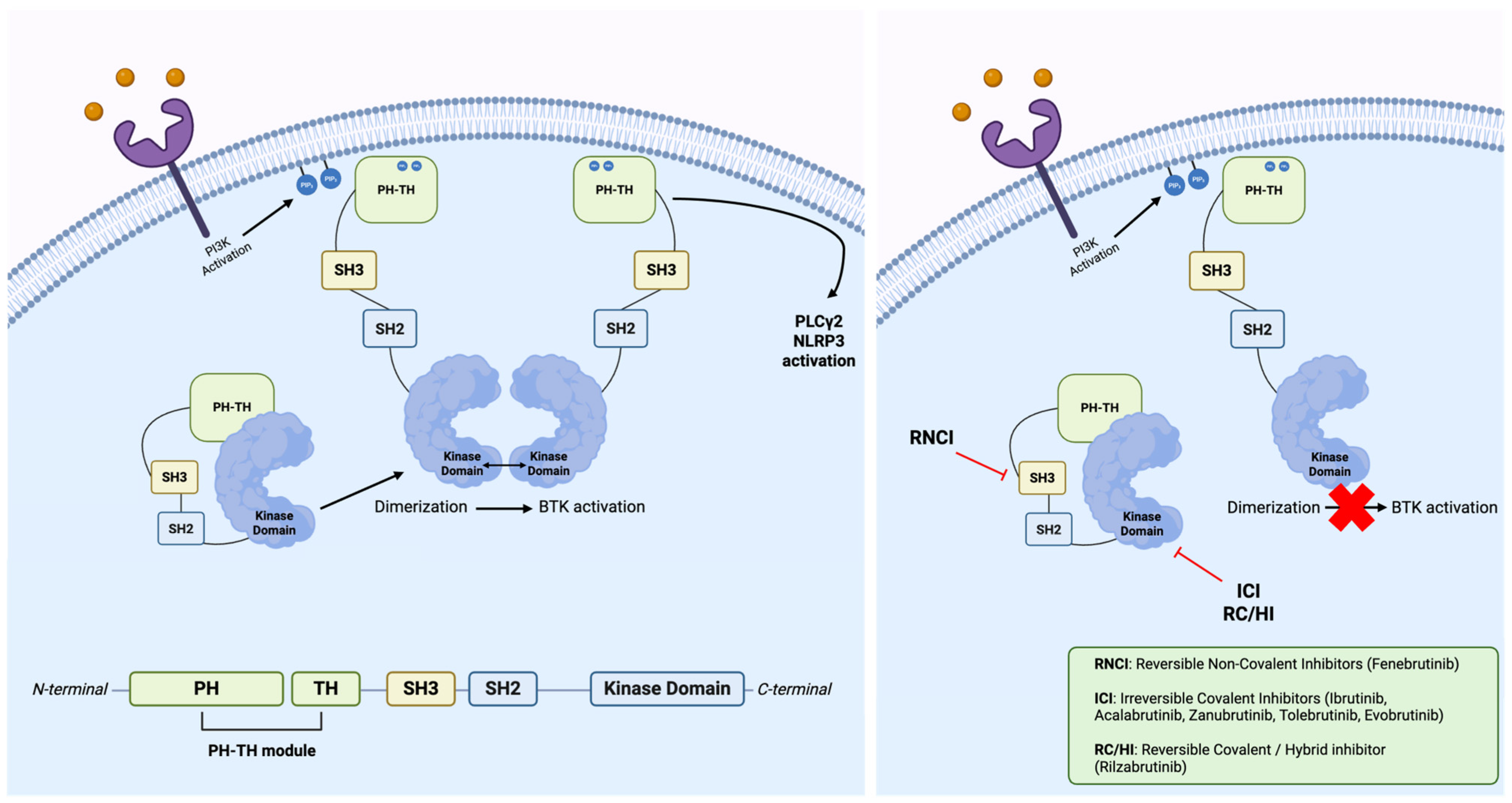
2. Bruton Tyrosine Kinase
Bruton Tyrosine Kinase’s Inhibitors
3. Bruton Tyrosine Kinase’s Inhibitors in Neurodegenerative Disease
3.1. Alzheimer’s Disease
3.2. Parkinson’s Disease
3.3. Multiple Sclerosis
3.4. Ischemic Stroke
3.5. Huntington’s Disease
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bhanumathy, K.K.; Balagopal, A.; Vizeacoumar, F.S.; Vizeacoumar, F.J.; Freywald, A.; Giambra, V. Protein Tyrosine Kinases: Their Roles and Their Targeting in Leukemia. Cancers 2021, 13, 184. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, M.; Algarzae, N.K.; Moussa, C. Tyrosine Kinases: Multifaceted Receptors at the Intersection of Several Neurodegenerative Disease-Associated Processes. Front. Dement. 2024, 3, 1458038. [Google Scholar] [CrossRef] [PubMed]
- Szilveszter, K.P.; Németh, T.; Mócsai, A. Tyrosine Kinases in Autoimmune and Inflammatory Skin Diseases. Front. Immunol. 2019, 10, 1862. [Google Scholar] [CrossRef] [PubMed]
- Thomson, R.J.; Moshirfar, M.; Ronquillo, Y. Tyrosine Kinase Inhibitors. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Boskabadi, S.J.; Dashti, A.; Karevan, S.; Kargar-Soleimanabad, S.; Salehifar, E. Clinical Uses and Safety Concerns of Tyrosine Kinase Inhibitors with a Focus on Novel Drugs: A Narrative Review. J. Oncol. Pharm. Pract. 2023, 12, 10781552231174790. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, D.J.; Witte, O.N. The Btk Subfamily of Cytoplasmic Tyrosine Kinases: Structure, Regulation and Function. Semin. Immunol. 1995, 7, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.T.; Gardberg, A.; Pereira, A.; Johnson, T.; Wu, Y.; Grenningloh, R.; Head, J.; Morandi, F.; Haselmayer, P.; Liu-Bujalski, L. Ability of Bruton’s Tyrosine Kinase Inhibitors to Sequester Y551 and Prevent Phosphorylation Determines Potency for Inhibition of Fc Receptor but Not B-Cell Receptor Signaling. Mol. Pharmacol. 2017, 91, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Konen, F.F.; Möhn, N.; Witte, T.; Schefzyk, M.; Wiestler, M.; Lovric, S.; Hufendiek, K.; Jendretzky, K.F.; Gingele, S.; Schwenkenbecher, P.; et al. Disease-Modifying Strategies: Targeting Protein Kinases in Multiple Sclerosis and Other Autoimmune Disorders. Autoimmun. Rev. 2025, 24, 103754. [Google Scholar] [CrossRef] [PubMed]
- Brunner, C.; Müller, B.; Wirth, T. Bruton’s Tyrosine Kinase Is Involved in Innate and Adaptive Immunity. Histol. Histopathol. 2005, 20, 945–955. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.M.; Cookson, M.R.; Bosch, L.V.D.; Zetterberg, H.; Holtzman, D.M.; Dewachter, I. Hallmarks of Neurodegenerative Diseases. Cell 2023, 186, 693–714. [Google Scholar] [CrossRef] [PubMed]
- D’Egidio, F.; Castelli, V.; d’Angelo, M.; Ammannito, F.; Quintiliani, M.; Cimini, A. Brain Incoming Call from Glia during Neuroinflammation: Roles of Extracellular Vesicles. Neurobiol. Dis. 2024, 201, 106663. [Google Scholar] [CrossRef] [PubMed]
- Keaney, J.; Gasser, J.; Gillet, G.; Scholz, D.; Kadiu, I. Inhibition of Bruton’s Tyrosine Kinase Modulates Microglial Phagocytosis: Therapeutic Implications for Alzheimer’s Disease. J. Neuroimmune Pharmacol. 2019, 14, 448–461. [Google Scholar] [CrossRef] [PubMed]
- Langlois, J.; Lange, S.; Ebeling, M.; Macnair, W.; Schmucki, R.; Li, C.; DeGeer, J.; Sudharshan, T.J.J.; Yong, V.W.; Shen, Y.-A.; et al. Fenebrutinib, a Bruton’s Tyrosine Kinase Inhibitor, Blocks Distinct Human Microglial Signaling Pathways. J. Neuroinflamm. 2024, 21, 276. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Bruton Tyrosine Kinase Inhibitors in B-Cell Malignancies: Their Use and Differential Features. Target. Oncol. 2022, 17, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, R.; Kirk, A.D. Long-Term Toxicity of Immunosuppressive Therapy. Transplant. Liver 2015, 1354–1363. [Google Scholar] [CrossRef]
- Wang, Q.; Pechersky, Y.; Sagawa, S.; Pan, A.C.; Shaw, D.E. Structural Mechanism for Bruton’s Tyrosine Kinase Activation at the Cell Membrane. Proc. Natl. Acad. Sci. USA 2019, 116, 9390–9399. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Vogan, E.M.; Nocka, L.M.; Rosen, C.E.; Zorn, J.A.; Harrison, S.C.; Kuriyan, J. Autoinhibition of Bruton’s Tyrosine Kinase (Btk) and Activation by Soluble Inositol Hexakisphosphate. eLife 2015, 4, e06074. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, T.; Kurosaki, M. Transphosphorylation of Bruton’s Tyrosine Kinase on Tyrosine 551 Is Critical for B Cell Antigen Receptor Function. J. Biol. Chem. 1997, 272, 15595–15598. [Google Scholar] [CrossRef] [PubMed]
- Tavakoli, G.M.; Yazdanpanah, N.; Rezaei, N. Targeting Bruton’s Tyrosine Kinase (BTK) as a Signaling Pathway in Immune-Mediated Diseases: From Molecular Mechanisms to Leading Treatments. Adv. Rheumatol. 2024, 64, 61. [Google Scholar] [CrossRef] [PubMed]
- Duarte, D.P.; Lamontanara, A.J.; La Sala, G.; Jeong, S.; Sohn, Y.-K.; Panjkovich, A.; Georgeon, S.; Kükenshöner, T.; Marcaida, M.J.; Pojer, F.; et al. Btk SH2-Kinase Interface Is Critical for Allosteric Kinase Activation and Its Targeting Inhibits B-Cell Neoplasms. Nat. Commun. 2020, 11, 2319. [Google Scholar] [CrossRef] [PubMed]
- L’Estrange-Stranieri, E.; Gottschalk, T.A.; Wright, M.D.; Hibbs, M.L. The Dualistic Role of Lyn Tyrosine Kinase in Immune Cell Signaling: Implications for Systemic Lupus Erythematosus. Front. Immunol. 2024, 15, 1395427. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Ghosh, A.; Greco, D.; Michaličková, D.; Slanař, O. Bruton’s Tyrosine Kinase: A Potential Novel Target for Neurological Disorders. Physiol. Res. 2025, 74, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Munshi, M.; Liu, X.; Chen, J.G.; Xu, L.; Tsakmaklis, N.; Demos, M.G.; Kofides, A.; Guerrera, M.L.; Jimenez, C.; Chan, G.G.; et al. SYK Is Activated by Mutated MYD88 and Drives Pro-Survival Signaling in MYD88 Driven B-Cell Lymphomas. Blood Cancer J. 2020, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Krämer, J.; Bar-Or, A.; Turner, T.J.; Wiendl, H. Bruton Tyrosine Kinase Inhibitors for Multiple Sclerosis. Nat. Rev. Neurol. 2023, 19, 289–304. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Mohammed, Z.; Singh, I. Bruton’s Tyrosine Kinase Drives Neuroinflammation and Anxiogenic Behavior in Mouse Models of Stress. J. Neuroinflamm. 2021, 18, 289. [Google Scholar] [CrossRef] [PubMed]
- Vermersch, P.; Airas, L.; Berger, T.; Deisenhammer, F.; Grigoriadis, N.; Hartung, H.-P.; Magyari, M.; Popescu, V.; Pozzilli, C.; Pugliatti, M.; et al. The Role of Microglia in Multiple Sclerosis: Implications for Treatment with Bruton’s Tyrosine Kinase Inhibitors. Front. Immunol. 2025, 16, 1495529. [Google Scholar] [CrossRef] [PubMed]
- Gruber, R.C.; Wirak, G.S.; Blazier, A.S.; Lee, L.; Dufault, M.R.; Hagan, N.; Chretien, N.; LaMorte, M.; Hammond, T.R.; Cheong, A.; et al. BTK Regulates Microglial Function and Neuroinflammation in Human Stem Cell Models and Mouse Models of Multiple Sclerosis. Nat. Commun. 2024, 15, 10116. [Google Scholar] [CrossRef] [PubMed]
- Karati, D.; Kumar, D. Tyrosine Kinase as Therapeutic Target of Neurodegenerative Disorders. Brain Disord. 2025, 17, 100193. [Google Scholar] [CrossRef]
- Rozkiewicz, D.; Hermanowicz, J.M.; Kwiatkowska, I.; Krupa, A.; Pawlak, D. Bruton’s Tyrosine Kinase Inhibitors (BTKIs): Review of Preclinical Studies and Evaluation of Clinical Trials. Molecules 2023, 28, 2400. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Luo, J.; Zhang, D.; Lu, Y.; Liao, W.; Zhang, J. Innovative Design and Potential Applications of Covalent Strategy in Drug Discovery. Eur. J. Med. Chem. 2025, 284, 117202. [Google Scholar] [CrossRef] [PubMed]
- Zain, R.; Vihinen, M. Structure-Function Relationships of Covalent and Non-Covalent BTK Inhibitors. Front. Immunol. 2021, 12, 694853. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A. BTK Inhibitors: Present and Future. Cancer J. 2019, 25, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Honigberg, L.A.; Smith, A.M.; Sirisawad, M.; Verner, E.; Loury, D.; Chang, B.; Li, S.; Pan, Z.; Thamm, D.H.; Miller, R.A.; et al. The Bruton Tyrosine Kinase Inhibitor PCI-32765 Blocks B-Cell Activation and Is Efficacious in Models of Autoimmune Disease and B-Cell Malignancy. Proc. Natl. Acad. Sci. USA 2010, 107, 13075–13080. [Google Scholar] [CrossRef] [PubMed]
- Estupiñán, H.Y.; Berglöf, A.; Zain, R.; Smith, C.I.E. Comparative Analysis of BTK Inhibitors and Mechanisms Underlying Adverse Effects. Front. Cell Dev. Biol. 2021, 9, 630942. [Google Scholar] [CrossRef] [PubMed]
- Lipsky, A.H.; Lamanna, N. Novel Combination Approaches with Targeted Agents in Frontline Chronic Lymphocytic Leukemia. Cancer 2023, 129, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Langrish, C.L.; Bradshaw, J.M.; Francesco, M.R.; Owens, T.D.; Xing, Y.; Shu, J.; LaStant, J.; Bisconte, A.; Outerbridge, C.; White, S.D.; et al. Preclinical Efficacy and Anti-Inflammatory Mechanisms of Action of the Bruton Tyrosine Kinase Inhibitor Rilzabrutinib for Immune-Mediated Disease. J. Immunol. 2021, 206, 1454–1468. [Google Scholar] [CrossRef] [PubMed]
- Rotstein, D.L. All Bruton’s Tyrosine Kinase Inhibitors Have Similar Efficacy and Risks: No. Mult. Scler. 2022, 28, 1500–1502. [Google Scholar] [CrossRef] [PubMed]
- Elamin, G.; Aljoundi, A.; Alahmdi, M.I.; Abo-Dya, N.E.; Soliman, M.E.S. Battling BTK Mutants with Noncovalent Inhibitors That Overcome Cys481 and Thr474 Mutations in Waldenström Macroglobulinemia Therapy: Structural Mechanistic Insights on the Role of Fenebrutinib. J. Mol. Model. 2022, 28, 355. [Google Scholar] [CrossRef] [PubMed]
- Robak, P.; Witkowska, M.; Wolska-Washer, A.; Robak, T. The Preclinical Discovery and Development of Orelabrutinib as a Novel Treatment Option for B-Cell Lymphoid Malignancies. Expert. Opin. Drug Discov. 2023, 18, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.; Berger, W.; Giménez-Arnau, A.; Hayama, K.; Jain, V.; Reich, A.; Haemmerle, S.; Lheritier, K.; Walsh, P.; Xia, S.; et al. Remibrutinib, a Novel BTK Inhibitor, Demonstrates Promising Efficacy and Safety in Chronic Spontaneous Urticaria. J. Allergy Clin. Immunol. 2022, 150, 1498–1506.e2. [Google Scholar] [CrossRef] [PubMed]
- Turner, T.J.; Brun, P.; Gruber, R.C.; Ofengeim, D. Comparative CNS Pharmacology of the Bruton’s Tyrosine Kinase (BTK) Inhibitor Tolebrutinib Versus Other BTK Inhibitor Candidates for Treating Multiple Sclerosis. Drugs R&D 2024, 24, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Cabanis, M.; Nicolas, O.; Vitse, O.; Jan, C.; Brun, P.; Soubayrol, P.; Smith, W.B.; Turner, T.J.; Krupka, E. A Phase I Trial Assessing the Safety, Pharmacokinetics, Cerebrospinal Fluid Penetrance, and Food Effect of BTK Inhibitor Tolebrutinib in Healthy Volunteers. Clin. Transl. Sci. 2024, 17, e13693. [Google Scholar] [CrossRef]
- Geladaris, A.; Torke, S.; Saberi, D.; Alankus, Y.B.; Streit, F.; Zechel, S.; Stadelmann-Nessler, C.; Fischer, A.; Boschert, U.; Häusler, D.; et al. BTK Inhibition Limits Microglia-Perpetuated CNS Inflammation and Promotes Myelin Repair. Acta Neuropathol. 2024, 147, 75. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-X.; Tian, Y.; Wang, Z.-T.; Ma, Y.-H.; Tan, L.; Yu, J.-T. The Epidemiology of Alzheimer’s Disease Modifiable Risk Factors and Prevention. J. Prev. Alzheimer’s Dis. 2021, 8, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Merighi, S.; Nigro, M.; Travagli, A.; Gessi, S. Microglia and Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 12990. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Ran, B.; Wang, Y.; Liu, L.; Li, W. PLCγ2 Impacts Microglia-Related Effectors Revealing Variants and Pathways Important in Alzheimer’s Disease. Front. Cell Dev. Biol. 2022, 10, 999061. [Google Scholar] [CrossRef] [PubMed]
- Bunney, T.D.; Kampyli, C.; Gregory, A.; Katan, M. Characterisation of Molecular Mechanisms for PLCγ2 Disease-Linked Variants. Adv. Biol. Regul. 2024, 94, 101053. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.N.R. Targeting the NLRP3 Inflammasome via BTK. Front. Cell Dev. Biol. 2021, 9, 630479. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Huang, Y.; Zhou, R. NLRP3 Inflammasome in Neuroinflammation and Central Nervous System Diseases. Cell Mol. Immunol. 2025, 22, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Jeon, S.G.; Kim, J.; Kang, R.J.; Kim, S.; Han, K.; Park, H.; Kim, K.; Sung, Y.M.; Nam, H.Y.; et al. Ibrutinib Modulates Aβ/Tau Pathology, Neuroinflammation, and Cognitive Function in Mouse Models of Alzheimer’s Disease. Aging Cell 2021, 20, e13332. [Google Scholar] [CrossRef] [PubMed]
- Gião, T.; Teixeira, T.; Almeida, M.R.; Cardoso, I. Choroid Plexus in Alzheimer’s Disease—The Current State of Knowledge. Biomedicines 2022, 10, 224. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.; Mechelli, A.; Natale, G.; Volpicelli-Daley, L.; Di Lazzaro, G.; Ghiglieri, V. Alpha-Synuclein in Parkinson’s Disease and Other Synucleinopathies: From Overt Neurodegeneration Back to Early Synaptic Dysfunction. Cell Death Dis. 2023, 14, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson Disease. Nat. Rev. Dis. Primers 2017, 3, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, M.; Lehtonen, S.; Jaronen, M.; Goldsteins, G.; Hämäläinen, R.H.; Koistinaho, J. Astrocyte Alterations in Neurodegenerative Pathologies and Their Modeling in Human Induced Pluripotent Stem Cell Platforms. Cell. Mol. Life Sci. 2019, 76, 2739–2760. [Google Scholar] [CrossRef] [PubMed]
- Brunner, C.; Betzler, A.C.; Brown, J.R.; Andreotti, A.H.; Grassilli, E. Editorial: Targeting Bruton Tyrosine Kinase. Front. Cell Dev. Biol. 2022, 10, 909655. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Li, Y.-J.; Ning, S.-B. Investigating the Molecular Mechanisms Underlying the Co-Occurrence of Parkinson’s Disease and Inflammatory Bowel Disease through the Integration of Multiple Datasets. Sci. Rep. 2024, 14, 17028. [Google Scholar] [CrossRef] [PubMed]
- Stevens, C.H.; Rowe, D.; Morel-Kopp, M.-C.; Orr, C.; Russell, T.; Ranola, M.; Ward, C.; Halliday, G.M. Reduced T Helper and B Lymphocytes in Parkinson’s Disease. J. Neuroimmunol. 2012, 252, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Isik, S.; Yeman Kiyak, B.; Akbayir, R.; Seyhali, R.; Arpaci, T. Microglia Mediated Neuroinflammation in Parkinson’s Disease. Cells 2023, 12, 1012. [Google Scholar] [CrossRef] [PubMed]
- Filippi, M.; Bar-Or, A.; Piehl, F.; Preziosa, P.; Solari, A.; Vukusic, S.; Rocca, M.A. Multiple Sclerosis. Nat. Rev. Dis. Primers 2018, 4, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Waubant, E.; Lucas, R.; Mowry, E.; Graves, J.; Olsson, T.; Alfredsson, L.; Langer-Gould, A. Environmental and Genetic Risk Factors for MS: An Integrated Review. Ann. Clin. Transl. Neurol. 2019, 6, 1905–1922. [Google Scholar] [CrossRef] [PubMed]
- Bjornevik, K.; Münz, C.; Cohen, J.I.; Ascherio, A. Epstein–Barr Virus as a Leading Cause of Multiple Sclerosis: Mechanisms and Implications. Nat. Rev. Neurol. 2023, 19, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Sintzel, M.B.; Rametta, M.; Reder, A.T. Vitamin D and Multiple Sclerosis: A Comprehensive Review. Neurol. Ther. 2017, 7, 59–85. [Google Scholar] [CrossRef] [PubMed]
- Bassani, C.; Molinari, M.; Romeo, V.; Martinelli, V.; Boschert, U.; Martino, G.; Muzio, L.; Farina, C. The Contribution of BTK Signaling in Myeloid Cells to Neuroinflammation. Front. Immunol. 2025, 16, 1595069. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Morch, M.T.; Gorter, R.; Lozinski, B.; Ghorbani, S.; Dong, Y.; Shen, Y.-A.; Harp, C.; Zandee, S.; Klement, W.; et al. Bruton Tyrosine Kinase in Lesions of Multiple Sclerosis and 3 of Its Models. Neurol. Neuroimmunol. Neuroinflamm. 2025, 12, e200413. [Google Scholar] [CrossRef] [PubMed]
- Evonuk, K.S.; Wang, S.; Mattie, J.; Cracchiolo, C.J.; Mager, R.; Ferenčić, Ž.; Sprague, E.; Carrier, B.; Schofield, K.; Martinez, E.; et al. Bruton’s Tyrosine Kinase Inhibition Reduces Disease Severity in a Model of Secondary Progressive Autoimmune Demyelination. Acta Neuropathol. Commun. 2023, 11, 115. [Google Scholar] [CrossRef] [PubMed]
- De Bondt, M.; Renders, J.; Petit de Prado, P.; Berghmans, N.; Pörtner, N.; Vanbrabant, L.; de Oliveira, V.L.S.; Duran, G.; Baeten, P.; Broux, B.; et al. Effect on Neutrophil Migration and Antimicrobial Functions by the Bruton’s Tyrosine Kinase Inhibitors Tolebrutinib, Evobrutinib, and Fenebrutinib. J. Leukoc. Biol. 2025, 117, qiae160. [Google Scholar] [CrossRef] [PubMed]
- Reidy, M.; Khan, M.; Mills, E.A.; Wu, Q.; Garton, J.; Draayer, D.E.; Zahoor, I.; Giri, S.; Axtell, R.C.; Mao-Draayer, Y. New Frontiers in Multiple Sclerosis Treatment: From Targeting Costimulatory Molecules to Bispecific Antibodies. Int. J. Mol. Sci. 2025, 26, 3880. [Google Scholar] [CrossRef] [PubMed]
- Damiani, G.; McCormick, T.S.; Leal, L.O.; Ghannoum, M.A. Recombinant Human Granulocyte Macrophage-Colony Stimulating Factor Expressed in Yeast (Sargramostim): A Potential Ally to Combat Serious Infections. Clin. Immunol. 2020, 210, 108292. [Google Scholar] [CrossRef] [PubMed]
- Steinmaurer, A.; Riedl, C.; König, T.; Testa, G.; Köck, U.; Bauer, J.; Lassmann, H.; Höftberger, R.; Berger, T.; Wimmer, I.; et al. The Relation between BTK Expression and Iron Accumulation of Myeloid Cells in Multiple Sclerosis. Brain Pathol. 2024, 34, e13240. [Google Scholar] [CrossRef] [PubMed]
- Papasouliotis, O.; Mitchell, D.; Girard, P.; Dangond, F.; Dyroff, M. Determination of a Clinically Effective Evobrutinib Dose: Exposure-Response Analyses of a Phase II Relapsing Multiple Sclerosis Study. Clin. Transl. Sci. 2022, 15, 2888–2898. [Google Scholar] [CrossRef] [PubMed]
- Montalban, X.; Vermersch, P.; Arnold, D.L.; Bar-Or, A.; Cree, B.A.C.; Cross, A.H.; Kubala Havrdova, E.; Kappos, L.; Stuve, O.; Wiendl, H.; et al. Safety and Efficacy of Evobrutinib in Relapsing Multiple Sclerosis (evolutionRMS1 and evolutionRMS2): Two Multicentre, Randomised, Double-Blind, Active-Controlled, Phase 3 Trials. Lancet Neurol. 2024, 23, 1119–1132. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Drulovic, J.; Dufek, M.; Budincevic, H.; Habek, M.; Caunt, M.; Sierzega, M.; Clayton, D.; Chen, Y.-F.; Ratchford, J.; et al. Fenebrutinib Maintains Low Disease Activity in Relapsing Multiple Sclerosis: Results from the FENopta Trial Open-Label Extension (P8-1.005). Neurology 2025, 104, 2248. [Google Scholar] [CrossRef]
- Oh, J.; Arnold, D.L.; Cree, B.A.C.; Ionete, C.; Kim, H.J.; Sormani, M.P.; Syed, S.; Chen, Y.; Maxwell, C.R.; Benoit, P.; et al. Tolebrutinib versus Teriflunomide in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2025, 392, 1893–1904. [Google Scholar] [CrossRef] [PubMed]
- Oudejans, E.; Luchicchi, A.; Strijbis, E.M.M.; Geurts, J.J.G.; van Dam, A.-M. Is MS Affecting the CNS Only? Lessons from Clinic to Myelin Pathophysiology. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e914. [Google Scholar] [CrossRef] [PubMed]
- Majumder, D. Ischemic Stroke: Pathophysiology and Evolving Treatment Approaches. Neurosci. Insights 2024, 19, 26331055241292600. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, M.G.; Paciaroni, M. Treatments in Ischemic Stroke: Current and Future. Eur. Neurol. 2022, 85, 349–366. [Google Scholar] [CrossRef] [PubMed]
- Salaudeen, M.A.; Bello, N.; Danraka, R.N.; Ammani, M.L. Understanding the Pathophysiology of Ischemic Stroke: The Basis of Current Therapies and Opportunity for New Ones. Biomolecules 2024, 14, 305. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Shichita, T.; Okada, M.; Komine, R.; Noguchi, Y.; Yoshimura, A.; Morita, R. Bruton’s Tyrosine Kinase Is Essential for NLRP3 Inflammasome Activation and Contributes to Ischaemic Brain Injury. Nat. Commun. 2015, 6, 7360. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Mo, Y.; Yue, E.-L.; Liu, Y.; Liu, K.-Y. Ibrutinib Ameliorates Cerebral Ischemia/Reperfusion Injury through Autophagy Activation and PI3K/Akt/mTOR Signaling Pathway in Diabetic Mice. Bioengineered 2021, 12, 7432–7445. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.-L.; Yang, S.; Zhou, L.-Q.; Chu, Y.-H.; Pang, X.-W.; You, Y.-F.; Zhang, H.; Zhang, L.-Y.; Zhu, L.-F.; Chen, L.; et al. Bruton’s Tyrosine Kinase Inhibition Ameliorated Neuroinflammation during Chronic White Matter Ischemia. J. Neuroinflamm. 2024, 21, 195. [Google Scholar] [CrossRef] [PubMed]
- Franke, M.; Bieber, M.; Kraft, P.; Weber, A.N.R.; Stoll, G.; Schuhmann, M.K. The NLRP3 Inflammasome Drives Inflammation in Ischemia/Reperfusion Injury after Transient Middle Cerebral Artery Occlusion in Mice. Brain Behav. Immun. 2021, 92, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Wang, N.; Liu, J.; Li, J.; Chang, L.; Yang, C.; Chen, Z.; Huang, W.; Wang, J.; Lang, X.; et al. Evobrutinib Mitigates Neuroinflammation after Ischemic Stroke by Targeting M1 Microglial Polarization via the TLR4/Myd88/NF-κB Pathway. Mol. Med. 2025, 31, 148. [Google Scholar] [CrossRef] [PubMed]
- Kander, E.M.; Zhao, Q.; Bhat, S.A.; Hirsch, J.; Byrd, J.C.; Ooka, L.; Wiczer, T.; Woyach, J.A.; Awan, F.T.; Rogers, K.A.; et al. Venous and Arterial Thrombosis in Patients with Haematological Malignancy During Treatment with Ibrutinib. Br. J. Haematol. 2019, 187, 399–402. [Google Scholar] [CrossRef] [PubMed]
- D’Egidio, F.; Castelli, V.; Cimini, A.; d’Angelo, M. Cell Rearrangement and Oxidant/Antioxidant Imbalance in Huntington’s Disease. Antioxidants 2023, 12, 571. [Google Scholar] [CrossRef] [PubMed]
- Wilton, D.K.; Mastro, K.; Heller, M.D.; Gergits, F.W.; Willing, C.R.; Fahey, J.B.; Frouin, A.; Daggett, A.; Gu, X.; Kim, Y.A.; et al. Microglia and Complement Mediate Early Corticostriatal Synapse Loss and Cognitive Dysfunction in Huntington’s Disease. Nat. Med. 2023, 29, 2866–2884. [Google Scholar] [CrossRef] [PubMed]
- Simmons, D.A.; James, M.L.; Belichenko, N.P.; Semaan, S.; Condon, C.; Kuan, J.; Shuhendler, A.J.; Miao, Z.; Chin, F.T.; Longo, F.M. TSPO–PET Imaging Using [18F]PBR06 Is a Potential Translatable Biomarker for Treatment Response in Huntington’s Disease: Preclinical Evidence with the p75NTR Ligand LM11A-31. Hum. Mol. Genet. 2018, 27, 2893–2912. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Li, S.; Li, X.-J.; Yin, P. Neuroinflammation in Huntington’s Disease: From Animal Models to Clinical Therapeutics. Front. Immunol. 2022, 13, 1088124. [Google Scholar] [CrossRef] [PubMed]
- Saba, J.; Couselo, F.L.; Bruno, J.; Carniglia, L.; Durand, D.; Lasaga, M.; Caruso, C. Neuroinflammation in Huntington’s Disease: A Starring Role for Astrocyte and Microglia. Curr. Neuropharmacol. 2022, 20, 1116–1143. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Jiang, D.; Yu, L.; Chen, Y.; Zhou, D.; Li, Y.; Wu, D.; Zhang, L.; Miao, L.; Ma, J.; et al. Evidence-Based Expert Consensus on Clinical Management of Safety of Bruton’s Tyrosine Kinase Inhibitors (2024). Chin. J. Cancer Res. 2024, 36, 240–256. [Google Scholar] [CrossRef] [PubMed]
- Gentile, G.; Poggio, T.; Catalano, A.; Voutilainen, M.; Lahnalampi, M.; Andrade-Martinez, M.; Ma, T.; Sankowski, R.; Goncharenko, L.; Tholen, S.; et al. Development of Combination Therapies with BTK Inhibitors and Dasatinib to Treat CNS-Infiltrating E2A-PBX1+/preBCR+ ALL. Blood Adv. 2024, 8, 2846–2860. [Google Scholar] [CrossRef] [PubMed]
- Grommes, C.; Tang, S.S.; Wolfe, J.; Kaley, T.J.; Daras, M.; Pentsova, E.I.; Piotrowski, A.F.; Stone, J.; Lin, A.; Nolan, C.P.; et al. Phase 1b Trial of an Ibrutinib-Based Combination Therapy in Recurrent/Refractory CNS Lymphoma. Blood 2019, 133, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Jaradat, J.H.; Alkhawaldeh, I.M.; Al-Bojoq, Y.; Ramadan, M.N.; Abuawwad, M.T.; Alabdallat, Y.J.; Nashwan, A.J. Efficacy and Safety of Ibrutinib in Central Nervous System Lymphoma: A Systematic Review and Meta-Analysis. Crit. Rev. Oncol. Hematol. 2025, 206, 104597. [Google Scholar] [CrossRef] [PubMed]
- Cree, B.; Hartung, H.-P. Update on Novel Multiple Sclerosis Treatments: From Dismal Defeat to Scintillating Success. Curr. Opin. Neurol. 2025, 38, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Nuesslein-Hildesheim, B.; Ferrero, E.; Schmid, C.; Huck, C.; Smith, P.; Tisserand, S.; Rubert, J.; Bornancin, F.; Eichlisberger, D.; Cenni, B. Remibrutinib (LOU064) Inhibits Neuroinflammation Driven by B Cells and Myeloid Cells in Preclinical Models of Multiple Sclerosis. J. Neuroinflamm. 2023, 20, 194. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Sun, X.; Lv, L.; Cui, Q.; Qian, J.; Xing, R.; Bai, X.; Chen, Y.; Liu, Q.; Lai, W.; et al. Efficacy of Individualized Orelabrutinib-Based Regimens in Relapsed or Refractory Central Nervous System Lymphoma. Front. Neurol. 2025, 16, 1570224. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, A.; Matsumoto, I.; Izutsu, K.; Nishikawa, R. Post-Marketing Surveillance of Tirabrutinib in 189 Patients with r/r Primary Central Nervous System Lymphoma. Future Oncol. 2025, 21, 1837–1847. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Egidio, F.; Kacem, H.; Lombardozzi, G.; d’Angelo, M.; Cimini, A.; Castelli, V. The Potential Therapeutic Role of Bruton Tyrosine Kinase Inhibition in Neurodegenerative Diseases. Appl. Sci. 2025, 15, 8239. https://doi.org/10.3390/app15158239
D’Egidio F, Kacem H, Lombardozzi G, d’Angelo M, Cimini A, Castelli V. The Potential Therapeutic Role of Bruton Tyrosine Kinase Inhibition in Neurodegenerative Diseases. Applied Sciences. 2025; 15(15):8239. https://doi.org/10.3390/app15158239
Chicago/Turabian StyleD’Egidio, Francesco, Housem Kacem, Giorgia Lombardozzi, Michele d’Angelo, Annamaria Cimini, and Vanessa Castelli. 2025. "The Potential Therapeutic Role of Bruton Tyrosine Kinase Inhibition in Neurodegenerative Diseases" Applied Sciences 15, no. 15: 8239. https://doi.org/10.3390/app15158239
APA StyleD’Egidio, F., Kacem, H., Lombardozzi, G., d’Angelo, M., Cimini, A., & Castelli, V. (2025). The Potential Therapeutic Role of Bruton Tyrosine Kinase Inhibition in Neurodegenerative Diseases. Applied Sciences, 15(15), 8239. https://doi.org/10.3390/app15158239