
NeuroTIS+: An Improved Method for Translation Initiation Site Prediction in Full-Length mRNA Sequence via Primary Structural Information


Abstract
1. Introduction
- The proposed method, NeuroTIS+, is an improved version of NeuroTIS, which preserves the basic framework of NeuroTIS and hence inherits the merits of explicitly modeling statistic dependencies among variables and automatic feature learning. Meanwhile, it assumes a stronger dependency relationship among codon labels and integrates novel frame information for TIS inference.
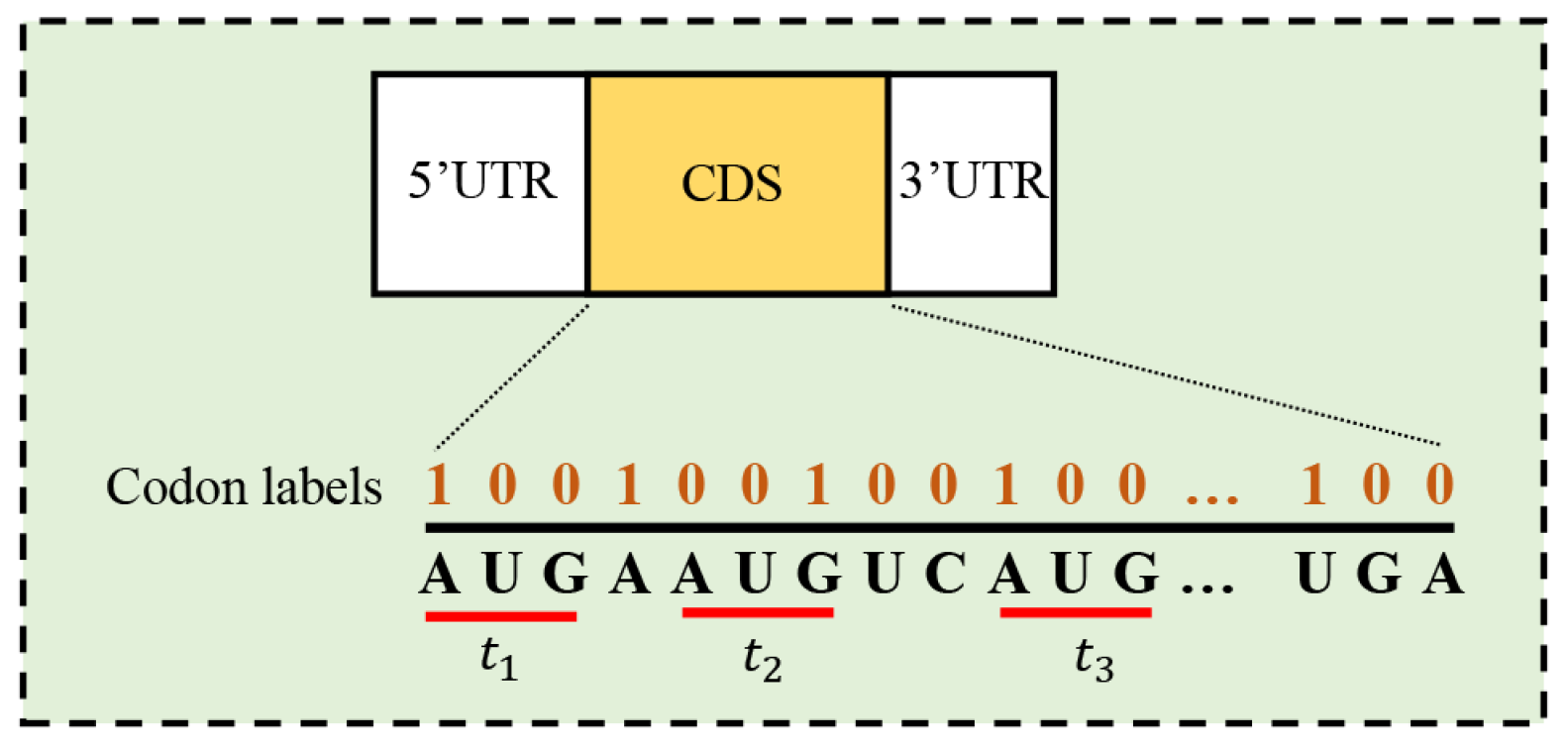
- We consider the primary structural information that a CDS is continuous and models codon label consistency by using a TCN, which can easily and naturally aggregate information across multiple codon labels through its convolutional layers. Moreover, a position embedding and a fast codon usage generating strategy for a sequence are also proposed to improve the prediction of coding sequences in mRNA sequences.
- We consider the heterogeneity of negative TISs and develop an adaptive grouping strategy for homogenous feature building, which effectively improves the prediction accuracy of TISs. Moreover, the adaptive grouping strategy stabilizes the learning process of CNNs.
2. Related Works
2.1. Codon Label Consistency
2.2. Non-Homogeneous Negative TIS
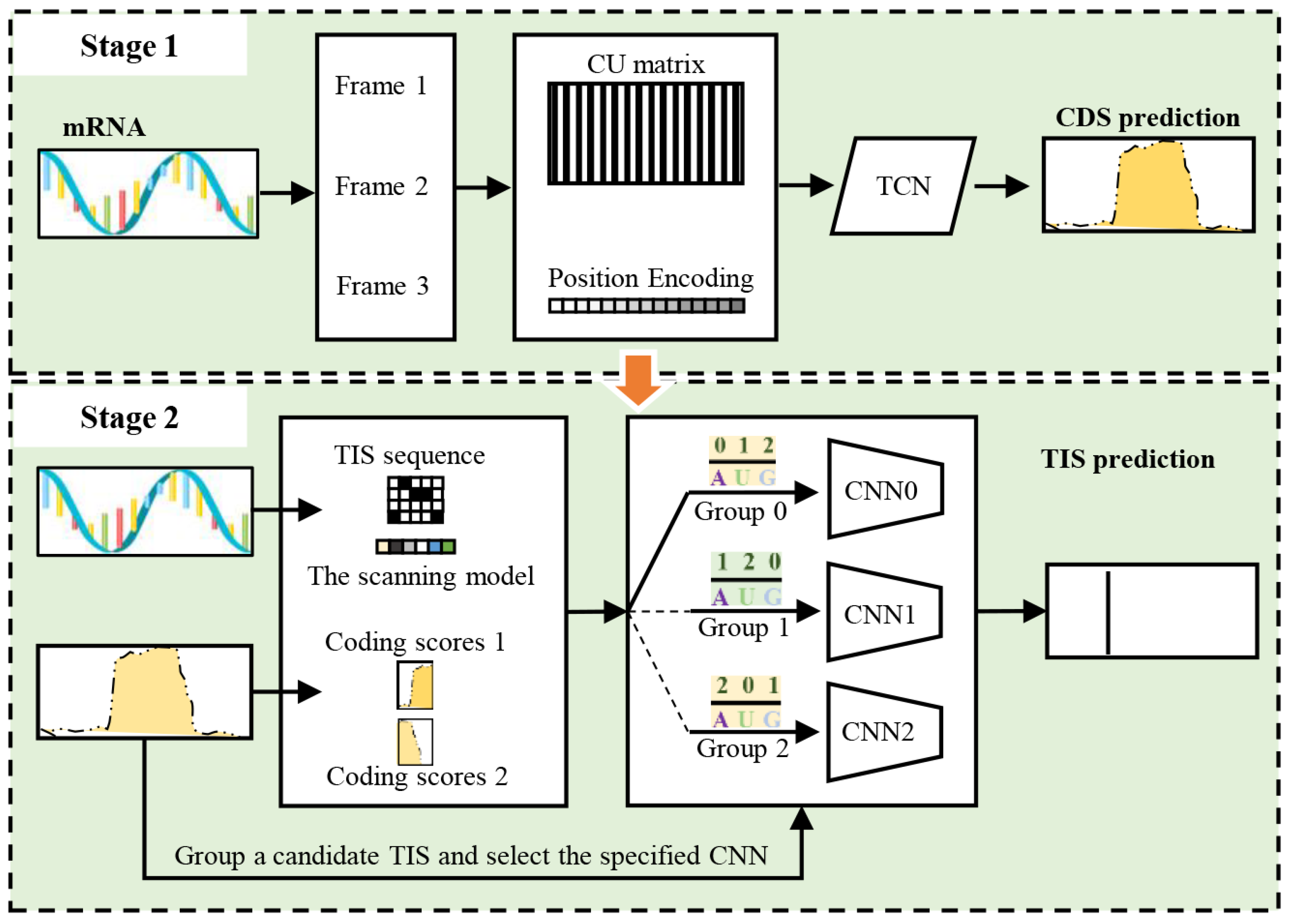
3. The Proposed Method
3.1. Preliminaries
3.2. NeuroTIS
3.3. NeuroTIS+
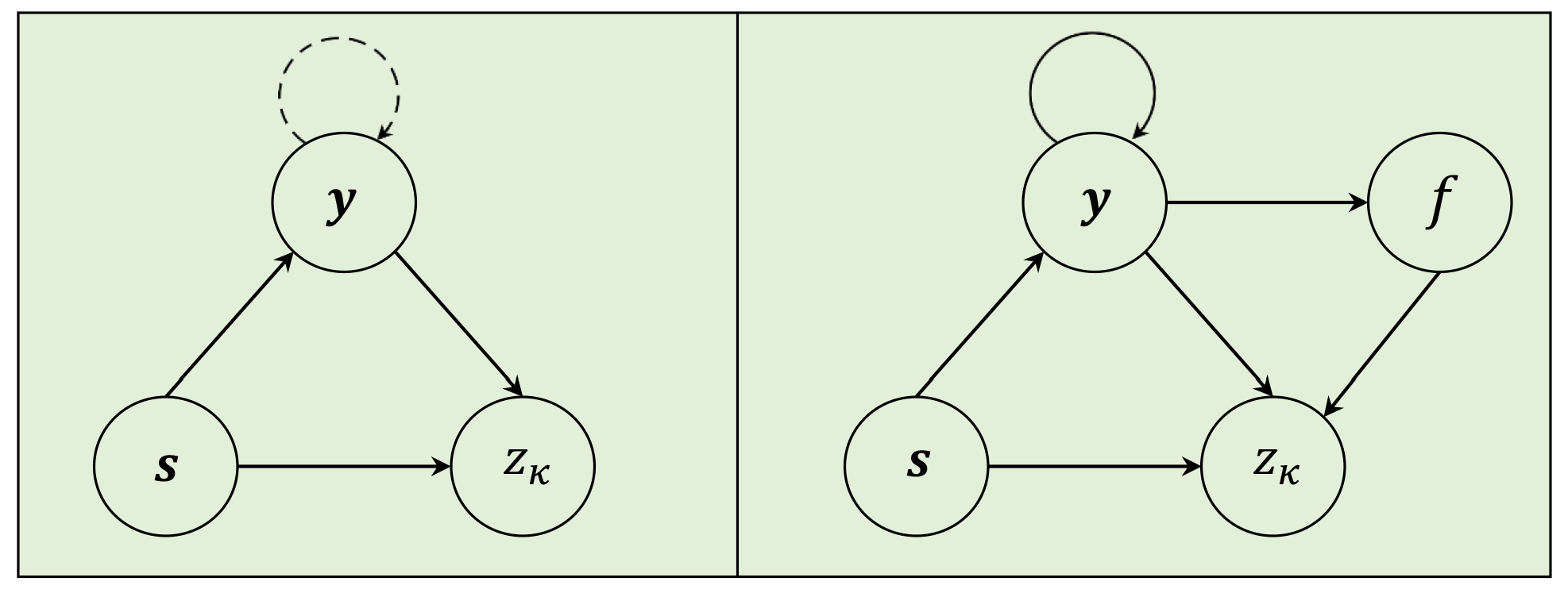
3.3.1. Dependency Network Representation
3.3.2. Temporal Convolutional Network for CDS Prediction
| Algorithm 1 Fast CU matrix generation strategy |
| Input: one reading frame of an mRNA sequence with length n and a sliding window. |
| Output: CU matrix with size of , in which each column is 64-dimensional codon usage statistics. |
| 1: pad the reading frame with ‘N’ on two sides. |
| 2: initialize an array of size 64, , an empty queue Q, |
| 3: calculate the index of each codon in sliding window, , and enqueue them into Q one by one, |
| 4: the 1st column of , initialize a scaler variable, , |
| 5: dequeue an element from Q, |
| 6: Slide the window with step 3 (one codon), and calculate the index of the codon , |
| 7: , |
| 8: , |
| 9: enqueue an element to Q, |
| 10: goto step 5 until the sliding window stops, |
| 11: return CU matrix . |
3.3.3. Frame-Specific CNN with Adaptive Grouping Strategy for TIS Prediction
4. Experiments
4.1. Datasets
4.2. Performance Measurements
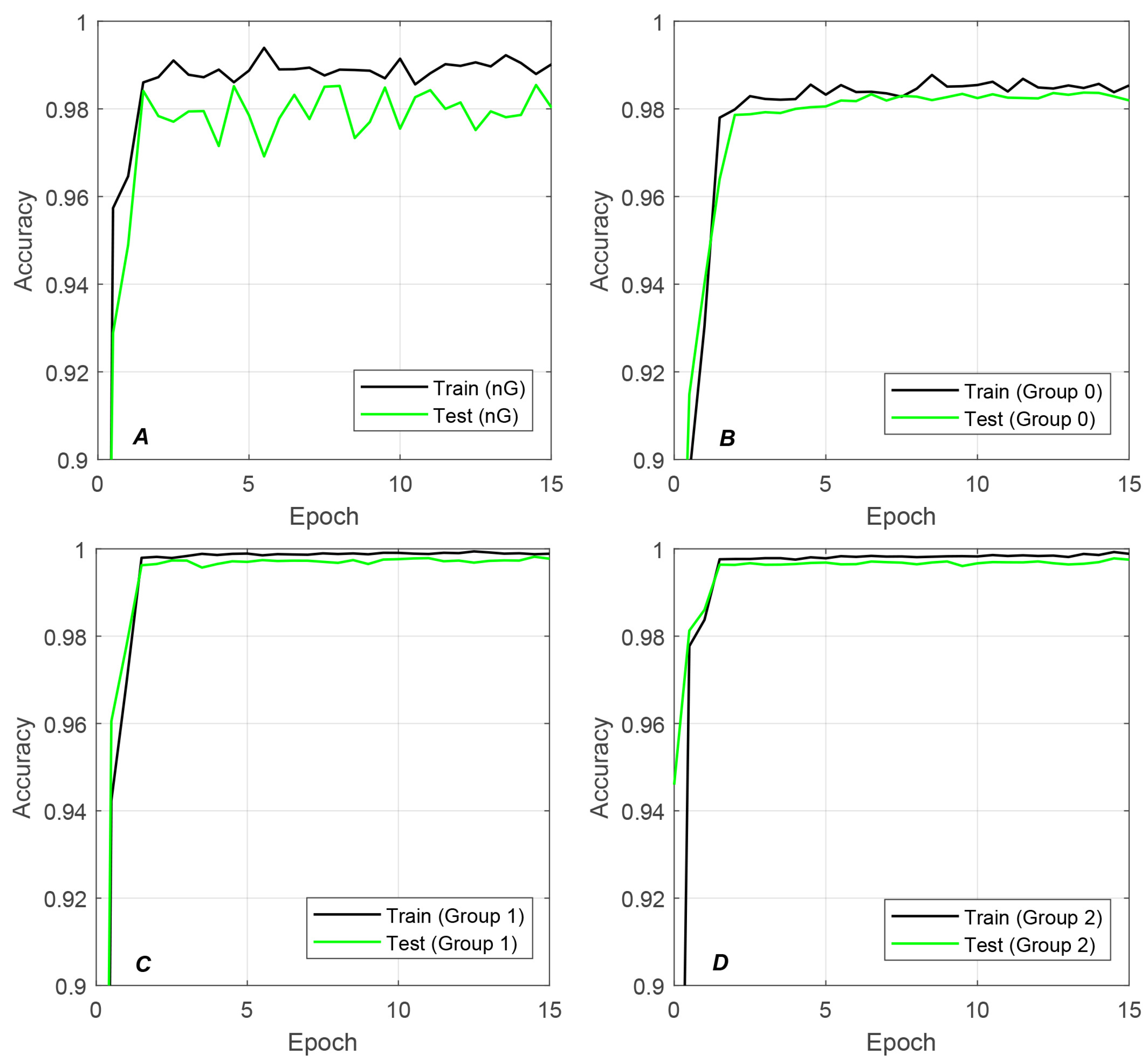
4.3. Significance of Adaptive Grouping Strategy
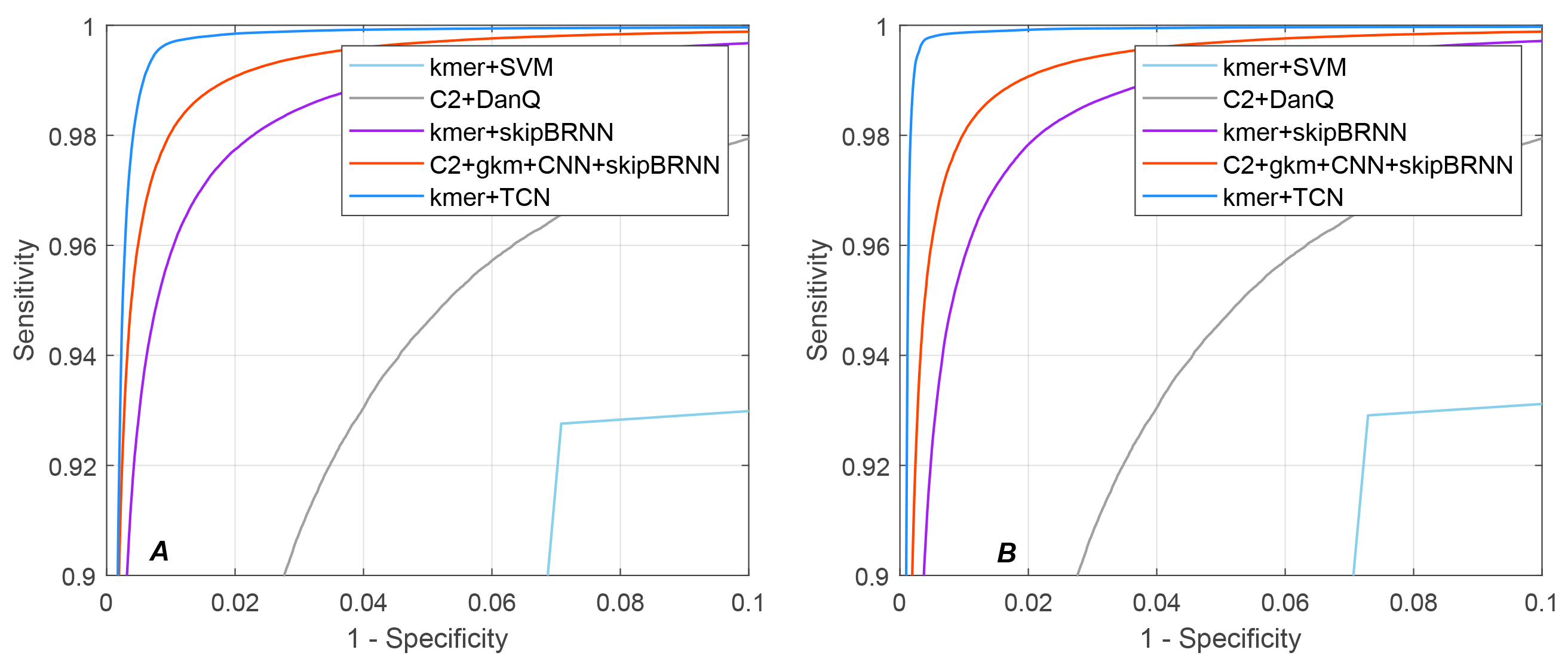
4.4. Performance Comparison for CDS Prediction
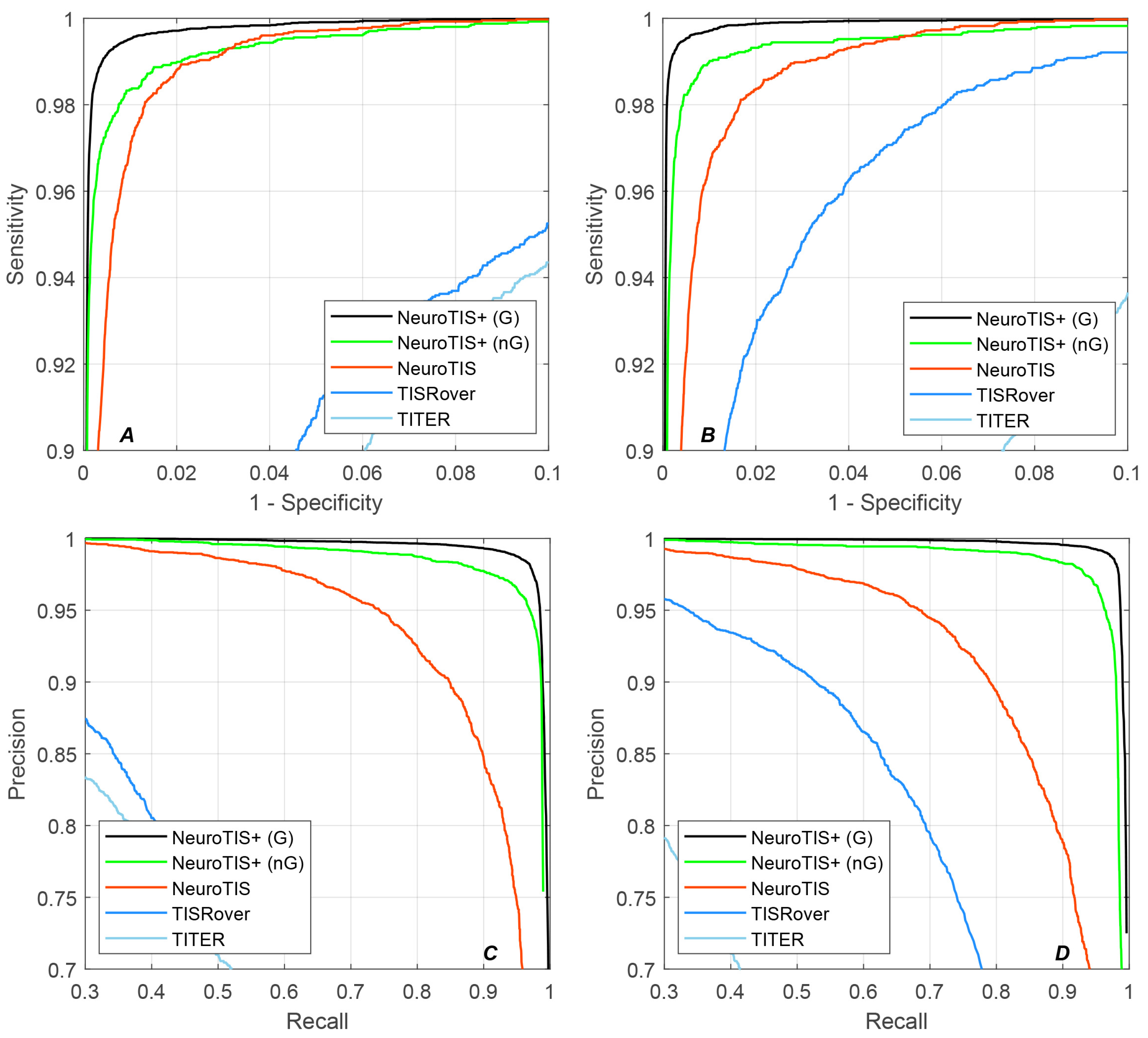
4.5. Performance Comparison for TIS Prediction
4.6. Performance on TISs Experimentally Validated via Ribosome Profiling
4.7. Time Cost of NeuroTIS+
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sonenberg, N.; Hinnebusch, A.G. Regulation of Translation Initiation in Eukaryotes: Mechanisms and Biological Targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef]
- Barbosa, C.; Peixeiro, I.; Romão, L. Gene expression regulation by upstream open reading frames and human disease. PLoS Genet. 2013, 9, e1003529. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Hu, H.; Jiang, T.; Zhang, L.; Zeng, J. TITER: Predicting translation initiation sites by deep learning. Bioinformatics 2017, 33, i234–i242. [Google Scholar] [CrossRef] [PubMed]
- Venket, R.; Louis, K.; Fantin, M.; Linda, R. A simple guide to de novo transcriptome assembly and annotation. Briefings Bioinform. 2022, 2, bbab563. [Google Scholar]
- Kozak, M. Translation of insulin-related polypeptides from messenger RNAs with tandemly reiterated copies of the ribosome binding site. Cell 1983, 34, 971–978. [Google Scholar] [CrossRef]
- Malys, N. Shine-Dalgarno sequence of bacteriophage T4: GAGG prevails in early genes. Mol. Biol. Rep. 2012, 39, 33–39. [Google Scholar] [CrossRef]
- Bernal, A.; Crammer, K.; Hatzigeorgiou, A.; Pereira, F. Global discriminative learning for higher-accuracy computational gene prediction. PLoS Comput. Biol. 2007, 3, e54. [Google Scholar] [CrossRef]
- Hinnebusch, A.G.; Ivanov, I.P.; Sonenberg, N. Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science 2016, 352, 1413–1416. [Google Scholar] [CrossRef]
- Boersma, S.; Khuperkar, D.; Verhagen, B.M.; Sonneveld, S.; Grimm, J.B.; Lavis, L.D.; Tanenbaum, M.E. Multi-color single-molecule imaging uncovers extensive heterogeneity in mRNA decoding. Cell 2019, 178, 458–472. [Google Scholar] [CrossRef]
- Khuperkar, D.; Hoek, T.A.; Sonneveld, S.; Verhagen, B.M.; Boersma, S.; Tanenbaum, M.E. Quantification of mRNA translation in live cells using single-molecule imaging. Nat. Protoc. 2020, 15, 1371–1398. [Google Scholar] [CrossRef]
- Pedersen, A.G.; Nielsen, H. Neural network prediction of translation initiation sites in eukaryotes: Perspectives for EST and genome analysis. In Proceedings of the International Conference on Intelligent Systems for Molecular Biology, Halkidiki, Greece, 21–26 June 1997. [Google Scholar]
- Rajapakse, J.C.; Ho, L.S. Markov encoding for detecting signals in genomic sequences. IEEE/ACM Trans. Comput. Biol. Bioinform. 2005, 2, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Zuallaert, J.; Kim, M.; Soete, A.; Saeys, Y.; Neve, W.D. TISRover: ConvNets learn biologically relevant features for effective translation initiation site prediction. Int. J. Data Min. Bioinform. 2018, 20, 267–284. [Google Scholar] [CrossRef]
- Zien, A.; Rätsch, G.; Mika, S.; Schölkopf, B.; Lengauer, T.; Müller, K.R. Engineering support vector machine kernels that recognize translation initiation sites. Bioinformatics 2000, 16, 799. [Google Scholar] [CrossRef]
- Li, H.; Jiang, T. A class of edit kernels for SVMs to predict translation initiation sites in eukaryotic mRNAs. In Proceedings of the Eighth International Conference on Resaerch in Computational Molecular Biology, San Diego, CA, USA, 27–31 March 2004. [Google Scholar]
- Chen, W.; Feng, P.M.; Deng, E.Z.; Lin, H.; Chou, K.C. iTIS-PseTNC: A sequence-based predictor for identifying translation initiation site in human genes using pseudo trinucleotide composition. Anal. Biochem. 2014, 462, 76–83. [Google Scholar] [CrossRef]
- Salamov, A.A. Assessing protein coding region integrity in cDNA sequencing projects. Bioinformatics 1998, 14, 384. [Google Scholar] [CrossRef]
- Li, G.; Leong, T.Y.; Zhang, L. Translation Initiation Sites Prediction with Mixture Gaussian Models. IEEE Trans. Knowl. Data Eng. 2005, 17, 1152–1160. [Google Scholar] [CrossRef]
- Nishikawa, T.; Ota, T.; Isogai, T. Prediction of Fullness of cDNA Fragment sequences by combining Statistical Information and Similarity with Protein Sequences. Bioinformatics 2000, 16, 960–967. [Google Scholar] [CrossRef]
- Hatzigeorgiou, A.; Mache, N.; Reczko, M. Functional site prediction on the DNA sequence by artificial neural networks. In Proceedings of the IEEE International Joint Symposia on Intelligence and Systems, Rockville, MD, USA, 4–5 November 1996; pp. 12–17. [Google Scholar]
- Hatzigeorgiou, A.G. Translation initiation start prediction in human cDNAs with high accuracy. Bioinformatics 2002, 18, 343–350. [Google Scholar] [CrossRef]
- Tzanis, G.; Berberidis, C.; Vlahavas, I. MANTIS: A data mining methodology for effective translation initiation site prediction. In Proceedings of the International Conference of the IEEE Engineering in Medicine and Biology Society, Lyon, France, 22–26 August 2007. [Google Scholar]
- Tzanis, G.; Berberidis, C.; Vlahavas, I. StackTIS: A stacked generalization approach for effective prediction of translation initiation sites. Comput. Biol. Med. 2012, 42, 61–69. [Google Scholar] [CrossRef]
- Wei, C.; Zhang, J.; Yuan, X.; He, Z.; Liu, G.; Wu, J. Neurotis: Enhancing the prediction of translation initiation sites in mrna sequences via a hybrid dependency network and deep learning framework. Knowl.-Based Syst. 2021, 212, 106459. [Google Scholar] [CrossRef]
- Kirkpatrick, J.; Pascanu, R.; Rabinowitz, N.; Veness, J.; Desjardins, G.; Rusu, A.A.; Milan, K.; Quan, J.; Ramalho, T.; Grabska-Barwinska, A.; et al. Overcoming catastrophic forgetting in neural networks. Proc. Natl. Acad. Sci. USA 2017, 114, 3521–3526. [Google Scholar] [CrossRef] [PubMed]
- Lea, C.; Flynn, M.D.; Vidal, R.; Reiter, A.; Hager, G.D. Temporal convolutional networks for action segmentation and detection. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Honolulu, HI, USA, 21–26 July 2017; pp. 156–165. [Google Scholar]
- Guo, Y.; Gu, S. Multi-Label Classification Using Conditional Dependency Networks. In Proceedings of the IJCAI 2011, Proceedings of the 22nd International Joint Conference on Artificial Intelligence, Barcelona, Spain, 16–22 July 2011. [Google Scholar]
- Li, S.; Farha, Y.A.; Liu, Y.; Cheng, M.M.; Gall, J. Ms-tcn++: Multi-stage temporal convolutional network for action segmentation. IEEE Trans. Pattern Anal. Mach. Intell. 2020, 45, 6647–6658. [Google Scholar] [CrossRef] [PubMed]
- Collobert, R.; Weston, J.; Bottou, L.; Karlen, M.; Kavukcuoglu, K.; Kuksa, P. Natural language processing (almost) from scratch. J. Mach. Learn. Res. 2011, 12, 2493–2537. [Google Scholar]
- Li, L.; Fan, Y.; Tse, M.; Lin, K.Y. A review of applications in federated learning. Comput. Ind. Eng. 2020, 149, 106854. [Google Scholar] [CrossRef]
- Huang, W.; Ye, M.; Du, B. Learn from others and be yourself in heterogeneous federated learning. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, New Orleans, LA, USA, 18–24 June 2022; pp. 10143–10153. [Google Scholar]
- Hu, G.Q.; Zheng, X.; Zhu, H.Q.; She, Z.S. Prediction of translation initiation site for microbial genomes with TriTISA. Bioinformatics 2009, 25, 123–125. [Google Scholar] [CrossRef]
- Pérez-Rodríguez, J.; Arroyo-Peña, A.G.; García-Pedrajas, N. Improving translation initiation site and stop codon recognition by using more than two classes. Bioinformatics 2014, 30, 2702–2708. [Google Scholar] [CrossRef]
- Schum, D.A. The Evidential Foundations of Probabilistic Reasoning by David A. Schum; Northwestern University Press: Evanston, IL, USA, 1994. [Google Scholar]
- Goodfellow, I.; Bengio, Y.; Courville, A. Deep Learning; MIT Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Mitchell, T.M.; Carbonell, J.G.; Michalski, R.S. Machine Learning; Springer: Berlin/Heidelberg, Germany, 1997. [Google Scholar]
- Davis, J.; Goadrich, M. The relationship between Precision-Recall and ROC curves. In Proceedings of the 23rd International Conference on Machine Learning, Pittsburgh, PA, USA, 25–29 June 2006; pp. 233–240. [Google Scholar]
- Daniel, Q.; Xiaohui, X. DanQ: A hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Res. 2016, 44, e107. [Google Scholar]
- Wei, C.; Zhang, J.; Yuan, X. Enhancing the prediction of protein coding regions in biological sequence via a deep learning framework with hybrid encoding. Digit. Signal Process. 2022, 123, 103430. [Google Scholar] [CrossRef]
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout: A Simple Way to Prevent Neural Networks from Overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]
- Treadgold, N.K.; Gedeon, T.D. Exploring constructive cascade networks. IEEE Trans. Neural Netw. 1999, 10, 1335–1350. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human | SN (%) | SP (%) | PRE (%) | ACC (%) | F1-Score | auROC | auPRC | MCC |
|---|---|---|---|---|---|---|---|---|
| TITER | 0.02 | 100 | - | 98.31 | - | 0.9788 | 0.6186 | - |
| TISRover | 92.52 | 93.77 | 20.26 | 93.75 | 0.3324 | 0.9760 | 0.3998 | 0.4167 |
| NeuroTIS | 98.19 | 98.54 | 56.29 | 98.53 | 0.7156 | 0.9985 | 0.9150 | 0.7377 |
| NeuroTIS+ (nG) | 98.38 | 98.94 | 84.29 | 98.91 | 0.9079 | 0.9989 | 0.9266 | 0.9052 |
| NeuroTIS+ (G) | 99.08 | 99.56 | 92.87 | 99.53 | 0.9588 | 0.9996 | 0.9385 | 0.9569 |
| Mouse | SN (%) | SP (%) | PRE (%) | ACC (%) | F1-Score | auROC | auPRC | MCC |
|---|---|---|---|---|---|---|---|---|
| TITER | 0.03 | 100 | - | 98.36 | - | 0.9766 | 0.5879 | - |
| TISRover | 95.29 | 96.74 | 32.52 | 96.72 | 0.4849 | 0.9936 | 0.7399 | 0.5463 |
| NeuroTIS | 98.12 | 98.31 | 48.90 | 98.30 | 0.6527 | 0.9982 | 0.9036 | 0.6865 |
| NeuroTIS+ (nG) | 98.63 | 99.26 | 86.96 | 99.23 | 0.9243 | 0.9991 | 0.9363 | 0.9223 |
| NeuroTIS+ (G) | 99.26 | 99.73 | 94.85 | 99.71 | 0.9701 | 0.9997 | 0.9460 | 0.9688 |
| Methods | Human | Mouse | ||||
|---|---|---|---|---|---|---|
| SN (%) | SP (%) | auROC | SN (%) | SP (%) | auROC | |
| kmer+SVM | 92.76 | 92.92 | - | 92.91 | 92.71 | - |
| C2+DanQ | 95.47 | 94.27 | 0.9889 | 95.32 | 94.37 | 0.9884 |
| kmer+skipBRNN | 98.25 | 97.39 | 0.9975 | 97.93 | 97.91 | 0.9973 |
| C2+gkm+CNN+skipBRNN | 99.08 | 97.97 | 0.9986 | 99.10 | 98.14 | 0.9985 |
| kmer+TCN | 99.64 | 99.67 | 0.9995 | 99.76 | 98.74 | 0.9988 |
| Dataset | Coding Number | TIS Number | Time Cost (min) | |
|---|---|---|---|---|
| kmer+TCN | Frame-Specific CNN | |||
| Human | 9,545,915 | 32,780 | 20 | 0.8 |
| Mouse | 7,883,216 | 17,420 | 15 | 0.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, W.; Wei, C. NeuroTIS+: An Improved Method for Translation Initiation Site Prediction in Full-Length mRNA Sequence via Primary Structural Information. Appl. Sci. 2025, 15, 7866. https://doi.org/10.3390/app15147866
Xiao W, Wei C. NeuroTIS+: An Improved Method for Translation Initiation Site Prediction in Full-Length mRNA Sequence via Primary Structural Information. Applied Sciences. 2025; 15(14):7866. https://doi.org/10.3390/app15147866
Chicago/Turabian StyleXiao, Wenqiu, and Chao Wei. 2025. "NeuroTIS+: An Improved Method for Translation Initiation Site Prediction in Full-Length mRNA Sequence via Primary Structural Information" Applied Sciences 15, no. 14: 7866. https://doi.org/10.3390/app15147866
APA StyleXiao, W., & Wei, C. (2025). NeuroTIS+: An Improved Method for Translation Initiation Site Prediction in Full-Length mRNA Sequence via Primary Structural Information. Applied Sciences, 15(14), 7866. https://doi.org/10.3390/app15147866