
BioSuiteT: A Unified Tool for Biological Sequence Analysis

 ,
,  , ,
, ,  , , and
, , and 
Simple Summary
Abstract
1. Background
- Compatibility and standardization: This can lead to compatibility issues [24], especially when integrating multiple tools into a workflow.
- Learning curves: Some tools have varying interfaces and functionalities, and this can create a steep learning curve for users, particularly those new to bioinformatics [22], especially if coding skills are required.
- Lack of community feedback and reviews: This helps users make informed decisions about which tools to use [22].
- Platform-specific tools: Tools are designed to work optimally with specific data types or experimental setups, which can limit their general applicability and require users to adapt their workflows accordingly [22].
- User-friendliness and accessibility: Many bioinformatics tools are not user-friendly, hindering their effective use [24], and most software tools lack a Graphical User Interface (GUI), making them difficult to use.
- 1.
- Challenges in selecting tools:
- a.
- Over 30 responses (62.5%) highlighted difficulties in choosing the right tool due to redundancy, poor documentation, or lack of clarity about functionalities.
- 2.
- Installation and usability issues:
- a.
- Around 12 responses (25%) directly pointed out problems with installing or using tools, citing a lack of standardization and technical complexity.
- 3.
- Barriers for non-programmers:
- a.
- At least 10 responses (20.8%) explicitly noted that users with limited programming expertise face significant barriers when using bioinformatics tools.
- 4.
- Recommendations for improvement:
- a.
- Unified platform: Around 10 responses (20.8%) advocate integrating tools into a common platform to simplify access and usability.
- b.
- User reviews: Five responses (10.4%) emphasized the need for platforms allowing users to review and rate tools, helping non-experts choose tools that align with their goals.
- c.
- Open-source frameworks: Seven responses (14.6%) suggested using open-source frameworks as the foundation for building elements for tool integration.
- 5.
- Time investment:
- a.
- Around 15 responses (31.25%) indicated that more time is spent installing and troubleshooting tools than on data analysis.
2. Materials and Methods
2.1. Data and Tools Source Selection
2.1.1. ExPASy
- Sequence analysis: Ability to analyze different types of biological sequences.
- Structure prediction: Prediction of the three-dimensional structure of proteins and nucleic acids.
- Homology search: Identification of similar sequences in databases.
- Primer design: Tools for efficiently designing primers for PCR.
- Gene expression Analysis: Evaluation of gene expression at the transcriptomic level.
- Functional Annotation: Assignment of biological functions to sequences.
- Databases: Access to a variety of biological databases.
2.1.2. NCBI Tools
- Sequence analysis: Ability to analyze different types of biological sequences.
- Biomedical databases: Repositories of biomedical data for research and analysis.
- Homology search: Identification of similar sequences in databases.
- Primer design: Tools for efficiently designing primers for PCR.
- Gene expression analysis: Evaluation of gene expression at the transcriptomic level.
- Functional annotation: Assignment of biological functions to sequences.
- Databases: NCBI focuses on biomedical information.
2.1.3. Galaxy
- Sequence analysis: Supports diverse sequence analyses.
- Homology search and sequence alignment: Integrates the BLAST tool for database comparisons.
- Gene expression analysis: This includes single-cell RNA-Seq workflows and corresponding visualizations.
2.1.4. Geneious
- Sequence analysis: Support for manipulation, annotation, and characterization of nucleotide/protein sequences.
- Transcription and reverse transcription: Integrates tools for DNA→RNA transcription and RNA→cDNA conversion.
- Sequence translation: Automatic translation of nucleotide sequences to amino acid sequences.
- Pairwise sequence alignment: Local/global alignment with configurable parameters.
2.1.5. Benchling
- Sequence analysis: Design, visualize, and analyze DNA/RNA/amino acid sequences with bulk cloning, alignment, and auto-annotation tools.
- Transcription and reverse transcription: Includes tools for DNA→RNA transcription and RNA→cDNA conversion, integrated into sequence workflows.
- Sequence translation: Automatic translation of nucleotide sequences to amino acid sequences.
- Pairwise sequence alignment: Local/global alignment with configurable parameters.
2.1.6. UGENE
- Sequence analysis: Create, edit, and annotate sequences; identify ORFs, repeats, and motifs; and perform restriction analysis with REBASE integration.
- BLAST integration: Local/remote BLAST+ searches against NCBI, UniProt, PDB, and other databases.
- Pairwise sequence alignment: Smith–Waterman algorithm for local alignment.
- PDB structure viewer and analysis: Visualize 3D molecular structures (PDB/MMDB formats) and predict protein secondary structures using GOR IV and PSIPRED.
2.1.7. Chimera
- Sequence analysis: Ability to analyze different types of biological sequences.
- PDB file analysis and viewer: To read PDB files, analyze the structure, obtain some data, and generate a 3D structure.
- Sequence analysis: Ability to analyze different types of biological sequences not directly by the sequence but with the structure.
- Interaction analysis: To see the interaction between structures.
2.1.8. BLAST
- Sequence analysis: Ability to analyze different types of biological sequences.
- Homology search: Identification of similar sequences in databases.
- Sequence alignment and translation: Aligns sequences and can translate nucleotide sequences during searches.
2.1.9. BioPython
- Sequence analysis: Ability to analyze different types of biological sequences.
- Homology search: Identification of similar sequences in databases.
- Sequence alignment and translation: Aligns sequences and can translate nucleotide sequences during searches.
- Database access: Accesses various biological databases using APIs like Entrez.
- Calculation and generation of phylogenetic trees: Includes modules for constructing and manipulating phylogenetic trees.
- Primer design: Partial support through libraries for designing primers.
- Functional annotation: Interfaces with annotation databases; can parse and analyze annotation data.
2.2. Most Used Tools and Functionalities in Bioinformatics
2.3. Comparison Between Bioinformatics Platforms and Software Tools and Libraries
2.4. Components for BioSuiteT
2.5. Development Framework for BioSuiteT and Modules
- 1.
- DNA Sequence Analysis:
- a.
- Properties analysis: BioSuiteT receives as input a sequence of DNA and, as output, delivers the complementary sequence, reverse complementary sequence, and the distribution of the amino acids with its plot (see Figure 5).
- 2.
- Protein Sequence Analysis:
- a.
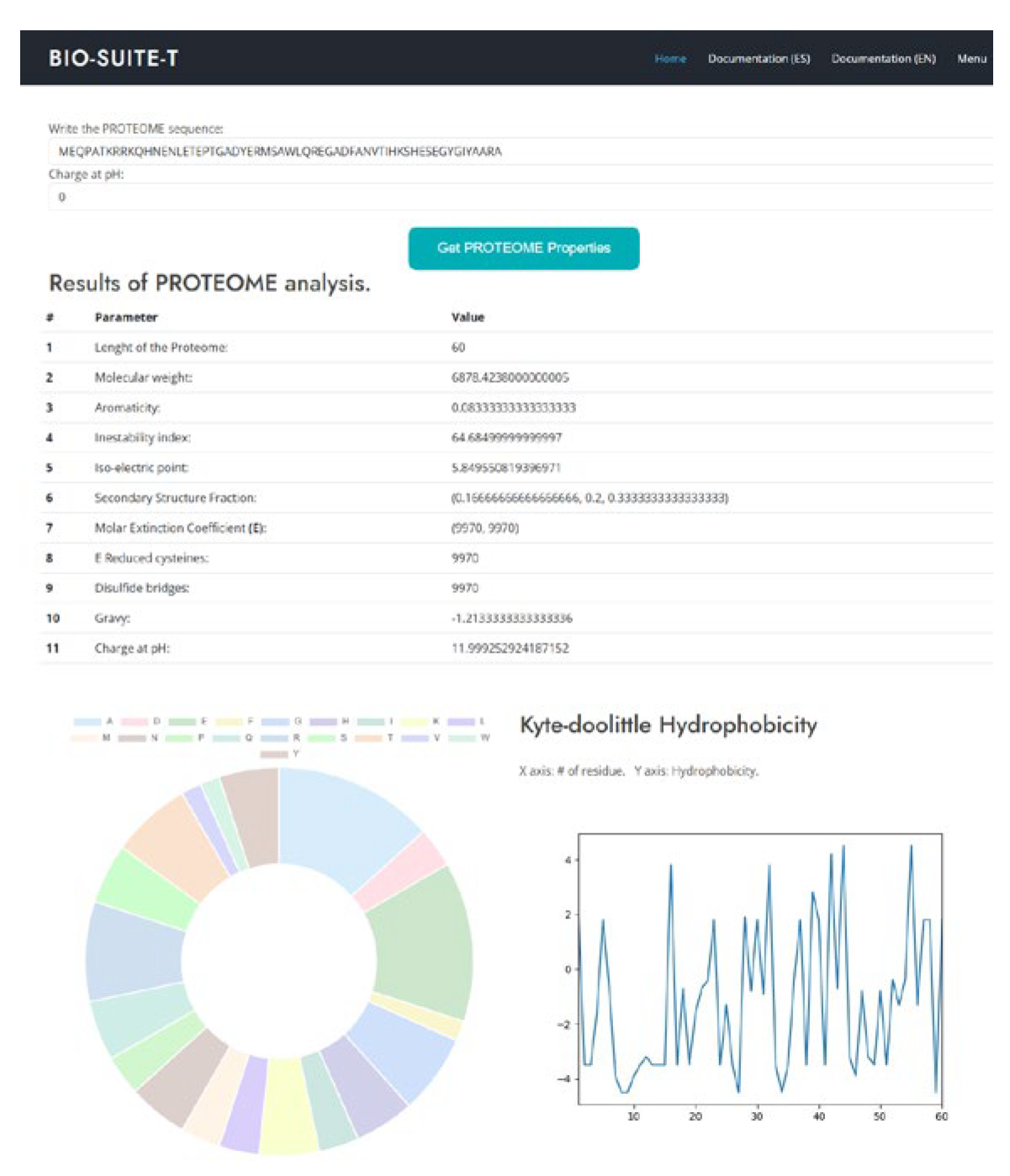
- Properties analysis: BioSuiteT receives as input a sequence of proteins and a pH level for calculating Charge; as output, it delivers the sequence length, molecular weight, aromaticity, instability index, isoelectric point, secondary structure, extinction coefficient, disulfide bridges, hydrophobicity, charge, and plots of amino acid distribution and Kyte Doolittle hydrophobicity (see Figure 6).
- 3.
- Transcription and Reverse Transcription:
- 4.
- Sequence Translation
- a.
- Sequence translation: BioSuiteT receives as input a sequence of mRNA and, as output, delivers the translated amino acid sequence using the respective codon table (BioSuiteT includes all the codon tables; see Figure 8).
- 5.
- Pairwise Sequence Aligner
- a.
- Pairwise sequence aligner: BioSuiteT receives as input two sequences for alignment and, as outputs, delivers aligned sequences with alignment statistics, such as match score, mismatch score, gap score, and information on the type of alignment (local and global). Also, BioSuiteT has all the penalization matrixes (Jones, Levin, McLachlan, MDM78, MegaBlast, NUC 4.4, and 23 more; this can be seen in Figure 9).
- 6.
- BLAST Tool
- a.
- Sequence search: BioSuiteT receives as input a sequence of proteins, nucleotides, or genes; as output, it delivers a list of organisms with similar sequences using BLAST (BioSuiteT includes BLAST-N, BLAST-P, T-BLAST-N, and BLAST-X; see Figure 10).
- 7.
- PDB Viewer
- a.
- Visualization: BioSuiteT receives as input a PDB file, and as output, a 3D protein structure visualization with multiple modes (sticks, cartoons, spheres, lines, cross, label alpha C’s, and variations in surfaces; this can be seen in Figure 11).
- 8.
- PDB Analysis
- a.
- Properties analysis: BioSuiteT receives as input a PDB file; as output, it delivers the properties of the structure such as structure name, deposition date, release date, structure resolution, structure keywords, structure method, structure reference, journal reference, author, compound, source, missing residues information, glycosylation information, model list, chain list, residues list, atom name and coordinates, β factor, element and atom distribution plots, and hetero atom information (see Figure 12).
- 9.
- Phylogenetic Trees
- a.
- Construction of phylogenetic trees: BioSuiteT receives as input a sequence of DNA and, as output, delivers the complementary sequence, reverse complementary sequence, and the distribution of the amino acids with its plot, as can be seen in Figure 13. This phylogenetic tree was constructed by using a CLUSTAL file, which contains an alignment of sequences. Based on this alignment, BioSuiteT, through its algorithms, generates the calculations and plots the phylogenetic tree.
- 10.
- MOTIFS Tool
- a.
- MOTIF analysis: BioSuiteT receives as input a list of MOTIFS and, as output, delivers the consensus sequences, degenerated sequences, reverse complementary sequences, and count matrixes as can be seen in Figure 14.
- 11.
- FungiRegEx (Regular Expression Identification)
- a.
- FungiRegEx module: BioSuiteT receives as input the regular expression to find and a selection of the database to look at the regular expression, as output, sequences that match, and the number of matches that the regular expression has found. The interface of this can be seen in Figure 15.
- 12.
- Stored Biological Data Retrieval
- a.
- Proteome and genome retrieval: BioSuiteT displays the data stored in the MongoDB database; see Figure 16.











2.6. Validation of BioSuiteT
2.6.1. Pairwise Sequence Aligner
- is the punctuation of coincidence/mismatch (e.g., +1 for match, −1 for mismatch).
- is the penalization for the gap (e.g., −2).
- BLOSUM and PAM substitution matrixes are integrated for protein analysis.
2.6.2. PDB Structure Analysis (3Dmol.js)
- Cartesian coordinates: Each atom in a PDB file is represented as .
- Surface rendering: Perspective projection equations are applied for interactive visualization:where is the focal length.
2.6.3. BLAST (Sequence Homology)
- are the sequence lengths.
- are parameters of the extreme distribution.
- is the alignment score.
2.6.4. MOTIFS Analysis
2.6.5. Statistical Results Validation
- Hypothesis testing: In differential analysis (e.g., gene expression), BioSuiteT applies statistical tests (t-test, ANOVA) with False Discovery Rate correction using the Benjamini–Hochberg procedure. The adjusted p-values are computed aswhere is the number of tests. These calculations are performed internally by the statistical libraries used and are not re-implemented within BioSuiteT.
- PCA (Principal Component Analysis): BioSuiteT includes PCA for dimensionality reduction, based on spectral decomposition of the covariance matrix:
2.7. Implementation
- Development Environment Setup
- Django Framework 4.1 as the web application framework.
- Python 3 is a programming language.
- MongoDB 6 for data storage.
- Front-end technologies: HTML5, CSS3, JS, and Bootstrap 5.
- Core Component Implementation
- Integration of BioPython library for the functionalities.
- Integration of 3Dmol.js for molecular visualization.
- Integration of BioPandas for structural data handling.
- FungiRegEx module for regular expression analysis.
- User Interface Development
- Responsive design implementation using Bootstrap.
- Interactive visualization components.
- Form validation and error handling.
- Testing and Quality Assurance
- Unit testing of individual components using the unittest library [53].
- User acceptance testing.
2.8. Concrete Usage Scenario
3. Results
3.1. BioSuiteT Implementation Results
- Core functionality integration
- Successfully integrated 12 functionalities (DNA analysis, proteome analysis, transcription, back transcription, translation, pairwise alignment, BLAST, PDB viewer, PDB analysis, phylogenetic trees, MOTIFS, and FungiRegEx) required for performing bioinformatic analysis, and one more to retrieve stored data.
- Technical Implementation
- Django-based architecture with MongoDB integration for efficient data handling.
- Responsive web interface for supporting multiple devices and screen sizes.
- Flexible deployment options allowing both local and server installation.
- Integration of well-proven and well-tested libraries, including BioPython, BioPandas, FungiRegEx, and 3Dmol.js.
- Implementation following the ISO/IEC 29110 standard using the VSEST 29110 tool.
- These results are summarized below in Table 3.
3.2. Performance Results
- Successfully demonstrated handling of up to 80 concurrent users.
- Maintained stable performance under varying load conditions.
- Efficient memory management of less than 2 GB.
- Reliable database operations through MongoDB.
- Consistent performance across different deployment scenarios.
3.3. Validation Results
- Tasks that included advanced workflows, such as sequence analysis, structure visualization, parameter identification, and execution of multi-step pipelines requiring the use of several integrated tools.
- Tasks focused on routine bioinformatics operations (e.g., BLAST searches, phylogenetic tree generation, data retrieval) to assess usability and accessibility for non-programmers.
- Tasks that involved reviewing system stability under load and validating backend integrations and automated installation processes.
- The quantification of key performance metrics is described below:
- A 95% reduction in technical support requirements was achieved by comparing the number of support requests submitted by non-programming users using BioSuiteT with those using standalone bioinformatics libraries. This was monitored over the testing period and confirmed through post-test surveys and usage logs.
- A 90% reduction in tool switching time, which is derived from time-tracking logs during user workflows and application log tracking. Participants performed identical multi-tool tasks using both BioSuiteT and traditional setups involving multiple separate tools. The time taken to switch between tools, reconfigure settings, and re-import/export data was measured and compared. The unified interface of BioSuiteT resulted in an average of 90% time savings during these transitions, equivalent to a reduction of approximately 465 s, on average, for completing a proposed task.
- User Testing
- 8 researchers, 5 experts directly involved in bioinformatics, 60 practitioners, and 8 developers participated in the testing process.
- Complete coverage and validation of all implemented functionalities.
- Thorough validation of user interface elements and interactions.
- Platform Performance
- No installation issues reported for the web-based version.
- 95% success rate for automated local installations across different environments.
- Less than 2 GB of memory usage during peak operation.
- Stable performance under a full load of 80 concurrent users.
- Usability and Efficiency Gains
- Non-programmers successfully completed the tasks without additional support.
- 95% reduction in technical support demand compared to installable tools.
- 90% reduction in tool-switching time due to the unified set of tools in a single interface.
4. Discussion
4.1. Addressing User Challenges
4.2. Integration and Unified Platform Benefits
4.3. Performance and Technical Considerations
4.4. Validation and User Expertise
4.5. Limitations and Future Work
- Scalability
- BioSuiteT was tested with limited concurrent users (80). While this capacity meets current research group requirements, expanding the platform’s reach would necessitate improvements in server infrastructure and load balancing.
- As biological datasets continue to grow and become complex, there is a need to improve and optimize databases to handle larger datasets more efficiently.
- Functionality
- BioSuiteT covers some of the current needs but presents expansion opportunities. There is a potential to integrate additional specialized bioinformatics tools, particularly in emerging areas of bioinformatics research.
- Current visualization capabilities are functional but could be enhanced to provide more sophisticated data representation options.
- Technical perspective
- Adding offline functionality to the web version would improve accessibility for users with unreliable or limited internet access. This feature would allow researchers to continue their work during network disruptions.
- The automated setup scripts, while effective, could be enhanced to handle a broader range of system configurations and dependencies.
- Memory usage optimization remains an ongoing challenge, with opportunities to improve efficiency when handling large amounts of information.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GUI | Graphical User Interface |
| PDB | Protein Data Bank |
| DNA | Deoxyribonucleic Acid |
| NCBI | National Center for Biotechnology Information |
| PCR | Polymerase Chain Reaction |
| ExPASy | Expert Protein Analysis System |
| BLAST | Basic Local Alignment Search Tool |
| MOTIFS | Specific sequences or structural elements in biological data |
| API | Application Programming Interface |
| ISO/IEC | International Organization for Standardization/International Electrotechnical Commission |
| MVT | Model–View–Template |
| 7Z | A file compression format |
References
- Vasileiou, D.; Karapiperis, C.; Baltsavia, I.; Chasapi, A.; Ahrén, D.; Janssen, P.; Iliopoulos, I.; Promponas, V.; Enright, A.; Ouzounis, C. CGG toolkit: Software components for a computational genomics. PLoS Comput. Biol. 2023, 19, e1011498. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, H.; Ahmed, H.; Hoque, N.; Roy, S.; Bhattacharyya, D. Big data analytics in bioinformatics: Architectures, techniques, tools and issues. Netw. Model. Anal. Health Inform. Bioinform. 2016, 5, 28. [Google Scholar] [CrossRef]
- Long, J.A. The ‘omics’ revolution: Use of genomic, transcriptomic, proteomic and metabolomic tools to predict male reproductive traits that impact fertility in livestock and poultry. Anim. Reprod. Sci. 2020, 220, 106354. [Google Scholar] [CrossRef] [PubMed]
- McArdle, A.; Menikou, S. What is proteomics? Arch. Dis. Child.—Educ. Pract. 2020, 106, 178–181. [Google Scholar] [CrossRef]
- Price, E.; Feyertag, F.; Evans, T.; Miskin, J.; Mitrophanous, K.; Dikicioglu, D. What is the real value of omics data? Enhancing research outcomes and securing long-term data excellence. Nucleic Acids Res. 2024, 52, 12130–12140. [Google Scholar] [CrossRef]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef]
- Biswapriya, M.; Langefeld, C.; Olivier, M.; Cox, L. Integrated omics: Tools, advances and future approaches. J. Mol. Endocrinol. 2019, 62, 21–45. [Google Scholar]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K.; Integration, M.-O.D. Multi-omics Data Integration, Interpretation, and Its Application. Bioinform. Biol. Insights 2020, 14, 1177932219899051. [Google Scholar] [CrossRef]
- Ahmed, A.; Allen, J.; Bhat, T.; Burra, P.; Fliege, C.; Hart, S.; Heldenbrand, J.; Hudson, M.; Deandre, D.; Kalmbach, M.; et al. Design considerations for workflow management systems use in production genomics research and the clinic. Sci. Rep. 2021, 11, 16–27. [Google Scholar] [CrossRef]
- Agamah, F.; Bayjanov, J.; Niehues, A.; Njoku, K.; Skelton, M.; Mazandu, G.; Ederveen, T.; Mulder, N.; Chimusa, E.; Hoen, P. Computational approaches for network-based integrative multi-omics analysis. Front. Mol. Biosci. 2022, 9, 967205. [Google Scholar] [CrossRef]
- Gerner, C.; Hill, M. Integrative Multi-Omics in Biomedical Research. Biomolecules 2021, 11, 1527. [Google Scholar] [CrossRef]
- Gasteiger, E. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [PubMed]
- Yaseen, M.; Shafi, A.; Masoodi, K. ExPASy portal. In Bioinformatics for EveryOne; Academic Press: Cambridge, MA, USA, 2022; pp. 119–125. [Google Scholar]
- Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information. Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 1 April 2025).
- National Institute of Health—National Library of Medicine. NCBI—About Us and Research. National Library of Medicine. 2024. Available online: https://www.nih.gov/about-nih/ (accessed on 18 December 2024).
- Cock, P.; Antao, T.; Chang, J.; Chapman, B.; Cox, C.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef] [PubMed]
- Cogent3 Team. Cogent3. Available online: https://cogent3.org/ (accessed on 1 May 2025).
- Armstrong, J.; Hickey, G.; Diekhans, M.; Fiddes, I.; Novak, A.; Alden, D.; Fang, Q.; Xie, D.; Feng, S.; Stiller, J.; et al. Progressive Cactus is a multiple-genome aligner for the thousand-genome era. Nature 2020, 587, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Knight, R.; Maxwell, P.; Birmingham, A.; Carnes, J.; Caporaso, G.; Easton, B.; Eaton, M.; Hamady, M.; Lindsay, H.; Liu, Z.; et al. PyCogent: A toolkit for making sense from sequence. Genome Biol. 2007, 8, R171. [Google Scholar] [CrossRef]
- Butterfield, A.; Vedagiri, V.; Lang, E.; Lawrence, C.; Wakefield, M.; Isaev, A.; Huttley, G. PyEvolve: A toolkit for statistical modelling of molecular evolution. BMC Bioinform. 2004, 5, 1. [Google Scholar] [CrossRef]
- Terrón-Macias, V.; Mejia, J.; Canseco-Pérez, M.; Muñoz, M.; Terrón-Hernández, M. FungiRegEx: A tool for patterns identification in Fungal Proteomic sequences using regular expressions. Appl. Sci. 2024, 14, 4429. [Google Scholar] [CrossRef]
- Cazier, J. Is There Too Many Bioinformatics Tools? Birmingham University: Birmingham, UK, 2013. Available online: https://www.researchgate.net/post/Is_there_too_many_bioinformatics_tools2 (accessed on 1 January 2025).
- Duck, G.; Nenadic, G.; Filannino, M.; Brass, A.; Robertson, D.; Stevens, R. A Survey of Bioinformatics Database and Software Usage through Mining the Literature. PLoS ONE 2016, 11, e0157989. [Google Scholar] [CrossRef]
- Ahmad, T.; Senaidy, A. Software Tools in Bioinformatics: A Survey on the Importance and Issues Faced in Implementation. Glob. Eng. Technol. Rev. 2013, 3, 6–10. [Google Scholar]
- Terrón-Macias, V.; Mejía, J. Propuesta de unificación de herramientas bioinformáticas para el análisis de secuencias proteómicas y genómicas de algas y hongos. Rev. Lbérica De Sist. Y Tecnol. De Inf. 2024, 53, 132–147. [Google Scholar] [CrossRef]
- Pettersen, E.; Goddard, T.; Huang, C.; Couch, G.; Greenblatt, D.; Meng, E.; Ferrin, T. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.; Gish, W.; Miller, W.; Myers, E.; Lipman, D. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- ExPASy. About Expasy. 1 January 2024. Available online: https://www.expasy.org/about#some-history (accessed on 31 December 2024).
- The Galaxy Community. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 Update. Nucleic Acids Res. 2022, 50, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Davies, K. From the Bench to Benchling. Genet. Eng. Biotechnol. News Edge 2020, 2, 303–309. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef]
- Bayat, A. Science, medicine, and the future Bioinformatics. Clin. Rev. 2002, 324, 1018–1022. [Google Scholar] [CrossRef]
- Ramsden, J. Bioinformatics: An Introduction, 4th ed.; Springer Nature: Buckingham, UK, 2023; pp. XXII, 404. [Google Scholar]
- Agnihotry, S.; Pathak, R.; Singh, D.; Tiwari, A.; Hussain, I. Protein structure prediction. In Bioinformatics: Methods and Applications; Academic Press: Cambridge, MA, USA, 2022; pp. 177–188. [Google Scholar]
- Zhang, C.; Freddolino, L. A large-scale assessment of sequence database search tools for homology-based protein function prediction. Brief. Bioinform. 2024, 25, 1–12. [Google Scholar] [CrossRef]
- Narayanan, P. Bioinformatics: A primer, United States of America; New Age International Publishers: New York, NY, USA, 2005; p. 248. [Google Scholar]
- Sherman, B.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.; Lane, C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, 216–221. [Google Scholar] [CrossRef]
- Abed, R.; Al-Najjar, Y. Bioinformatics Storing Databases. Tech. Biochem. 2021, 2, 96–105. [Google Scholar] [CrossRef]
- Huddleston, J.; Hadfield, J.; Sibley, T.; Lee, J.; Fay, K.; Ilcisin, M.; Harkins, E.; Bedford, T.; Neher, R.; Hodcroft, E. Augur: A bioinformatics toolkit for phylogenetic analyses of human pathogens. J. Open Source Softw. 2021, 6, 2906. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.; Skjærven, L.; Yao, X.-Q. The Bio3D packages for structural bioinformatics. Protein Sci. 2020, 30, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Mathon, L.; Valentini, A.; Guérin, P.E.; Normandeau, E.; Noel, C.; Lionnet, C.; Boulanger, E.; Thuiller, W.; Bernatchez, L.; Mouillot, D.; et al. Benchmarking bioinformatic tools for fast and accurate eDNA metabarcoding species identification. Mol. Ecol. Resour. 2021, 21, 2565–2579. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Song, Z.; Zhong, X.; Huang, M.; Shen, D.; Gao, P.; Qian, X.; Wang, M.; He, X.; Wang, T.; et al. Sangerbox: A comprehensive, interaction-friendly clinical bioinformatics analysis platform. iMeta 2022, 1, e36. [Google Scholar] [CrossRef]
- Wang, Y.; Geer, L.; Chappey, C.; Kans, J.; Bryant, S. Cn3D: Sequence and structure views for Entrez. Trends Biochem. Sci. 2000, 25, 300–302. [Google Scholar] [CrossRef]
- Raschka, S. BioPandas: Working with molecular structures in pandas DataFrames. J. Open Source Softw. 2017, 2, 279. [Google Scholar] [CrossRef]
- Rego, N.; Koes, D. 3Dmol.js: Molecular visualization with WebGL. Bioinformatics 2014, 31, 1322–1324. [Google Scholar] [CrossRef]
- Mejía, J.; Terrón-Macias, V.; Muñoz, M.; Terrón-Hernández, M.; Canseco-Perez, M. VSEST 29110 Tool: Using ChatGPT to evaluate the implementation of the ISO/IEC 29110 work products. IEEE Access 2024, 12, 120935–120948. [Google Scholar] [CrossRef]
- Chen, S.; Ahmmed, S.; Lal, K.; Dem, C. DJANGO Web Development Framework: Powering the modern web. Am. J. Trade Policy 2020, 1, 99–106. [Google Scholar] [CrossRef]
- Kumar, M.; Nandal, R. Role of Python in Rapid Web Application Development Using Django. In Proceedings of the International Conference on Innovative Computing and Communication, New Delhi, India, 8 March 2024; pp. 1–13. [Google Scholar]
- Chauhan, A. A Review on Various Aspects of MongoDb Databases. Int. J. Eng. Res. Technol. 2019, 8, 90–92. [Google Scholar]
- Manohar, P.; Singh, S. Protein Sequence Alignment: A Review. World Appl. Program. 2012, 2, 141–145. [Google Scholar]
- Pandy, G.; Pugazhenthi, V.; Murugan, A. Advances in Software Testing in 2024: Experimental Insights, Frameworks, and Future Directions. Int. J. Adv. Res. Comput. Commun. Eng. 2024, 13, 40–45. [Google Scholar]
- Lukasczyk, S.; Kroiß, F.; Fraser, G. Automated Unit Test Generation for Python. Search-Based Softw. Eng. 2020, 12420, 9–24. [Google Scholar]
- Gentile, A. Basics of DJango_Model-View-Template (MVT) Architecture. 30 November 2023. Available online: https://angelogentileiii.medium.com/basics-of-django-model-view-template-mvt-architecture-8585aecffbf6 (accessed on 18 December 2024).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functionalities | Platforms | Software Tools and Libraries | |||||||
|---|---|---|---|---|---|---|---|---|---|
| ExPASy | NCBI Tools | Galaxy | Geneious | Benchling | UGENE | Chimera | BLAST | BioPython | |
| Sequence analysis | ✔ | ✔ | ✔ | ✔ | ✔ | ✔ | Partial | ✔ | ✔ |
| Structure prediction | ✔ | ✔ | ✖ | ✖ | ✖ | ✖ | ✖ | ✖ | Partial |
| Homology search and sequence alignment | ✔ | ✔ | ✔ | ✔ | ✔ | ✔ | ✖ | ✔ | ✔ |
| Primer design | ✔ | ✔ | ✔ | ✔ | ✖ | ✔ | ✖ | ✖ | Partial |
| Gene expression analysis | ✔ | ✔ | ✔ | ✔ | ✖ | ✔ | ✖ | ✖ | Partial |
| Functional annotation | ✔ | ✔ | ✔ | ✔ | Partial | ✔ | ✖ | ✖ | Partial |
| Databases and data retrieval | ✔ | ✔ | ✔ | ✔ | ✔ | ✔ | ✖ | ✖ | ✔ |
| Phylogenetic analysis | ✔ | ✔ | ✔ | ✔ | ✖ | ✔ | ✖ | ✖ | ✔ |
| Structural visualization and analysis | Partial | Partial | ✖ | Limited | ✖ | ✔ | ✔ | ✖ | Limited |
| Translation and transcription | ✔ | ✔ | ✔ | ✔ | ✔ | ✔ | ✖ | ✖ | ✔ |
| Genome and species exploration | Partial | ✔ | ✔ | Partial | ✖ | ✔ | ✖ | ✖ | Partial |
| Reverse transcription | ✔ | ✖ | ✖ | ✖ | ✖ | ✖ | ✖ | ✖ | ✔ |
| Interaction analysis | Partial | Partial | ✖ | ✖ | ✖ | ✖ | ✔ | ✖ | Partial |
| Variants and mutations | Limited | ✔ | ✔ | ✔ | ✖ | ✔ | Partial | ✖ | Partial |
| Component | Role |
|---|---|
| BioPython v.1.81 | Core sequence analysis |
| BioPandas v0.4.1 | Structural data handling |
| FungiRegEx v1.0 | Regular expression finding |
| 3Dmol.js | PDB structural visualization |
| Python v3.10 | Programming language |
| Django v4.1.3 | Framework for web application development |
| User Challenge | % Affected Users | BioSuiteT Solution | Impact |
|---|---|---|---|
| Tool installation complexity | 25% | Web-based access; automated local scripts for execution. | Web: 0 min setup; local: 85% faster installation. |
| Programming expertise barrier | 20.8% | Unified GUI; no-code interface. | Non-programmers completed tasks without support. |
| Tool fragmentation | 20.8% | Tools are integrated into a single environment. | Reduction in tool switching. |
| Time spent troubleshooting | 31.25% | Single environment; detailed documentation about how to execute the web application locally. | Reduction in support requests. |
| Demand for unified frameworks | 14.6% | Django-based integration. | Full-tool interoperability. |
| Functionalities | Platforms | ||||||
|---|---|---|---|---|---|---|---|
| ExPASy | NCBI Tools | BioSuiteT | Galaxy | Geneious | Benchling | UGENE | |
| DNA sequence analysis | ✔ | ✔ | ✔ | ✔ | ✔ | ✔ | ✔ |
| Protein sequence analysis | ✔ | ✔ | ✔ | ✔ | ✔ | ✔ | ✔ |
| Transcription | ✔ | ✔ | ✔ | ✔ | ✔ | ✔ | ✔ |
| Reverse transcription | ✔ | ✖ | ✔ | ✖ | ✖ | ✖ | ✖ |
| Sequence translation | ✔ | ✔ | ✔ | ✔ | ✔ | ✔ | ✔ |
| Pairwise sequence alignment | ✔ | ✔ | ✔ | ✔ | ✔ | ✔ | ✔ |
| BLAST integration | ✔ | ✔ | ✔ | ✔ | ✔ | ✔ | ✔ |
| PDB structure viewer | ✔ | ✔ | ✔ | ✖ | Limited | ✖ | ✖ |
| PDB analysis | ✖ | ✖ | Partial | ✖ | ✖ | ✖ | ✖ |
| Phylogenetic trees | ✔ | ✔ | ✔ | ✔ | ✔ | ✖ | ✔ |
| MOTIFS analysis | ✔ | ✔ | ✔ | ✔ | ✔ | Partial | ✔ |
| Regular expression search | ✖ | ✖ | ✔ | ✖ | ✔ | ✖ | ✔ |
| Biological data storage | ✖ | ✖ | ✔ | ✔ | ✔ | ✔ | ✔ |
| GUI | ✔ | ✔ | ✔ | ✔ | ✔ | ✔ | ✔ |
| No installation required (web version) | ✔ | ✔ | ✔ | ✔ | ✖ | ✔ | ✖ |
| Unified environment | Partial | Partial | ✔ | ✔ | ✔ | ✔ | ✔ |
| Type of GUI | Web | Web | Web/Local | Web | Desktop | Web | Desktop |
| Installation | No required | No required | Optional | Required | Required | No required | Required |
| Modularity | No | No | Yes | Yes | No | No | No |
| paid? | Free | Free | Free | Free | Paid | Paid | Free |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terron-Macias, V.; Mejia, J.; Muñoz, M.; Terron-Hernandez, M.; Canseco-Perez, M.; Berrones-Hernández, R.; Quiñonez, Y. BioSuiteT: A Unified Tool for Biological Sequence Analysis. Appl. Sci. 2025, 15, 6565. https://doi.org/10.3390/app15126565
Terron-Macias V, Mejia J, Muñoz M, Terron-Hernandez M, Canseco-Perez M, Berrones-Hernández R, Quiñonez Y. BioSuiteT: A Unified Tool for Biological Sequence Analysis. Applied Sciences. 2025; 15(12):6565. https://doi.org/10.3390/app15126565
Chicago/Turabian StyleTerron-Macias, Victor, Jezreel Mejia, Mirna Muñoz, Miguel Terron-Hernandez, Miguel Canseco-Perez, Roberto Berrones-Hernández, and Yadira Quiñonez. 2025. "BioSuiteT: A Unified Tool for Biological Sequence Analysis" Applied Sciences 15, no. 12: 6565. https://doi.org/10.3390/app15126565
APA StyleTerron-Macias, V., Mejia, J., Muñoz, M., Terron-Hernandez, M., Canseco-Perez, M., Berrones-Hernández, R., & Quiñonez, Y. (2025). BioSuiteT: A Unified Tool for Biological Sequence Analysis. Applied Sciences, 15(12), 6565. https://doi.org/10.3390/app15126565