
In Silico Development of SARS-CoV-2 Non-Covalent Mpro Inhibitors: A Review



Abstract
1. Introduction
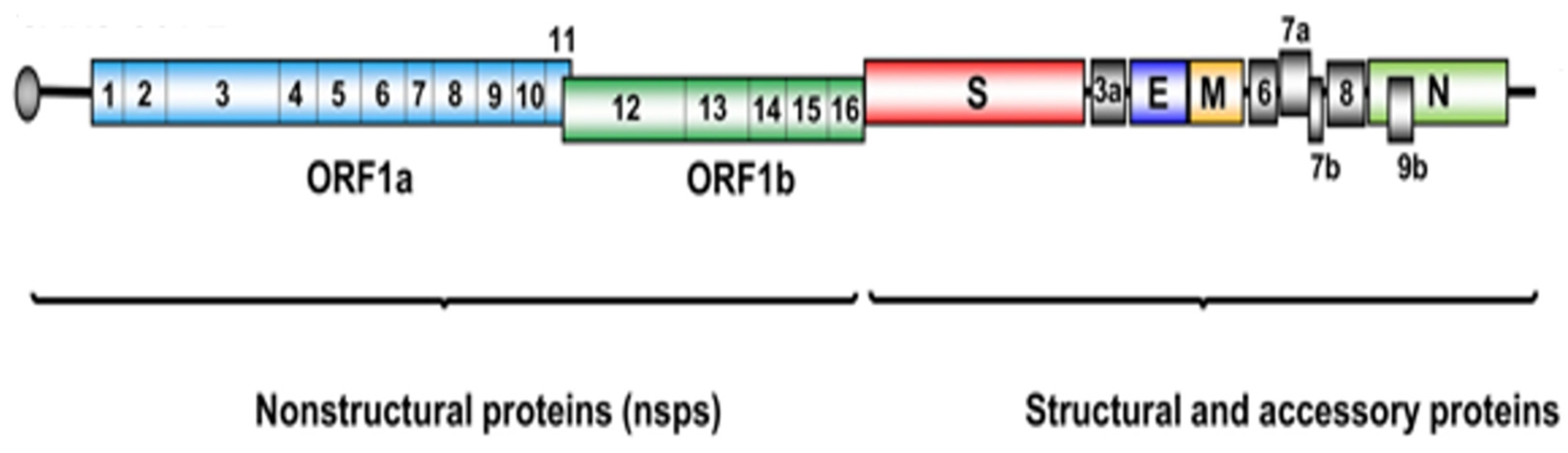
1.1. The Main Protease (Mpro) Enzyme of SARS-CoV-2
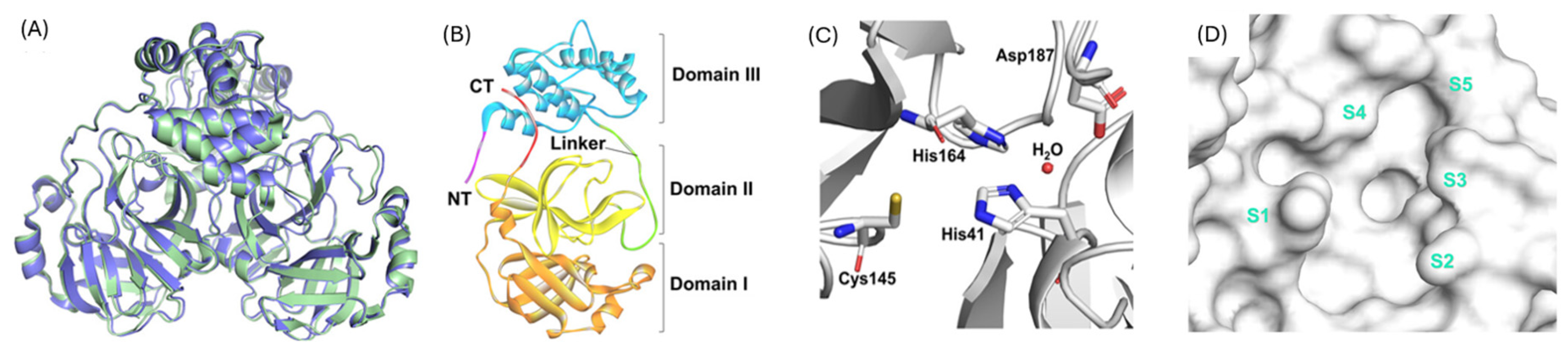
1.2. The Need for Novel Inhibitors
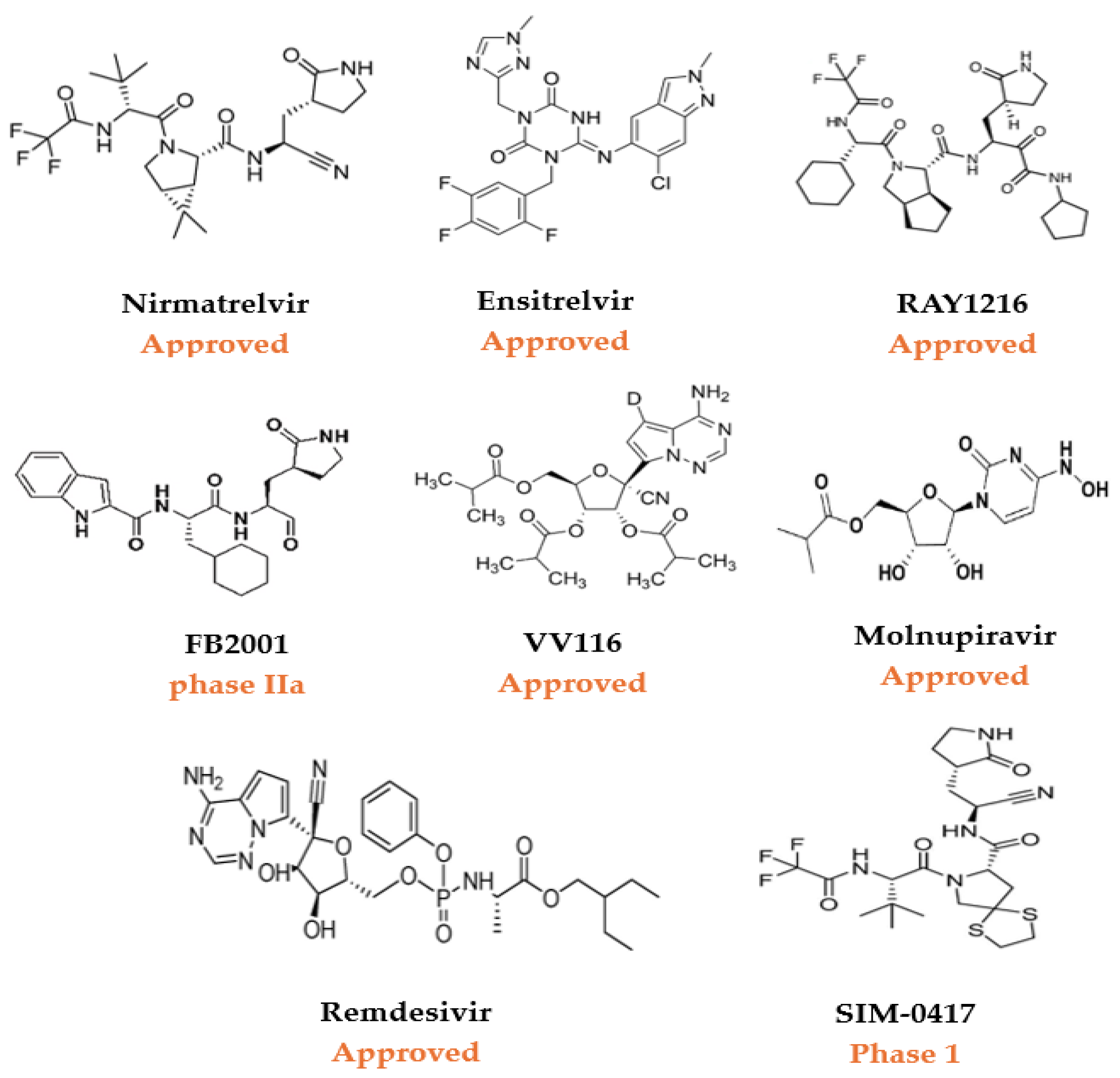
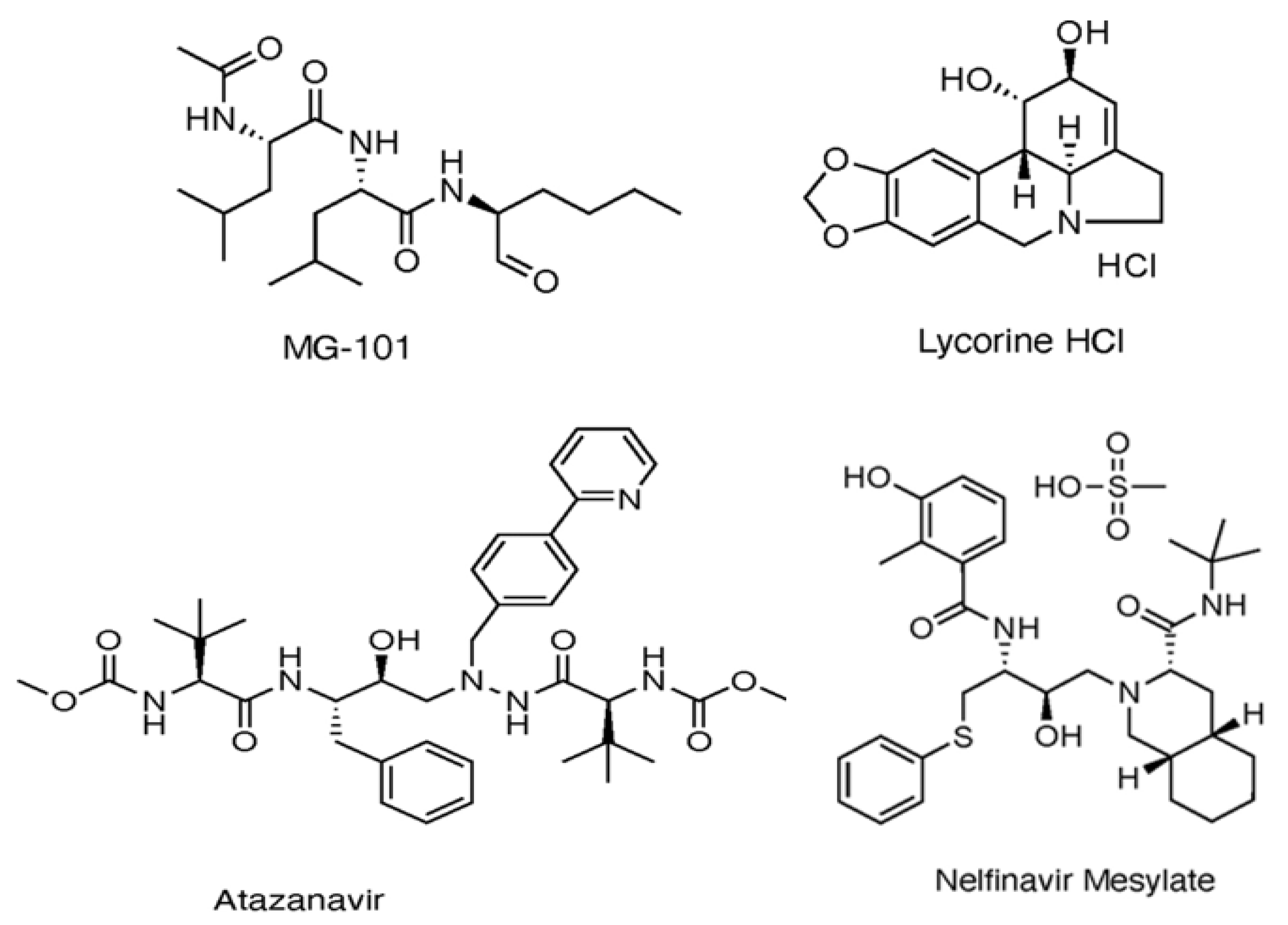
1.3. The Role of Drug Repurposing
2. Mpro Inhibitors for SARS-CoV-2
2.1. Overview of Mpro Inhibitors
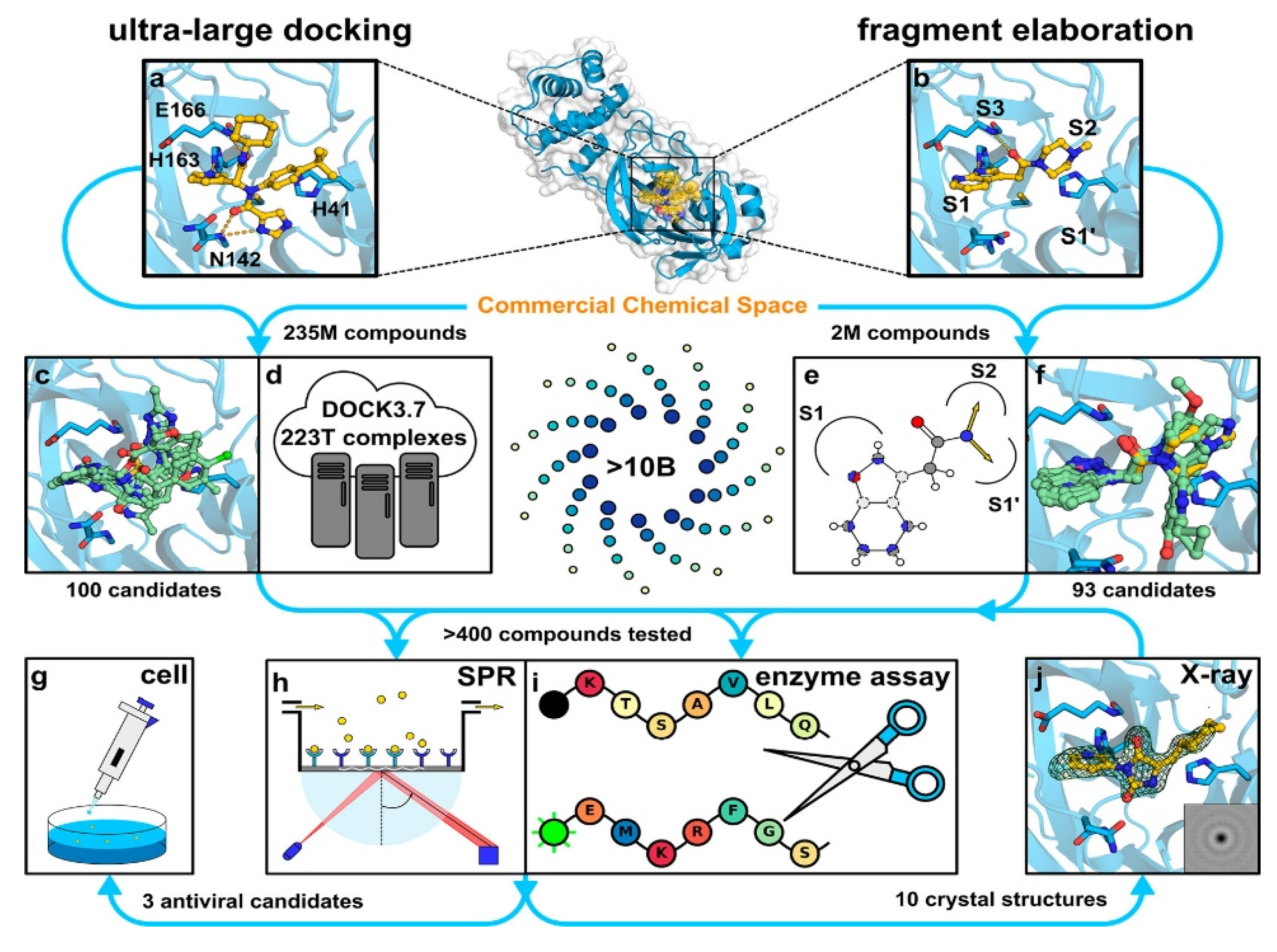
2.2. Discovery Strategies for Non-Covalent Inhibitors of Mpro
2.3. Computer-Aided Drug Design
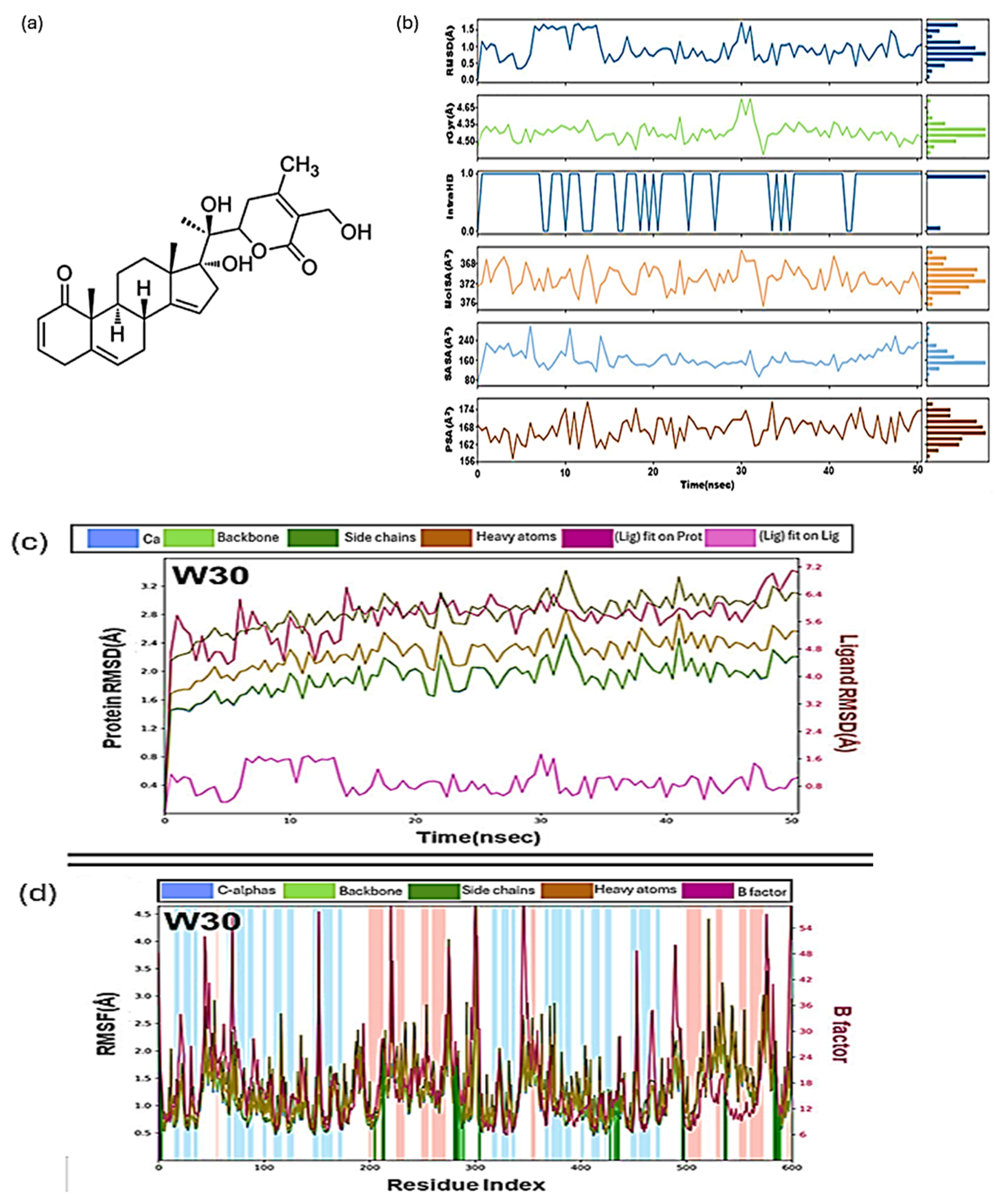
2.4. Molecular Dynamics (MD) in Drug Development
2.5. Leveraging Quantum Mechanics for ADME-Optimised Drugs
3. Summary and Outlook
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kumar, D.; Chauhan, G.; Kalra, S.; Kumar, B.; Gill, M.S. A perspective on potential target proteins of COVID-19: Comparison with SARS-CoV for designing new small molecules. Bioorgan. Chem. 2020, 104, 104326. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liang, C.; Xin, L.; Ren, X.; Tian, L.; Ju, X.; Li, H.; Wang, Y.; Zhao, Q.; Liu, H. The development of Coronavirus 3C-Like protease (3CLpro) inhibitors from 2010 to 2020. Eur. J. Med. Chem. 2020, 206, 112711. [Google Scholar] [CrossRef]
- Sharma, P.; Vijayan, V.; Pant, P.; Sharma, M.; Vikram, N.; Kaur, P.; Singh, T.; Sharma, S. Identification of potential drug candidates to combat COVID-19: A structural study using the main protease (mpro) of SARS-CoV-2. J. Biomol. Struct. Dyn. 2021, 39, 6649–6659. [Google Scholar] [CrossRef]
- Krammer, F. SARS-CoV-2 vaccines in development. Nature 2020, 586, 516–527. [Google Scholar] [CrossRef]
- Astuti, I. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 407–412. [Google Scholar] [CrossRef]
- Chakraborty, C.; Bhattacharya, M.; Sharma, A.R.; Chatterjee, S.; Agoramoorthy, G.; Lee, S.-S. Structural Landscape of nsp Coding Genomic Regions of SARS-CoV-2-ssRNA Genome: A Structural Genomics Approach Toward Identification of Druggable Genome, Ligand-Binding Pockets, and Structure-Based Druggability. Mol. Biotechnol. 2024, 66, 641–662. [Google Scholar] [CrossRef]
- Mondol, W.C. Exploring the Potential of Organic Molecules in the Treatment of COVID-19; Brac University: Dhaka, Bangladesh, 2021. [Google Scholar]
- Chan, C.C.-Y.; Guo, Q.; Chan, J.F.-W.; Tang, K.; Cai, J.-P.; Chik, K.K.-H.; Huang, Y.; Dai, M.; Qin, B.; Ong, C.P.; et al. Identification of novel small-molecule inhibitors of SARS-CoV-2 by chemical genetics. Acta Pharm. Sin. B 2024, 14, 4028–4044. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Jiang, X.-M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F. Structure-based design, synthesis and biological evaluation of peptidomimetic aldehydes as a novel series of antiviral drug candidates targeting the SARS-CoV-2 main protease. bioRxiv 2020. [Google Scholar] [CrossRef]
- Lamers, M.M.; Haagmans, B.L. SARS-CoV-2 pathogenesis. Nat. Rev. Microbiol. 2022, 20, 270–284. [Google Scholar] [CrossRef]
- Brant, A.C.; Tian, W.; Majerciak, V.; Yang, W.; Zheng, Z.-M. SARS-CoV-2: From its discovery to genome structure, transcription, and replication. Cell Biosci. 2021, 11, 136. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Geng, M.; Peng, Y.; Meng, L.; Lu, S. Molecular immune pathogenesis and diagnosis of COVID-19. J. Pharm. Anal. 2020, 10, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Adelusi, T.I.; Oyedele, A.-Q.K.; Monday, O.E.; Boyenle, I.D.; Idris, M.O.; Ogunlana, A.T.; Ayoola, A.M.; Fatoki, J.O.; Kolawole, O.E.; David, K.B. Dietary polyphenols mitigate SARS-CoV-2 main protease (Mpro)–Molecular dynamics, molecular mechanics, and density functional theory investigations. J. Mol. Struct. 2022, 1250, 131879. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lee, J.-Y.; Yang, J.-S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef]
- Muratov, E.N.; Amaro, R.; Andrade, C.H.; Brown, N.; Ekins, S.; Fourches, D.; Isayev, O.; Kozakov, D.; Medina-Franco, J.L.; Merz, K.M. A critical overview of computational approaches employed for COVID-19 drug discovery. Chem. Soc. Rev. 2021, 50, 9121–9151. [Google Scholar] [CrossRef]
- Han, S.H.; Goins, C.M.; Arya, T.; Shin, W.-J.; Maw, J.; Hooper, A.; Sonawane, D.P.; Porter, M.R.; Bannister, B.E.; Crouch, R.D.; et al. Structure-Based Optimization of ML300-Derived, Noncovalent Inhibitors Targeting the Severe Acute Respiratory Syndrome Coronavirus 3CL Protease (SARS-CoV-2 3CLpro). J. Med. Chem. 2022, 65, 2880–2904. [Google Scholar] [CrossRef]
- Unoh, Y.; Uehara, S.; Nakahara, K.; Nobori, H.; Yamatsu, Y.; Yamamoto, S.; Maruyama, Y.; Taoda, Y.; Kasamatsu, K.; Suto, T.; et al. Discovery of S-217622, a Noncovalent Oral SARS-CoV-2 3CL Protease Inhibitor Clinical Candidate for Treating COVID-19. J. Med. Chem. 2022, 65, 6499–6512. [Google Scholar] [CrossRef]
- Duan, X.; Lacko, L.A.; Chen, S. Druggable targets and therapeutic development for COVID-19. Front. Chem. 2022, 10, 963701. [Google Scholar] [CrossRef]
- Liu, X.; Ren, X.; Hua, M.; Liu, F.; Ren, X.; Sui, C.; Li, Q.; Luo, F.; Jiang, Z.; Xia, Z. Progress of SARS-CoV-2 Main protease peptide-like inhibitors. Chem. Biol. Drug Des. 2024, 103, e14425. [Google Scholar] [CrossRef]
- Xu, Y.-S.; Chigan, J.-Z.; Li, J.-Q.; Ding, H.-H.; Sun, L.-Y.; Liu, L.; Hu, Z.; Yang, K.-W. Hydroxamate and thiosemicarbazone: Two highly promising scaffolds for the development of SARS-CoV-2 antivirals. Bioorgan. Chem. 2022, 124, 105799. [Google Scholar] [CrossRef]
- Tumskiy, R.S.; Tumskaia, A.V. Multistep rational molecular design and combined docking for discovery of novel classes of inhibitors of SARS-CoV-2 main protease 3CLpro. Chem. Phys. Lett. 2021, 780, 138894. [Google Scholar] [CrossRef]
- Ebenezer, O.; Shapi, M. Promising inhibitors against main protease of SARS-CoV-2 from medicinal plants: In silico identification. Acta Pharm. 2022, 72, 159–169. [Google Scholar] [CrossRef]
- Hu, Q.; Xiong, Y.; Zhu, G.-H.; Zhang, Y.-N.; Zhang, Y.-W.; Huang, P.; Ge, G.-B. The SARS-CoV-2 main protease (Mpro): Structure, function, and emerging therapies for COVID-19. MedComm 2022, 3, e151. [Google Scholar] [CrossRef]
- Fernandes, Q.; Inchakalody, V.P.; Merhi, M.; Mestiri, S.; Taib, N.; Moustafa Abo El-Ella, D.; Bedhiafi, T.; Raza, A.; Al-Zaidan, L.; Mohsen, M.O.; et al. Emerging COVID-19 variants and their impact on SARS-CoV-2 diagnosis, therapeutics and vaccines. Ann. Med. 2022, 54, 524–540. [Google Scholar] [CrossRef]
- Cooper, M.S.; Zhang, L.; Ibrahim, M.; Zhang, K.; Sun, X.; Röske, J.; Göhl, M.; Brönstrup, M.; Cowell, J.K.; Sauerhering, L. Diastereomeric Resolution Yields Highly Potent Inhibitor of SARS-CoV-2 Main Protease. J. Med. Chem. 2022, 65, 13328–13342. [Google Scholar] [CrossRef]
- Zhu, J.; Zhang, H.; Lin, Q.; Lyu, J.; Lu, L.; Chen, H.; Zhang, X.; Zhang, Y.; Chen, K. Progress on SARS-CoV-2 3CLpro Inhibitors: Inspiration from SARS-CoV 3CLpro Peptidomimetics and Small-Molecule Anti-Inflammatory Compounds. Drug Des. Dev. Ther. 2023, 16, 1067–1082. [Google Scholar] [CrossRef]
- Zhou, K.; Chen, D. Conventional Understanding of SARS-CoV-2 Mpro and Common Strategies for Developing Its Inhibitors. Chem.—A Eur. J. 2023, 24, e202300301. [Google Scholar] [CrossRef]
- Song, L.; Gao, S.; Ye, B.; Yang, M.; Cheng, Y.; Kang, D.; Yi, F.; Sun, J.-P.; Menéndez-Arias, L.; Neyts, J. Medicinal chemistry strategies towards the development of non-covalent SARS-CoV-2 Mpro inhibitors. Acta Pharm. Sin. B 2023, 14, 87–109. [Google Scholar] [CrossRef]
- Mengist, H.M.; Dilnessa, T.; Jin, T. Structural basis of potential inhibitors targeting SARS-CoV-2 main protease. Front. Chem. 2021, 9, 622898. [Google Scholar] [CrossRef]
- Alexandris, N.; Lagoumintzis, G.; Chasapis, C.T.; Leonidas, D.D.; Papadopoulos, G.E.; Tzartos, S.J.; Tsatsakis, A.; Eliopoulos, E.; Poulas, K.; Farsalinos, K. Nicotinic cholinergic system and COVID-19: In silico evaluation of nicotinic acetylcholine receptor agonists as potential therapeutic interventions. Toxicol. Rep. 2021, 8, 73–83. [Google Scholar] [CrossRef]
- Li, Q.; Kang, C. Progress in developing inhibitors of SARS-CoV-2 3C-like protease. Microorganisms 2020, 8, 1250. [Google Scholar] [CrossRef]
- Lockbaum, G.J.; Reyes, A.C.; Lee, J.M.; Tilvawala, R.; Nalivaika, E.A.; Ali, A.; Kurt Yilmaz, N.; Thompson, P.R.; Schiffer, C.A. Crystal structure of SARS-CoV-2 main protease in complex with the non-covalent inhibitor ML188. Viruses 2021, 13, 174. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Li, W.; Zhou, X.; Zhang, J.; Li, J. Crystal structures of coronaviral main proteases in complex with the non-covalent inhibitor X77. Int. J. Biol. Macromol. 2024, 276, 133706. [Google Scholar] [CrossRef]
- Zagórska, A.; Czopek, A.; Fryc, M.; Jończyk, J. Inhibitors of SARS-CoV-2 Main Protease (Mpro) as Anti-Coronavirus Agents. Biomolecules 2024, 14, 797. [Google Scholar] [CrossRef]
- Nandi, A.; Asadi, M.; Zhang, A.; Chu, Z.T.; Warshel, A. Mechanistic Insights into Nitrile and Alkyne Covalent Inhibitors of the SARS-CoV-2 Main Protease. ACS Catal. 2025, 15, 1158–1169. [Google Scholar] [CrossRef]
- Zhang, R.; Yan, H.; Zhou, J.; Yan, G.; Liu, X.; Shang, C.; Chen, Y. Improved fluorescence-based assay for rapid screening and evaluation of SARS-CoV-2 main protease inhibitors. J. Med. Virol. 2024, 96, e29498. [Google Scholar] [CrossRef]
- Narayanan, A.; Narwal, M.; Majowicz, S.A.; Varricchio, C.; Toner, S.A.; Ballatore, C.; Brancale, A.; Murakami, K.S.; Jose, J. Identification of SARS-CoV-2 inhibitors targeting Mpro and PLpro using in-cell-protease assay. Commun. Biol. 2022, 5, 169. [Google Scholar] [CrossRef]
- Jayawardena, T.U.; Merindol, N.; Liyanage, N.S.; Desgagné-Penix, I. Unveiling Amaryllidaceae alkaloids: From biosynthesis to antiviral potential–a review. Nat. Prod. Rep. 2024, 41, 721–747. [Google Scholar] [CrossRef]
- Frediansyah, A.; Tiwari, R.; Sharun, K.; Dhama, K.; Harapan, H. Antivirals for COVID-19: A critical review. Clin. Epidemiol. Glob. Health 2021, 9, 90–98. [Google Scholar] [CrossRef]
- Mao, L.; Shaabani, N.; Zhang, X.; Jin, C.; Xu, W.; Argent, C.; Kushnareva, Y.; Powers, C.; Stegman, K.; Liu, J. Olgotrelvir, a dual inhibitor of SARS-CoV-2 Mpro and cathepsin L, as a standalone antiviral oral intervention candidate for COVID-19. Med 2024, 5, 42–61.e23. [Google Scholar] [CrossRef]
- Pang, X.; Xu, W.; Liu, Y.; Li, H.; Chen, L. The research progress of SARS-CoV-2 main protease inhibitors from 2020 to 2022. Eur. J. Med. Chem. 2023, 257, 115491. [Google Scholar] [CrossRef]
- Thakur, A.; Sharma, G.; Badavath, V.N.; Jayaprakash, V.; Merz, K.M., Jr.; Blum, G.; Acevedo, O. Primer for Designing Main Protease (Mpro) Inhibitors of SARS-CoV-2. J. Phys. Chem. Lett. 2022, 13, 5776–5786. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Liu, H.; Sun, Q.; Liang, H.; Li, C.; Deng, X.; Liu, Y.; Lai, L. Potent inhibitors of SARS-CoV-2 3C-like protease derived from N-substituted isatin compounds. Eur. J. Med. Chem. 2020, 206, 112702. [Google Scholar] [CrossRef]
- She, Z.; Yao, Y.; Wang, C.; Li, Y.; Xiong, X.; Liu, Y. Mpro-targeted anti-SARS-CoV-2 inhibitor-based drugs. J. Chem. Res. 2023, 47, 17475198231184799. [Google Scholar] [CrossRef]
- Yang, L.; Wang, Z. Bench-to-bedside: Innovation of small molecule anti-SARS-CoV-2 drugs in China. Eur. J. Med. Chem. 2023, 257, 115503. [Google Scholar] [CrossRef]
- Kitamura, N.; Sacco, M.D.; Ma, C.; Hu, Y.; Townsend, J.A.; Meng, X.; Zhang, F.; Zhang, X.; Ba, M.; Szeto, T.; et al. Expedited Approach toward the Rational Design of Noncovalent SARS-CoV-2 Main Protease Inhibitors. J. Med. Chem. 2022, 65, 2848–2865. [Google Scholar] [CrossRef]
- Kühl, N.; Lang, J.; Leuthold, M.M.; Klein, C.D. Discovery of potent benzoxaborole inhibitors against SARS-CoV-2 main and dengue virus proteases. Eur. J. Med. Chem. 2022, 240, 114585. [Google Scholar] [CrossRef]
- Zhang, J.-W.; Xiong, Y.; Wang, F.; Zhang, F.-M.; Yang, X.; Lin, G.-Q.; Tian, P.; Ge, G.; Gao, D. Discovery of 9,10-dihydrophenanthrene derivatives as SARS-CoV-2 3CLpro inhibitors for treating COVID-19. Eur. J. Med. Chem. 2022, 228, 114030. [Google Scholar] [CrossRef]
- Kneller, D.W.; Li, H.; Galanie, S.; Phillips, G.; Labbé, A.; Weiss, K.L.; Zhang, Q.; Arnould, M.A.; Clyde, A.; Ma, H.; et al. Structural, Electronic, and Electrostatic Determinants for Inhibitor Binding to Subsites S1 and S2 in SARS-CoV-2 Main Protease. J. Med. Chem. 2021, 64, 17366–17383. [Google Scholar] [CrossRef]
- Gao, S.; Sylvester, K.; Song, L.; Claff, T.; Jing, L.; Woodson, M.; Weiße, R.H.; Cheng, Y.; Schäkel, L.; Petry, M.; et al. Discovery and Crystallographic Studies of Trisubstituted Piperazine Derivatives as Non-Covalent SARS-CoV-2 Main Protease Inhibitors with High Target Specificity and Low Toxicity. J. Med. Chem. 2022, 65, 13343–13364. [Google Scholar] [CrossRef]
- Luttens, A.; Gullberg, H.; Abdurakhmanov, E.; Vo, D.D.; Akaberi, D.; Talibov, V.O.; Nekhotiaeva, N.; Vangeel, L.; De Jonghe, S.; Jochmans, D.; et al. Ultralarge Virtual Screening Identifies SARS-CoV-2 Main Protease Inhibitors with Broad-Spectrum Activity against Coronaviruses. J. Am. Chem. Soc. 2022, 144, 2905–2920. [Google Scholar] [CrossRef]
- Zhu, W.; Xu, M.; Chen, C.Z.; Guo, H.; Shen, M.; Hu, X.; Shinn, P.; Klumpp-Thomas, C.; Michael, S.G.; Zheng, W. Identification of SARS-CoV-2 3CL protease inhibitors by a quantitative high-throughput screening. ACS Pharmacol. Transl. Sci. 2020, 3, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Rothan, H.A.; Teoh, T.C. Cell-Based High-Throughput Screening Protocol for Discovering Antiviral Inhibitors Against SARS-CoV-2 Main Protease (3CLpro). Mol. Biotechnol. 2021, 63, 240–248. [Google Scholar] [CrossRef]
- Drayman, N.; DeMarco, J.K.; Jones, K.A.; Azizi, S.-A.; Froggatt, H.M.; Tan, K.; Maltseva, N.I.; Chen, S.; Nicolaescu, V.; Dvorkin, S. Masitinib is a broad coronavirus 3CL inhibitor that blocks replication of SARS-CoV-2. Science 2021, 373, 931–936. [Google Scholar] [CrossRef]
- Xu, T.; Xu, M.; Zhu, W.; Chen, C.Z.; Zhang, Q.; Zheng, W.; Huang, R. Efficient Identification of Anti-SARS-CoV-2 Compounds Using Chemical Structure- and Biological Activity-Based Modeling. J. Med. Chem. 2022, 65, 4590–4599. [Google Scholar] [CrossRef]
- Gentile, F.; Fernandez, M.; Ban, F.; Ton, A.-T.; Mslati, H.; Perez, C.F.; Leblanc, E.; Yaacoub, J.C.; Gleave, J.; Stern, A. Automated discovery of noncovalent inhibitors of SARS-CoV-2 main protease by consensus Deep Docking of 40 billion small molecules. Chem. Sci. 2021, 12, 15960–15974. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Flury, P.; Krüger, N.; Su, H.; Schäkel, L.; Barbosa Da Silva, E.; Eppler, O.; Kronenberger, T.; Nie, T.; Luedtke, S.; et al. Small-Molecule Thioesters as SARS-CoV-2 Main Protease Inhibitors: Enzyme Inhibition, Structure–Activity Relationships, Antiviral Activity, and X-ray Structure Determination. J. Med. Chem. 2022, 65, 9376–9395. [Google Scholar] [CrossRef]
- Mahdi, M.; Mótyán, J.A.; Szojka, Z.I.; Golda, M.; Miczi, M.; Tőzsér, J. Analysis of the efficacy of HIV protease inhibitors against SARS-CoV-2′s main protease. Virol. J. 2020, 17, 190. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhu, Q.; Zhou, X.; Li, W.; Yin, X.; Li, J. Structural Basis for the Inhibition of SARS-CoV-2 Mpro D48N Mutant by Shikonin and PF-07321332. Viruses 2024, 16, 65. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Mangiavacchi, F.; Botwina, P.; Menichetti, E.; Bagnoli, L.; Rosati, O.; Marini, F.; Fonseca, S.F.; Abenante, L.; Alves, D.; Dabrowska, A.; et al. Seleno-Functionalization of Quercetin Improves the Non-Covalent Inhibition of Mpro and Its Antiviral Activity in Cells against SARS-CoV-2. Int. J. Mol. Sci. 2021, 22, 7048. [Google Scholar]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Cui, M.; Zheng, C.; Zhang, P.; Ren, S.; Bao, J.; Gao, D.; Sun, R.; Wang, M.; Lin, J.; et al. Both Baicalein and Gallocatechin Gallate Effectively Inhibit SARS-CoV-2 Replication by Targeting Mpro and Sepsis in Mice. Inflammation 2022, 45, 1076–1088. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Yao, S.; Zhao, W.; Li, M.; Liu, J.; Shang, W.; Xie, H.; Ke, C.; Gao, M.; Yu, K. Discovery of baicalin and baicalein as novel, natural product inhibitors of SARS-CoV-2 3CL protease in vitro. bioRxiv 2020. [Google Scholar] [CrossRef]
- Duong, C.Q.; Nguyen, P.T.V. Exploration of SARS-CoV-2 Mpro Noncovalent Natural Inhibitors Using Structure-Based Approaches. ACS Omega 2023, 8, 6679–6688. [Google Scholar] [CrossRef]
- Verma, S.; Patel, C.N.; Chandra, M. Identification of novel inhibitors of SARS-CoV-2 main protease (Mpro) from Withania sp. by molecular docking and molecular dynamics simulation. J. Comput. Chem. 2021, 42, 1861–1872. [Google Scholar] [CrossRef]
- Yang, W.; Wang, Y.; Han, D.; Tang, W.; Sun, L. Recent advances in application of computer-aided drug design in anti-COVID-19 Virials Drug Discovery. Biomed. Pharmacother. 2024, 173, 116423. [Google Scholar] [CrossRef]
- Gaudêncio, S.P.; Pereira, F. A Computer-Aided Drug Design Approach to Predict Marine Drug-Like Leads for SARS-CoV-2 Main Protease Inhibition. Mar. Drugs 2020, 18, 633. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wan, J.; Wang, G. A survey on computational methods in discovering protein inhibitors of SARS-CoV-2. Brief. Bioinform. 2021, 23, bbab416. [Google Scholar] [CrossRef]
- Ngo, S.T.; Nguyen, T.H.; Tung, N.T.; Vu, V.V.; Pham, M.Q.; Mai, B.K. Characterizing the ligand-binding affinity toward SARS-CoV-2 Mpro via physics-and knowledge-based approaches. Phys. Chem. Chem. Phys. 2022, 24, 29266–29278. [Google Scholar] [CrossRef]
- Sayed, A.M.; Alhadrami, H.A.; El-Gendy, A.O.; Shamikh, Y.I.; Belbahri, L.; Hassan, H.M.; Abdelmohsen, U.R.; Rateb, M.E. Microbial natural products as potential inhibitors of SARS-CoV-2 main protease (Mpro). Microorganisms 2020, 8, 970. [Google Scholar] [CrossRef]
- Amin, S.A.; Banerjee, S.; Singh, S.; Qureshi, I.A.; Gayen, S.; Jha, T. First structure–activity relationship analysis of SARS-CoV-2 virus main protease (Mpro) inhibitors: An endeavor on COVID-19 drug discovery. Mol. Divers. 2021, 25, 1827–1838. [Google Scholar] [CrossRef] [PubMed]
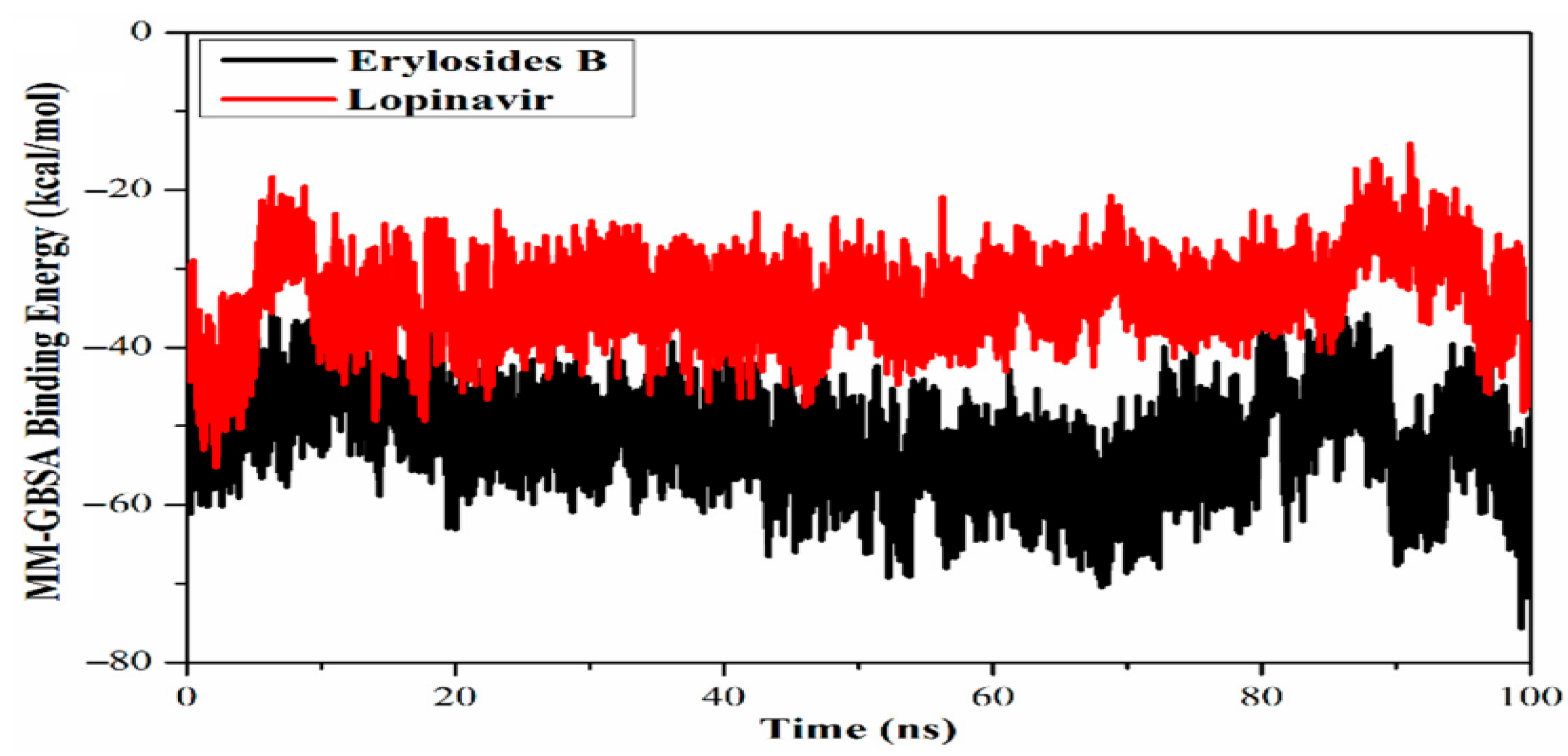
- Ibrahim, M.A.A.; Abdelrahman, A.H.M.; Mohamed, T.A.; Atia, M.A.M.; Al-Hammady, M.A.M.; Abdeljawaad, K.A.A.; Elkady, E.M.; Moustafa, M.F.; Alrumaihi, F.; Allemailem, K.S.; et al. In Silico Mining of Terpenes from Red-Sea Invertebrates for SARS-CoV-2 Main Protease (Mpro) Inhibitors. Molecules 2021, 26, 2082. [Google Scholar] [CrossRef] [PubMed]
- Hicks, E.G.; Kandel, S.E.; Lampe, J.N. Identification of Aloe-derived natural products as prospective lead scaffolds for SARS-CoV-2 main protease (Mpro) inhibitors. Bioorgan. Med. Chem. Lett. 2022, 66, 128732. [Google Scholar] [CrossRef]
- Adem, Ş.; Eyupoglu, V.; Ibrahim, I.M.; Sarfraz, I.; Rasul, A.; Ali, M.; Elfiky, A.A. Multidimensional in silico strategy for identification of natural polyphenols-based SARS-CoV-2 main protease (Mpro) inhibitors to unveil a hope against COVID-19. Comput. Biol. Med. 2022, 145, 105452. [Google Scholar] [CrossRef]
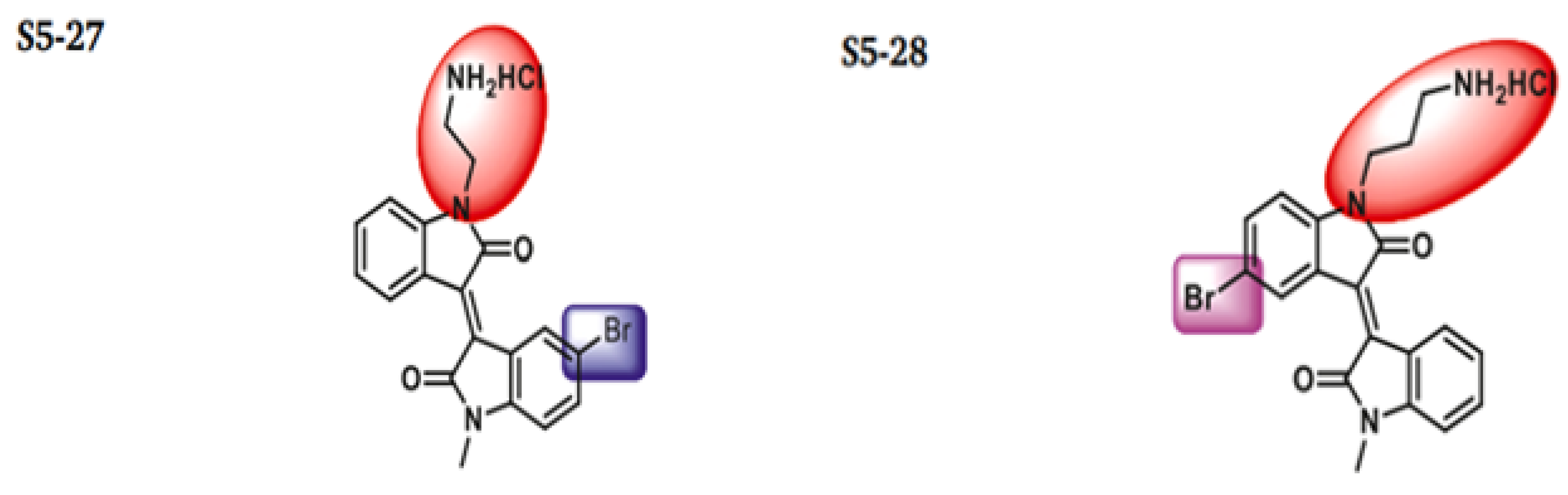
- Altincekic, N.; Jores, N.; Löhr, F.; Richter, C.; Ehrhardt, C.; Blommers, M.J.J.; Berg, H.; Öztürk, S.; Gande, S.L.; Linhard, V.; et al. Targeting the Main Protease (Mpro, nsp5) by Growth of Fragment Scaffolds Exploiting Structure-Based Methodologies. ACS Chem. Biol. 2024, 19, 563–574. [Google Scholar] [CrossRef]
- Janin, Y.L. On the origins of SARS-CoV-2 main protease inhibitors. RSC Med. Chem. 2024, 15, 81–118. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chen, M.; Yuan, F.; Zhang, J.; Song, J.; Yang, L. Exploring the covalent inhibition mechanisms of inhibitors with two different warheads acting on SARS-CoV-2 Mpro by QM/MM simulations. Comput. Theor. Chem. 2024, 1242, 114979. [Google Scholar] [CrossRef]
- Gao, Q.; Liu, S.; Zhou, Y.; Fan, J.; Ke, S.; Zhou, Y.; Fan, K.; Wang, Y.; Zhou, Y.; Xia, Z.; et al. Discovery of meisoindigo derivatives as noncovalent and orally available Mpro inhibitors: Their therapeutic implications in the treatment of COVID-19. Eur. J. Med. Chem. 2024, 273, 116498. [Google Scholar] [CrossRef]
- Chen, R.; Gao, Y.; Liu, H.; Li, H.; Chen, W.; Ma, J. Advances in research on 3C-like protease (3CL pro) inhibitors against SARS-CoV-2 since 2020. RSC Med. Chem. 2023, 14, 9–21. [Google Scholar] [CrossRef]
- Kuzikov, M.; Costanzi, E.; Reinshagen, J.; Esposito, F.; Vangeel, L.; Wolf, M.; Ellinger, B.; Claussen, C.; Geisslinger, G.; Corona, A. Identification of inhibitors of SARS-CoV-2 3CL-pro enzymatic activity using a small molecule in vitro repurposing screen. ACS Pharmacol. Transl. Sci. 2021, 4, 1096–1110. [Google Scholar] [CrossRef]
- Liu, X.; Li, Z.; Liu, S.; Sun, J.; Chen, Z.; Jiang, M.; Zhang, Q.; Wei, Y.; Wang, X.; Huang, Y.-Y. Potential therapeutic effects of dipyridamole in the severely ill patients with COVID-19. Acta Pharm. Sin. B 2020, 10, 1205–1215. [Google Scholar] [CrossRef] [PubMed]
- Hung, H.-C.; Ke, Y.-Y.; Huang, S.Y.; Huang, P.-N.; Kung, Y.-A.; Chang, T.-Y.; Yen, K.-J.; Peng, T.-T.; Chang, S.-E.; Huang, C.-T. Discovery of M protease inhibitors encoded by SARS-CoV-2. Antimicrob. Agents Chemother. 2020, 64, e00872-20. [Google Scholar] [CrossRef]
- Cáceres, C.J.; Cardenas-Garcia, S.; Carnaccini, S.; Seibert, B.; Rajao, D.S.; Wang, J.; Perez, D.R. Efficacy of GC-376 against SARS-CoV-2 virus infection in the K18 hACE2 transgenic mouse model. Sci. Rep. 2021, 11, 9609. [Google Scholar] [CrossRef] [PubMed]
- Motiwale, M.; Yadav, N.S.; Kumar, S.; Kushwaha, T.; Choudhir, G.; Sharma, S.; Singour, P.K. Finding potent inhibitors for COVID-19 main protease (Mpro): An in silico approach using SARS-CoV-3CL protease inhibitors for combating CORONA. J. Biomol. Struct. Dyn. 2022, 40, 1534–1545. [Google Scholar] [CrossRef]
- Salo-Ahen, O.M.H.; Alanko, I.; Bhadane, R.; Bonvin, A.M.J.J.; Honorato, R.V.; Hossain, S.; Juffer, A.H.; Kabedev, A.; Lahtela-Kakkonen, M.; Larsen, A.S.; et al. Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development. Processes 2021, 9, 71. [Google Scholar] [CrossRef]
- Sarfraz, M.; Rauf, A.; Keller, P.; Qureshi, A.M. N,N′-dialkyl-2-thiobarbituric acid based sulfonamides as potential SARS-CoV-2 main protease inhibitors. Can. J. Chem. 2021, 99, 330–345. [Google Scholar] [CrossRef]
- Yang, J.; Lin, X.; Xing, N.; Zhang, Z.; Zhang, H.; Wu, H.; Xue, W. Structure-Based Discovery of Novel Nonpeptide Inhibitors Targeting SARS-CoV-2 Mpro. J. Chem. Inf. Model. 2021, 61, 3917–3926. [Google Scholar] [CrossRef]
- Mohaptra, R.K.; Dhama, K.; El-Arabey, A.A.; Sarangi, A.K.; Tiwari, R.; Bin Emran, T.; Azam, M.; Raval, M.K.; Seidel, V.; Abdalla, M. Repurposing benzimidazole and benzothiazole derivatives as potential inhibitors of SARS-CoV-2: DFT, QSAR, molecular docking, molecular dynamics simulation, and in-silico pharmacokinetic and toxicity studies. J. King Saud Univ.-Sci. 2021, 33, 101637. [Google Scholar] [CrossRef] [PubMed]
- Omage, F.B.; Madabeni, A.; Tucci, A.R.; Nogara, P.A.; Bortoli, M.; Rosa, A.d.S.; Neuza dos Santos Ferreira, V.; Teixeira Rocha, J.B.; Miranda, M.D.; Orian, L. Diphenyl Diselenide and SARS-CoV-2: In silico Exploration of the Mechanisms of Inhibition of Main Protease (Mpro) and Papain-like Protease (PLpro). J. Chem. Inf. Model. 2023, 63, 2226–2239. [Google Scholar] [CrossRef]
- Citarella, A.; Scala, A.; Piperno, A.; Micale, N. SARS-CoV-2 Mpro: A potential target for peptidomimetics and small-molecule inhibitors. Biomolecules 2021, 11, 607. [Google Scholar] [CrossRef]
- Wang, F.; Vasilyev, V. In Silico Tuning of Binding Selectivity for New SARS-CoV-2 Main Protease Inhibitors. 2024. Available online: https://papers.ssrn.com/sol3/papers.cfm?abstract_id=4981058 (accessed on 13 May 2025).
- Elgohary, A.M.; Elfiky, A.A.; Pereira, F.; Abd El-Aziz, T.M.; Sobeh, M.; Arafa, R.K.; El-Demerdash, A. Investigating the structure-activity relationship of marine polycyclic batzelladine alkaloids as promising inhibitors for SARS-CoV-2 main protease (Mpro). Comput. Biol. Med. 2022, 147, 105738. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Vasilyev, V. Accelerating optical reporting for conformation of tyrosine kinase inhibitors in solutions. Int. J. Quantum Chem. 2021, 121, e26765. [Google Scholar] [CrossRef]
- Ruud, K.; Helgaker, T.; Bak, K.L.; Jørgensen, P.; Olsen, J. Accurate magnetizabilities of the isoelectronic series BeH−, BH, and CH+. The MCSCF-GIAO approach. Chem. Phys. 1995, 195, 157–169. [Google Scholar] [CrossRef]
- Habgood, M.; James, T.; Heifetz, A. Conformational Searching with Quantum Mechanics. In Quantum Mechanics in Drug Discovery; Heifetz, A., Ed.; Springer: New York, NY, USA, 2020; pp. 207–229. [Google Scholar]
- Alagawani, S.; Vasilyev, V.; Wang, F. Optical spectra of EGFR inhibitor AG-1478 for benchmarking DFT functionals. Electron. Struct. 2023, 5, 024011. [Google Scholar] [CrossRef]



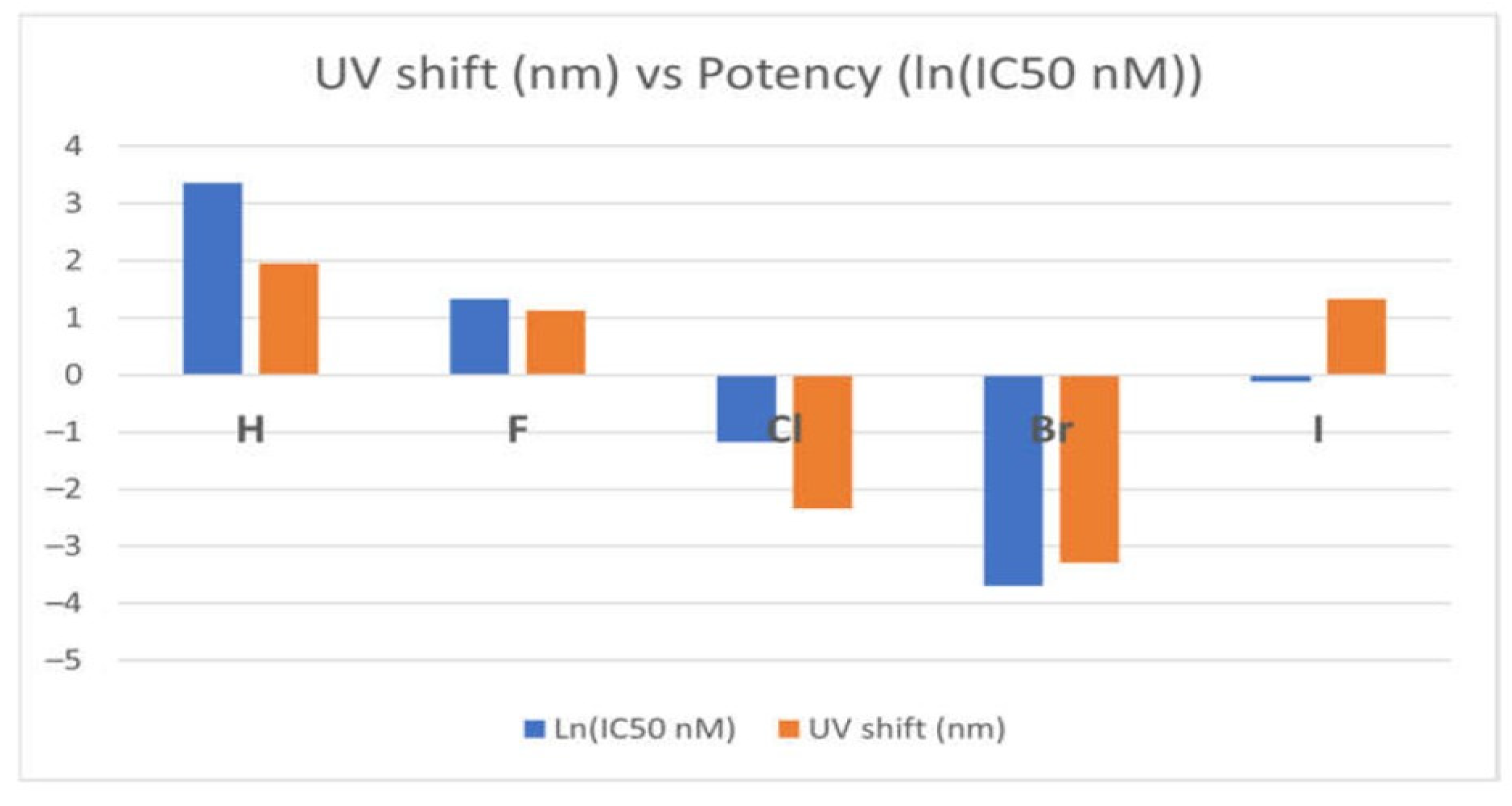

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Structure | Strategies for the Discovery of Non-Covalent Mpro Inhibitors | Mpro IC50 (μm) | Ref. |
|---|---|---|---|---|
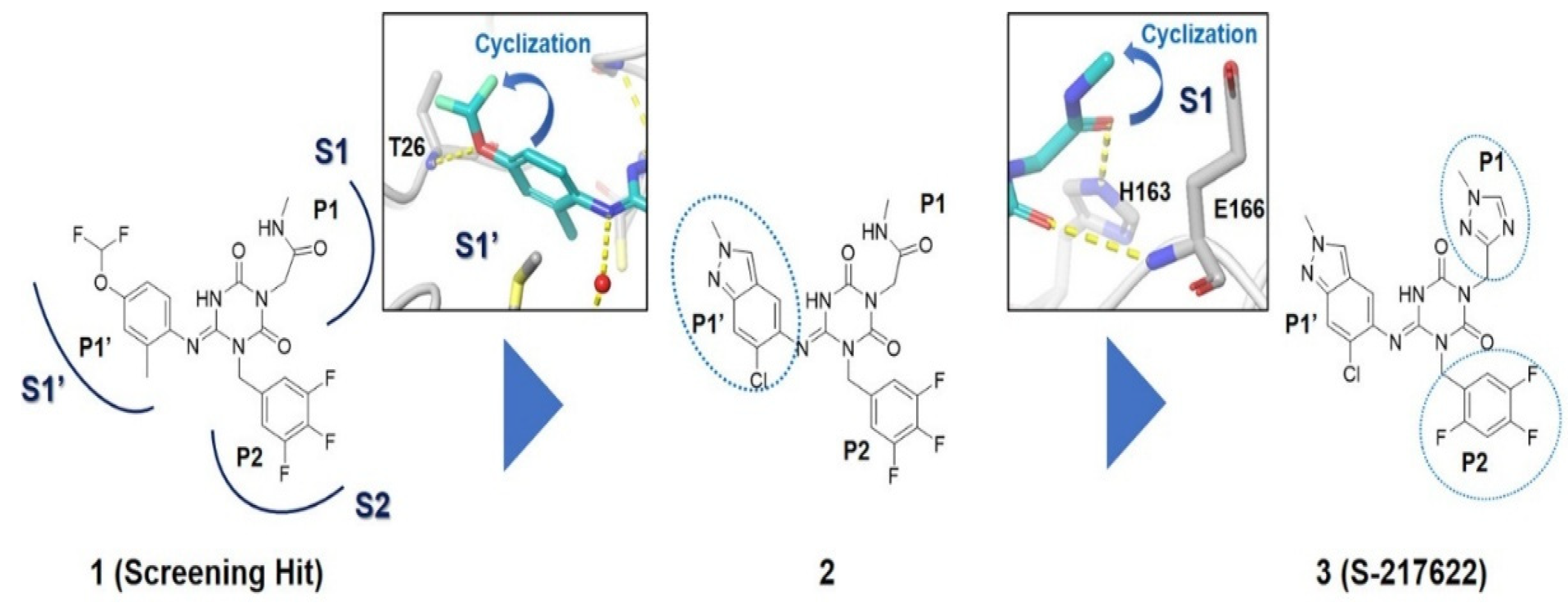
| S-217622 | 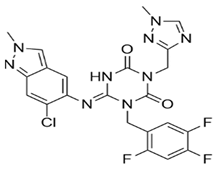 | Docking-based virtual screening followed by biological screening using an in-house compound library. | 0.013 | [17] |
| Mprosevir (WU-04) |  | DNA-encoded library screening. | 0.072 | [44] |
| ML188 | 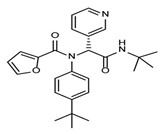 | Structure-based drug design generated through the Ugi reaction. | 2.5 | [32] |
| 23R |  | Coupling structure-based drug design with the one-pot Ugi four-component reaction. | 0.20 | [46] |
| CCF981 | 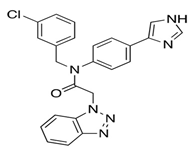 | Structure-based optimisations. | 0.068 | [16] |
| N-substituted isatin derivative compound 26 | 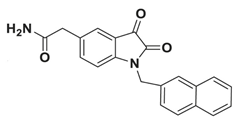 | In-house synthetic compound library screening. | 0.045 | [43] |
| Benzoxaborole-based inhibitor compound 18 | 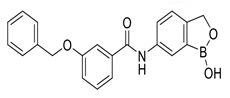 | Biochemical assay screenings. | 6.1 | [47] |
| 9,10-dihydrophenanthrene derivatives C1 C2 | 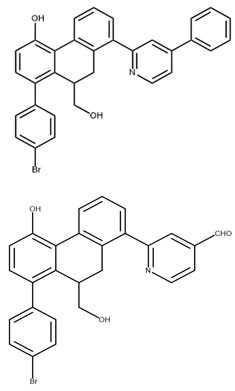 | Fluorescence resonance energy transfer (FRET) biochemical assay. | 1.55 1.81 | [48] |
| HL-3-68 |  | Large-scale virtual screening and structural optimisation. | 0.29 | [49] |
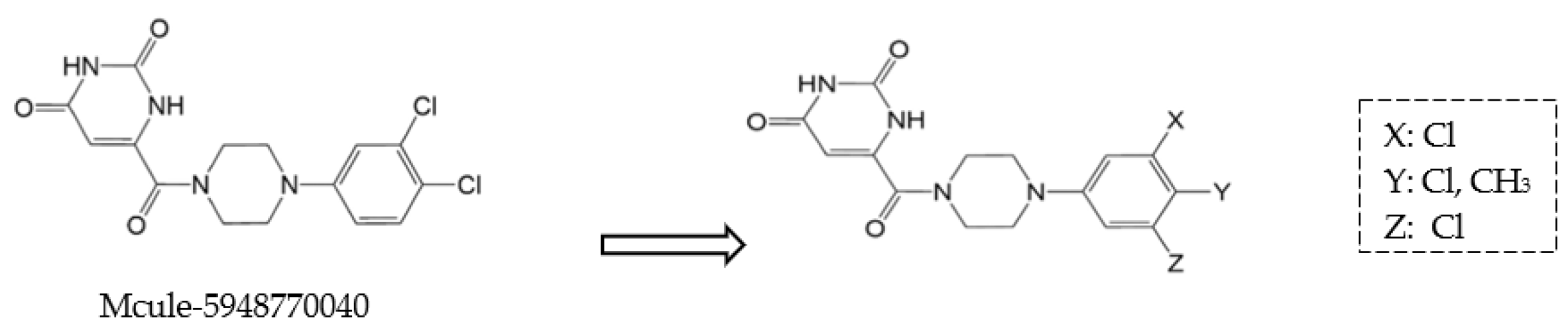
| Mcule-CSR-494190-S1 |  | Large-scale virtual screening with structural optimisation. | 0.29 | [41] |
| GC-14 | 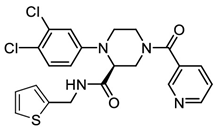 | Structure-based rational design combined with multi-site binding and privileged structure assembly strategies. | 0.40 | [50] |
| Imidazolidine-2,4-dione-based inhibitor compound 19 | 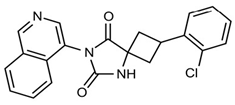 | Virtual screening combined with structural optimisation. | 0.077 | [51] |
| Walrycin B | 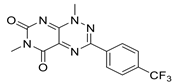 | High-throughput screening (HTS) using FRET assays. | 0.26 | [52] |
| Quinazoline derivative QZ4 | 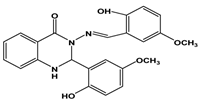 | Small in-house library screening using a GFP cell-based assay. | 6.5 | [53] |
| Masitinib |  | Cell-based HTS. | 2.5 | [54] |
| Z-DQMD-FMK | 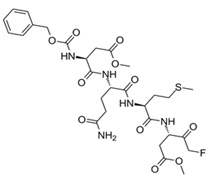 | Machine learning-driven virtual screening. | 0.92 | [55] |
| Z4927220858 | 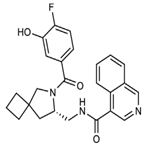 | Deep docking methods. | ~10 | [56] |
| Inhibitor | Structure | IC50 (µm) |
|---|---|---|
| Lopinavir+Ritonavir | 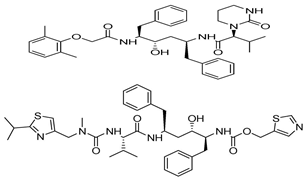 | 10.9 |
| Ritonavir | 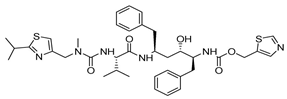 | 13.7 |
| Darunavir | 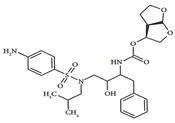 | 36.1 |
| Atazanavir | 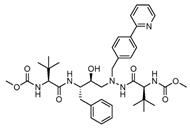 | 60.7 |
| Lopinavir | 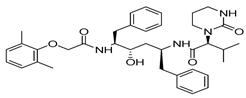 | Indeterminable |
| Inhibitor | Structure | Strategies for the Discovery of Non-Covalent Mpro Inhibitors | Mpro IC50 (μm) |
|---|---|---|---|
| Shikonin | 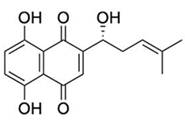 | High-throughput screening. | 0.397 |
| Quercetin | 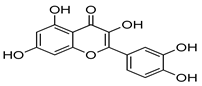 | In vitro screening. | 7.4 192 |
| Selenoquercetin analogues compound 2d | 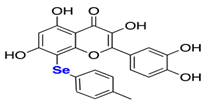 | Screening a small library of flavonols and flavone derivatives. | 8 |
| Baicalein | 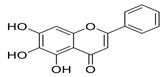 | Enzymatic assays and a phenotypic assay using Vero E6 cells. | 0.39 |
| Calycosin-7-O-β-glucopyranoside | 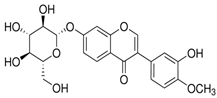 | The structure-based 3D pharmacophore model was examined by induced-fit molecular docking. | 0.549 |
| Compound | MolDock Score | HBond Score |
|---|---|---|
| N3 (Reference) | −162.17 | −8.19 |
| Nelfinavir | −147.38 | −6.87 |
| Hesperidin | −178.59 | −20.26 |
| Rutin | −176.27 | −21.24 |
| Diosmin | −174.13 | −27.26 |
| Apigenin | −171.01 | −10.19 |
| Complexes | ΔEele a | ΔEvdW b | ΔGpol c | ΔGnonpol d | ΔGcalc e |
|---|---|---|---|---|---|
Z1244904919-Mpro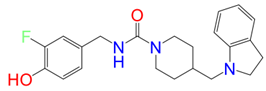 | –18.76 | –46.88 | –65.63 | –3.81 | –45.72 |
Z1759961356-Mpro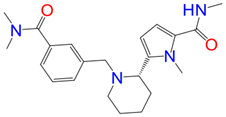 | –6.03 | –52.51 | –58.54 | –4.25 | –48.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alagawani, I.; Wang, F. In Silico Development of SARS-CoV-2 Non-Covalent Mpro Inhibitors: A Review. Appl. Sci. 2025, 15, 6544. https://doi.org/10.3390/app15126544
Alagawani I, Wang F. In Silico Development of SARS-CoV-2 Non-Covalent Mpro Inhibitors: A Review. Applied Sciences. 2025; 15(12):6544. https://doi.org/10.3390/app15126544
Chicago/Turabian StyleAlagawani, Islam, and Feng Wang. 2025. "In Silico Development of SARS-CoV-2 Non-Covalent Mpro Inhibitors: A Review" Applied Sciences 15, no. 12: 6544. https://doi.org/10.3390/app15126544
APA StyleAlagawani, I., & Wang, F. (2025). In Silico Development of SARS-CoV-2 Non-Covalent Mpro Inhibitors: A Review. Applied Sciences, 15(12), 6544. https://doi.org/10.3390/app15126544