1. Introduction
Organ-on-chip platforms have rapidly gained recognition as highly predictive and physiologically relevant tools for modeling human tissues and diseases in vitro, offering enhanced translational potential compared to conventional cell culture methods. To fully characterize these microphysiological systems, it is critical to assess both nuclear and mitochondrial genetic integrity, as well as epigenetic modifications that can influence cellular responses to drugs or environmental stimuli. While profiling absorption, distribution, metabolism, and excretion (ADME) genes remains central to understanding pharmacokinetics and toxicity in organ-on-chip setups, investigating mitochondrial genome perturbations—including sequence variations and methylation patterns—provides a more comprehensive picture of cellular metabolism and stress responses.
The integrity of the mitochondrial genome (mtDNA) is crucial for cellular energy production, metabolism [
1,
2,
3,
4], and overall cell health [
5,
6,
7,
8,
9,
10]. Unlike the nuclear genome, mtDNA exists in multiple copies per cell and is particularly susceptible to accumulating sequence variations and molecular damage [
1,
2,
3,
4,
11,
12,
13,
14,
15] leading to heteroplasmy (the coexistence of different mtDNA types within a cell) and epigenetic modifications such as DNA methylation [
16,
17,
18]. Accurately characterizing these mtDNA features is vital for understanding cellular function, aging, and disease pathogenesis, but it poses significant technical challenges [
15].
The human mitochondrial genome is circular, consisting of 37 genes, has no introns, and is represented by naked double-stranded DNA, more susceptible to molecular modification and damage [
13,
14,
15]. MtDNA defects were mainly studied using Sanger sequencing, Southern blot, and long and quantitative PCR. However, these technologies are expensive and limited in speed, throughput, and sensitivity.
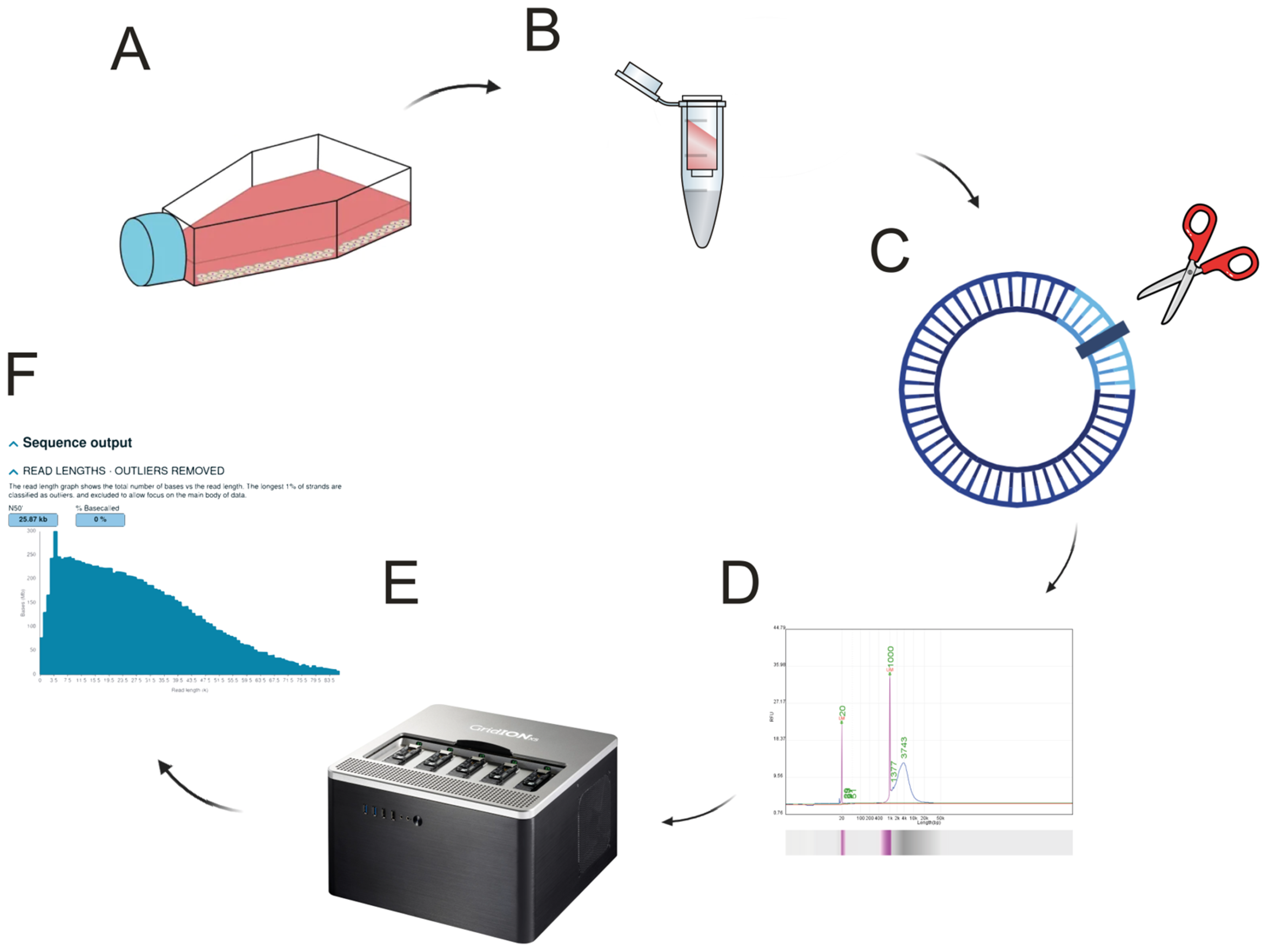
Recently, next-generation sequencing (NGS) has been more and more explored to study mtDNA defects. In this study, we employed third-generation sequencing technology from Oxford Nanopore, which utilizes flow cells containing arrays of nanopores embedded in an electro-resistant membrane. Each nanopore is coupled to an electrode connected to a channel and sensor chip that continuously monitors the ionic current passing through the pore. As a DNA or RNA molecule translocates through the nanopore, characteristic disruptions in the current produce a so-called “squiggle” signal. This signal is then deciphered by real-time base-calling algorithms to reconstruct the nucleotide sequence. Unlike short-read sequencing methods, which constrain analyses to limited read lengths, nanopore sequencing accommodates continuous, long-read data. Such extended read lengths enable complete resolution of repetitive regions, identification of structural variants, and discrimination between different isoforms, thereby offering a more comprehensive genomic characterization. Long-read sequencing is based on a simplified protocol that requires nanopore adaptor ligation of the sequenced DNA strand. Therefore, opening the circular mitochondrial DNA is crucial for this protocol to work. DNA shearing is one way to produce many linearized strands of DNA, but unless ultrasound or enzyme approaches are applied, it results in non-uniform length of the future sequencing reads.
To avoid amplification-related biases and to enable the simultaneous detection of base modifications (e.g., methylation) alongside the nucleotide sequence, we employed another ONT technology referred to as Adaptive Sampling. This software-controlled method is unique to nanopore sequencing and selectively enriches the desired region without PCR amplification. By providing a FASTA file specifying the target region, the adaptive sampling algorithm examines the first 400–500 bp of each DNA fragment as it enters the nanopore. If the partial sequence aligns to the region of interest, the read is permitted to continue translocating for full sequencing; otherwise, it is rapidly rejected, freeing the nanopore for a new molecule. This approach preserves the native genomic DNA structure while specifically enhancing coverage of the target region.
The protocol was initially optimized using the human choriocarcinoma cell line JAR, with evaluations of two commercial restriction endonucleases, PvuII-HF and BamHI-HF. Subsequently, the optimized protocol was applied to human adipose-derived mesenchymal stem cells (HAdMSCs), which were selected for their regenerative potential, versatility, abundance, and capacity to differentiate into multiple lineages—properties that make them valuable for therapeutic applications in regenerative medicine and pharmacogenomic studies. It is noted that this cell population is also frequently referred to in the literature as adipose stem cells (ASC) or adipose-derived stem cells (ADSC). This study demonstrates a straightforward, reproducible, and reliable protocol for the targeted sequencing of mitochondrial DNA without amplification requirements, while simultaneously sequencing nuclear genes of interest.
Here, we test the hypothesis that combining restriction-enzyme linearization of the circular mitochondrial genome with real-time Oxford Nanopore adaptive sampling (AS) yields a single-tube, PCR-free workflow that can simultaneously profile mtDNA heteroplasmy and, potentially, DNA methylation in AdMSC at single-molecule resolution. Specifically, we set out to quantify the on-target enrichment and coverage uniformity that AS affords relative to conventional whole-genome sequencing, benchmark the sensitivity and accuracy of heteroplasmy against orthogonal controls, and provide an analysis pipeline strategy that phases genetic information to facilitate mitochondrial studies in regenerative-medicine models. By validating this streamlined protocol in primary human AdMSC, we aim to equip the stem-cell community with a cost-effective tool for interrogating mitochondrial dysfunction in metabolic disease and ageing.
2. Materials and Methods
2.1. Cell Line
Human choriocarcinoma cell line JAR was obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in RPMI-1640 medium with HEPES modification supplemented with L-glutamine (Sigma-Aldrich, R5886, Milwaukee, WI, USA), 10% fetal bovine serum (FBS), and 1% penicillin/streptomycin solution. Cells were maintained at 37 °C in a humidified incubator with 5% CO2 atmosphere. For DNA and RNA analyses, cell pellets containing approximately 1–2 × 106 cells from a single T75 flask were harvested and stored at −80 °C.
Human adipose-derived mesenchymal stem cells (HAdMSC) cell line was purchased from Innoprot (Derio, Spain) and cultured in mesenchymal stem cell medium containing 5% fetal bovine serum (FBS), 1% mesenchymal stem cell growth supplements and 1% penicillin/streptomycin solution, all provided by Innoprot. Cells were cultured in Corning™ CellBIND™ surface cell culture flasks—T75—and maintained at 37 °C in a humidified incubator with 5% CO
2. When they reached 80–90% confluency, they were cryopreserved in complete medium +15% FBS and 15% DMSO following the slow-freezing protocol. For DNA and RNA analysis, a dry pellet from one T75 flask, approximately 5 × 10
5 cells, was directly frozen at −80 °C (
Figure 1).
2.2. DNA Isolation
Total DNA was isolated using ZymoResearch Quick-DNA Miniprep Kit (Irvine, CA, USA) according to the manufacturer’s instructions. Briefly, one tube with a dry pellet Jar passage 31 and HAdMSC passage 5 was resuspended in 100 µL Elution Buffer (EB) for a total of 130 µL in 1.5 mL Eppendorf LoBind tube. Then, 130 µL BioFluid & Cell Buffer and 13 µL Proteinase K were added. The mixture was vortexed for 10–15 s and was incubated at 55 °C for 10 min. After the incubation, 280 µL Genomic Binding Buffer was added to the digested sample, which was vortexed again for 10–15 s. The mixture was transferred to a Zymo-Spin™ IIC-XLR Column in a collection tube and centrifuged at ≥12,000× g for 1 min. The collection tube with the flow through was discarded. The Zymo-Spin™ IIC-XLR Column was placed onto a new collection tube and 400 µL of DNA Pre-Wash Buffer was added. They were centrifuged at ≥12,000× g for one minute again, after which 700 µL of g-DNA Wash Buffer was added and they were centrifuged again under the same conditions. Finally, the spin column was transferred to a clean microcentrifuge tube and 50 µL Elution Buffer was added directly to the matrix. The sample was incubated for 5 min at room temperature, then centrifuged at ≥14,000× g for 1 min. The eluted DNA was stored at −80 °C for future use.
2.3. DNA Quantification and Quality Control
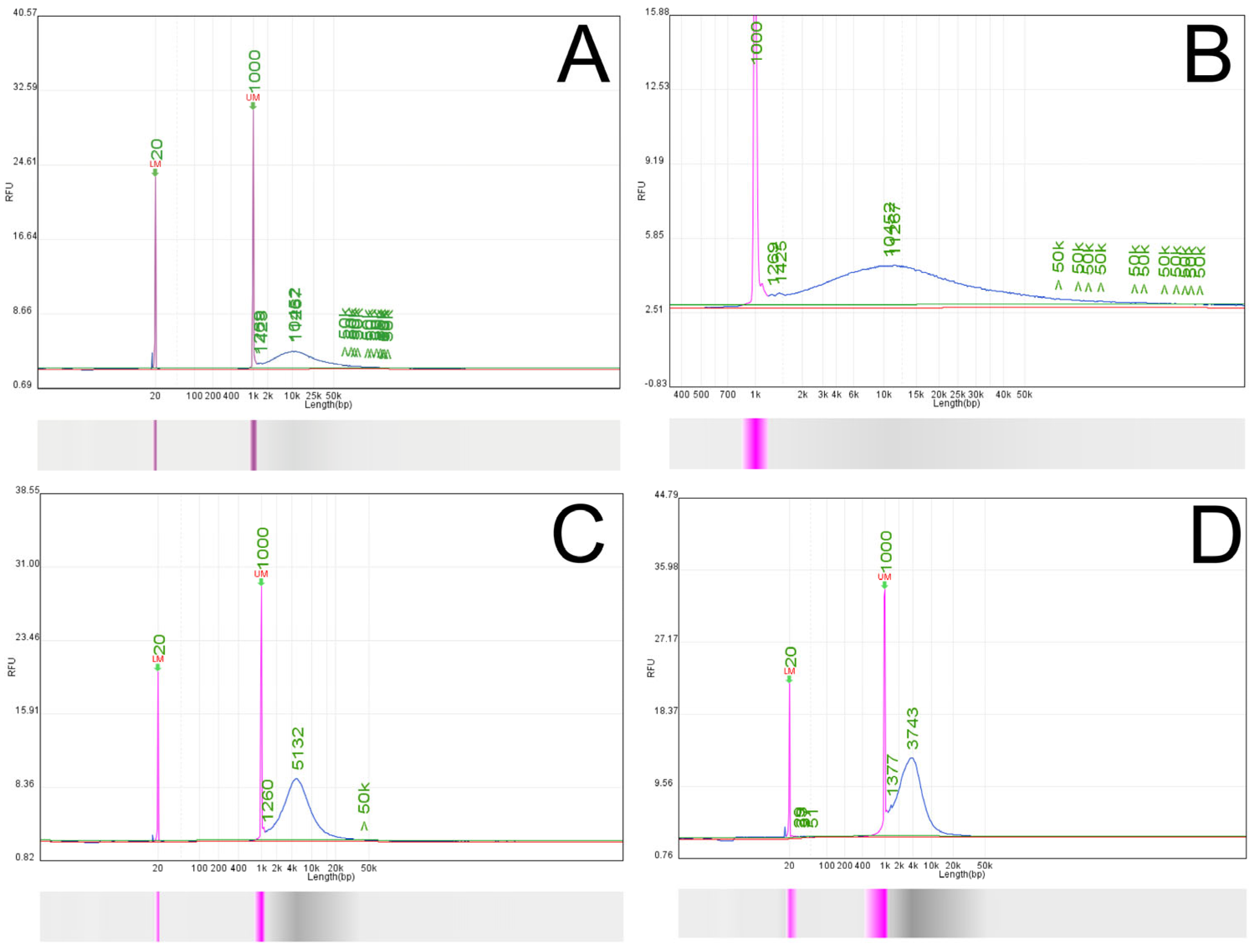
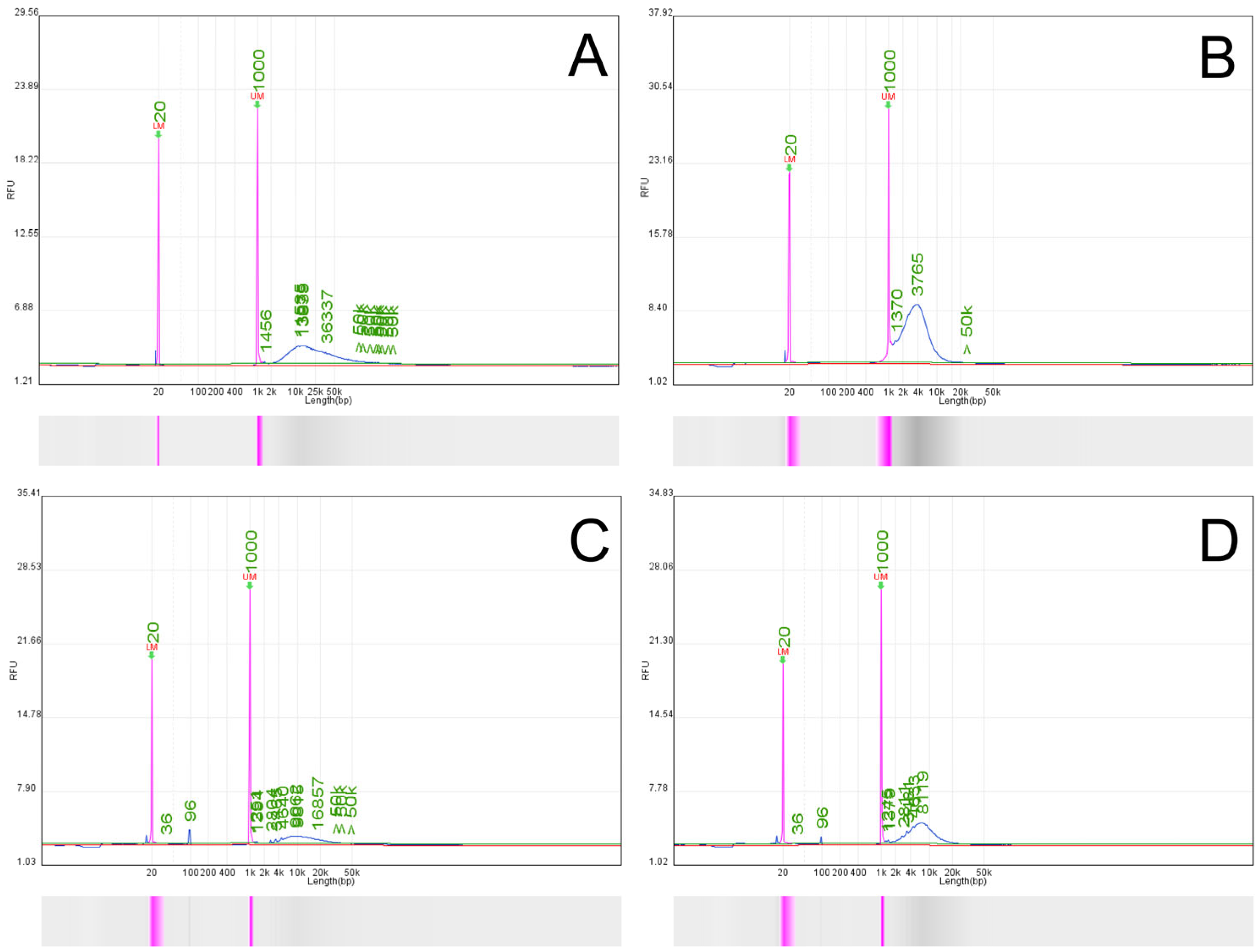
The quantity and quality of the extracted sample were assessed on a Qubit Fluorometer and Qsep-100 Bio-Fragment Analyzer using Qubit dsDNA HS Assay Kit and High Sensitivity KiloBase N3 cartridge (Waltham, MA, USA). The Qubit Fluorometer was first calibrated with standards 1 and 2 after mixing 190 µL working solution with 10 µL of the standard; the solution was then briefly vortexed and left for 2 min at room temperature. The sample solution was prepared by mixing 1 µL DNA sample in 199 µL working solution, vortexed briefly, and left for 2 min at room temperature. For the bio-fragment analysis, a 2 ng/µL concentration of the gDNA has been chosen. For this purpose, 0.6 µL gDNA was mixed with 19.4 µL Dilution Buffer. On the buffer tray, a 20 µL 20–1000 bp Alignment marker (109-100 A) was placed on position MA1, and a 40× diluted 500 bp–23 kB Size marker (C109700) was positioned on MA3. The calibration and the fragment analysis were run on a 4 kV current.
2.4. Enzymatic Treatment
Nanopores can only capture linearized DNA, so any mtDNA molecule must be sufficiently degraded to lose its native circular conformation before sequencing [
19]. The restriction endonuclease BamHI-HF or PvuII-HF, purchased from New England Biolabs (Boston, MA, USA) (R3136S and R3151SS), performed the linearization step. The enzymes recognize 5′-G^GATCC-3′ and 5′-CAG^CTG-3′, in the presence of CutSmart Buffer, as previously described [
20]. Briefly, a starting amount of 1300 ng (23 µL) gDNA was transferred to a 1.5 mL Eppendorf LoBind tube. Then, the following reagents were added: 5 µL 10× rCutSmart Buffer, 20.9 µL molecular grade water, and 1.1 µL PvuII-HF. The reaction was incubated at 37 °C for 1 h. The efficiency of this enzymatic digestion step was assessed by comparing the fragment size distribution profile of the treated sample against an aliquot of the original untreated native gDNA using capillary electrophoresis (Qsep-100 Bio-Fragment Analyzer, BiOptic Inc., New Taipei City, Taiwan), as presented in
Section 3.1 (
Figure 2 and
Figure 3). The starting amount of gDNA is higher than the 1 µg gDNA requested for starting the ligation protocol of Oxford Nanopore Technologies (Oxford, UK) because there is a decrease in the final quantity of the DNA after the digestion and the cleaning step.
Exonuclease V was purchased from New England Biolabs (M0345S) to test the genomic DNA cleavage. The reaction was prepared as described in the protocol; 1 µg DNA was mixed with 5 µL NebBuffer (10×), 5 µL ATP (10 mM), 1 µL Exonuclease V (10 units), and filled with molecular grade water up to 50 µL. It was then incubated at 37 °C for 30 min and heat-inactivated at 70 °C for 30 min.
2.5. DNA Purification by AMPure Beads
After the digested DNA is quantified and assessed for quality, we recommend conducting a clean-up step using Agencourt AMPure XP beads [
21]. In a nutshell, 90 µL of the resuspended AMPure XP beads were added to the sample. The volume was determined from the following equation: (Volume of Agencourt AMPure XP per reaction) = 1.8 × (Reaction Volume). The reaction was incubated for 5 min on a Hula mixer. Then, 500 µL 70% ethanol was freshly prepared. The reaction was placed on EpiCypher 1.5 mL Tubes Magnetic Separation Rack for approximately 2 min until the solution became clear. With the tube still on the magnetic rack, the cleared solution was aspirated and discarded. Two washing steps were performed with 200 µL 70% ethanol while keeping the tube on the magnet. Finally, the sample was removed from the rack, eluted in 49 µL molecular grade water, and incubated for 10 min on a Hula mixer. Once the incubation finished, the tube was placed again onto the EpiCypher rack, and the cleared solution was transferred to a new 1.5 LoBind tube. The cleaned DNA was once again quantified and quality checked.
2.6. ONT Library Preparation and Sequencing on the GridION Instrument
Two distinct libraries were prepared for comparative analysis in this study. (i) WGS Baseline Library: Approximately 1 µg of untreated native genomic DNA (gDNA) served as the input for standard library preparation. This library represents the baseline genomic material sequenced via WGS. (ii) Adaptive Sampling (AS) Library: Approximately 1 µg of gDNA that had been previously treated with PvuII-HF (as described in
Section 2.4) served as the input for this library. This library was subjected to targeted enrichment via adaptive sampling during sequencing. Each DNA sample was prepared independently for Oxford Nanopore Technologies (ONT) (Oxford, UK) sequencing on R9.4.1 flow cells using the Ligation Sequencing Kit (SQK-LSK109, ONT). Library preparation commenced with DNA repair and end-preparation. In a 0.2 mL PCR tube, 48 µL containing 1 µg of input DNA was mixed with 1 µL DNA control sample (CS, provided with kit), 3.5 µL NEBNext FFPE DNA Repair Buffer (New England Biolabs [NEB]), 2 µL NEBNext FFPE DNA Repair Mix (NEB), 3.5 µL NEBNext Ultra II End-prep Reaction Buffer (NEB), and 3 µL NEBNext Ultra II End-prep Enzyme Mix (NEB). The reaction was incubated at 20 °C for 5 min, followed by 65 °C for 5 min. The reaction mixture was purified using AMPure XP beads (Beckman Coulter, Singapore). Subsequently, 1 µL of the purified, repaired, and end-prepped DNA was quantified using a Qubit fluorometer (Invitrogen, Waltham, MA, USA) to confirm DNA recovery before proceeding.
For adapter ligation, the remaining purified DNA (~60 µL) was transferred to a 1.5 mL tube and mixed with 25 µL Ligation Buffer (LNB, kit component), 10 µL NEBNext Quick T4 DNA Ligase (NEB), and 5 µL Adapter Mix (AMX, kit component). This ligation mixture was incubated at room temperature (approximately 20–25 °C) for 10 min. The adapter-ligated DNA was then purified using AMPure XP beads, utilizing the Short Fragment Buffer (SFB, kit component) during the wash steps as per the manufacturer’s protocol to ensure retention of all fragments for both libraries. Finally, the purified library DNA was eluted in 15 µL Elution Buffer (EB, kit component). The final adapter-ligated libraries were quantified using a Qubit fluorometer, and their size distribution and quality were assessed using a Qsep100 instrument (BiOptic Inc., Taiwan, China) before proceeding to flow cell loading. As there is no requirement in the Ligation Sequencing Kit SQK-LSK109 (ONT, Oxford, UK) for fragment analysis prior library loading, no such has been performed and an average fragment length of 4 kB was taken considering the initial fragment analysis after digestion with PvuII-HF. Library with an approximate concentration of 50 fmol was loaded onto the flow cell.
2.7. Priming and Loading the SpotON Flow Cell for GridION
Prior to loading each library, a corresponding R9.4.1 SpotON flow cell was primed. The flow cell priming mix was prepared by adding 30 µL of Flush Tether (FLT) directly into one tube of Flush Buffer (FB) (both kit components). A volume of 800 µL of the complete priming mix was loaded into the flow cell via the priming port, followed by a 5 min incubation period. During this incubation, the final loading mix for each library was prepared separately. A target amount of 40 fmol of the final adapter-ligated library (corresponding to approximately 250 ng, depending on average fragment size) was used. For each library, 12 µL of the DNA library was mixed with 37.5 µL Sequencing Buffer (SQB, kit component) and 25.5 µL Loading Beads (LB, kit component) in a new tube. Immediately after preparation, 75 µL of this final loading mix was carefully loaded onto the flow cell dropwise via the SpotON sample port.
2.8. Sequencing on GridION and Adaptive Sampling
Before initiating the sequencing run, a flow cell check was performed using the MinKNOW software (ONT, Oxford, UK) to assess the number of available nanopores; R9.4.1 flow cells with fewer than approximately 800 active pores were typically not used, following recommendations. After confirming the flow cell quality, priming, and loading, the sequencing run was started via the MinKNOW interface. Adaptive sampling (ReadUntil) was enabled by selecting the corresponding option in the software setup for the AS Library. A BED file containing the genomic coordinates of the reference mitochondrial genome sequence, and another 121 ADME genes was uploaded to direct the instrument to selectively sequence reads originating from this target region. For the WGS baseline library (prepared from untreated native DNA), sequencing proceeded using a standard WGS run protocol without the adaptive sampling feature enabled. The following software versions from Oxford Nanopore were used during the sequencing run: MinKNOW24.02.16, Bream7.9.8, Configuration5.9.18, Dorado 7.3.11, and MinKNOW Core5.9.12.
2.9. Bioinformatic Analysis
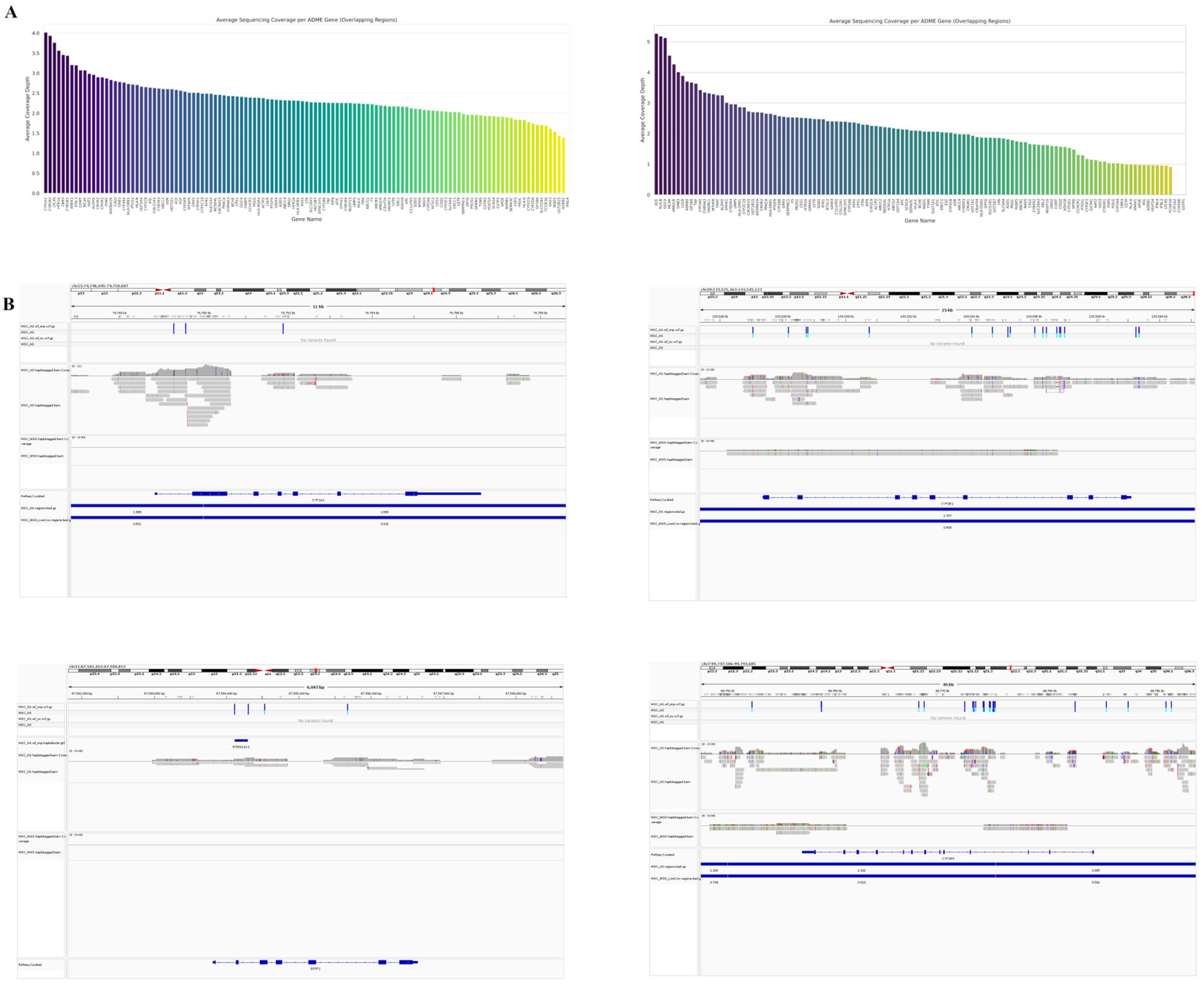
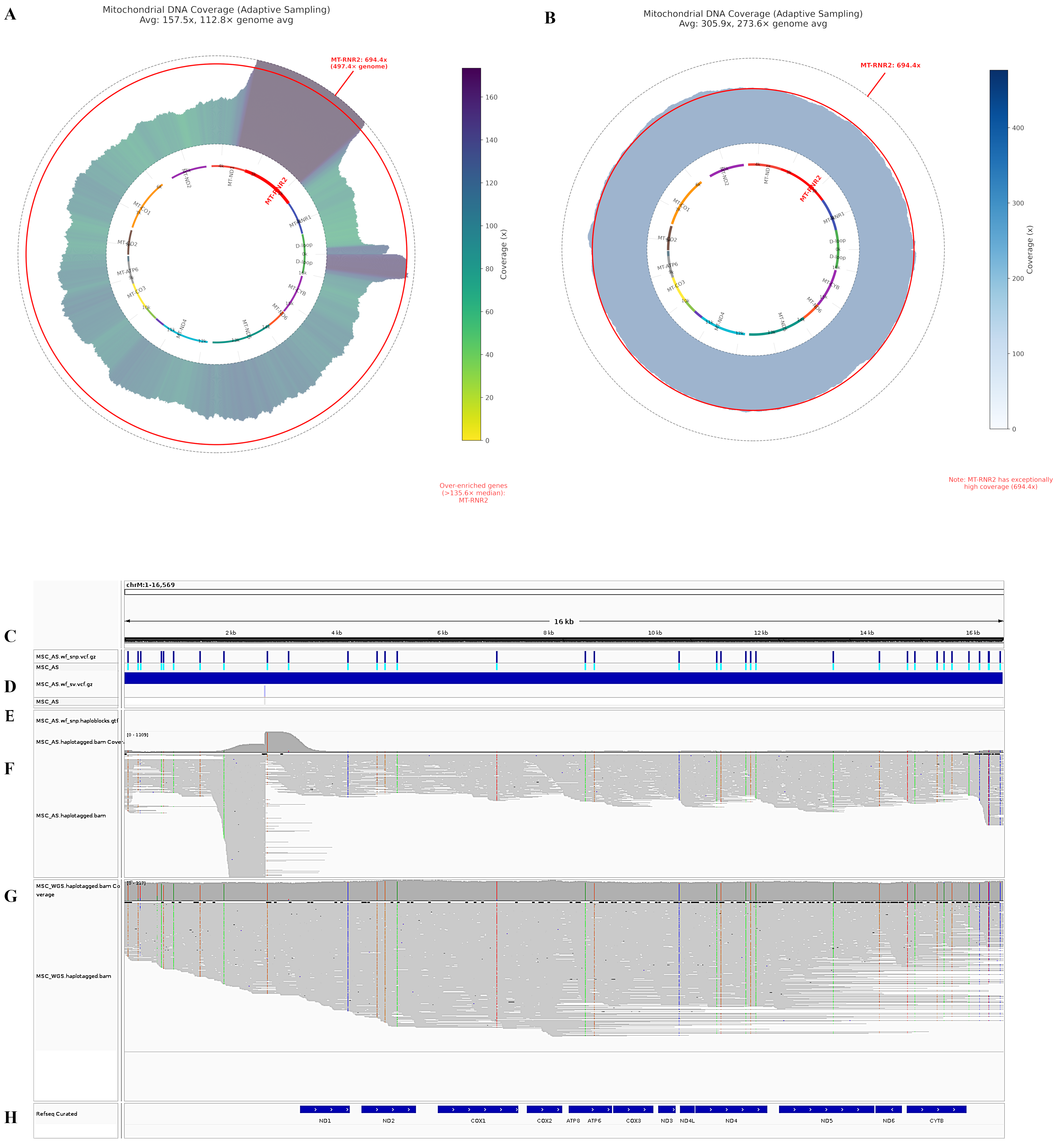
Oxford Nanopore bioinformatic workflow Nextflow was used for super-accuracy model re-basecalling (wf-basecall, Dorado v0.7.0, SUP model) and human genome alignment (wf-alignment, Ensembl reference Hs.GRCh38 (release 110)) of the two produced sets of POD5 files for the libraries. Python code, using pysam and mathplotlib, and NumPy was used to generate circus-like visualization displaying mtDNA coverage depth throughout the mitochondrial genome, with integrated gene annotations and statistical highlighting of over-enriched regions relative to the nuclear genome. Statistical significance for enrichment was determined by calculating a threshold based on 2× the median enrichment across all mtDNA genes, where enrichment was defined as the ratio of a gene’s mean coverage to the average nuclear genome coverage sampled across multiple chromosomes.
Mitochondrial DNA methylation analysis was performed through a multi-step pipeline for analysis of the two WGS and adaptive sampling BAM files (obtained above). Cytosine modifications were then extracted with a custom Python 3.10 notebook that uses pysam v0.22.0, NumPy v1.26, SciPy v1.11 (‘scipy.stats.spearmanr’), Matplotlib v3.8 and tqdm v4.66. The algorithm (i) retains only C→5-mC or C→5-hmC calls whose ‘ML’ posterior probability ≥ 0.80, (ii) maps read coordinates to reference positions, (iii) classifies a site as non-CpG when the reference dinucleotide is C H (H = A, C, T), and (iv) bins methylated versus total CH calls in 100 bp windows to calculate % 5-modC. Mean read-depth and window-level methylation are plotted along the 16.6 kb mitogenome, and a methylation-versus-coverage scatter-plot—annotated with Spearman ρ—confirms that the residual signal is independent of sequencing depth.
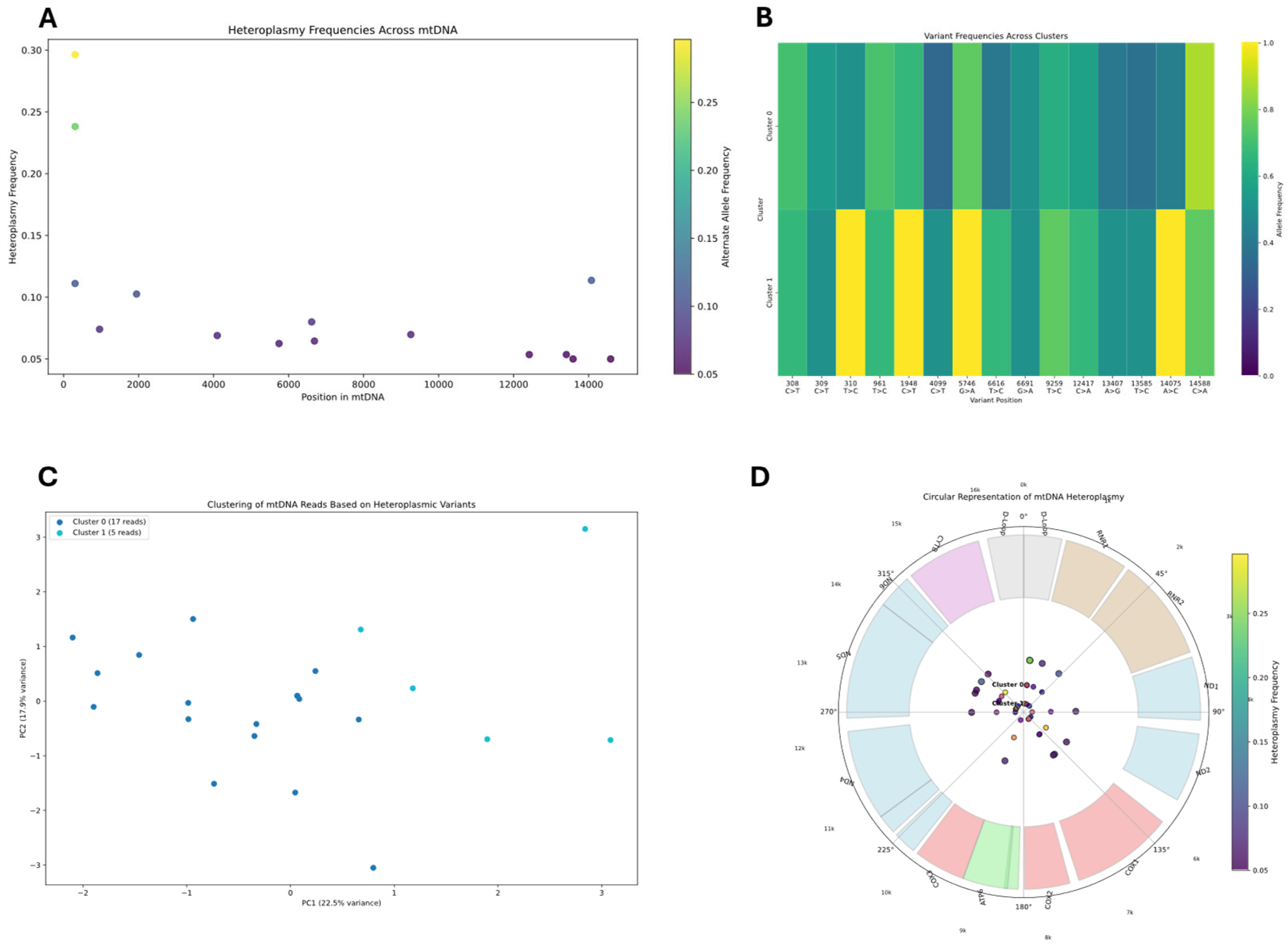
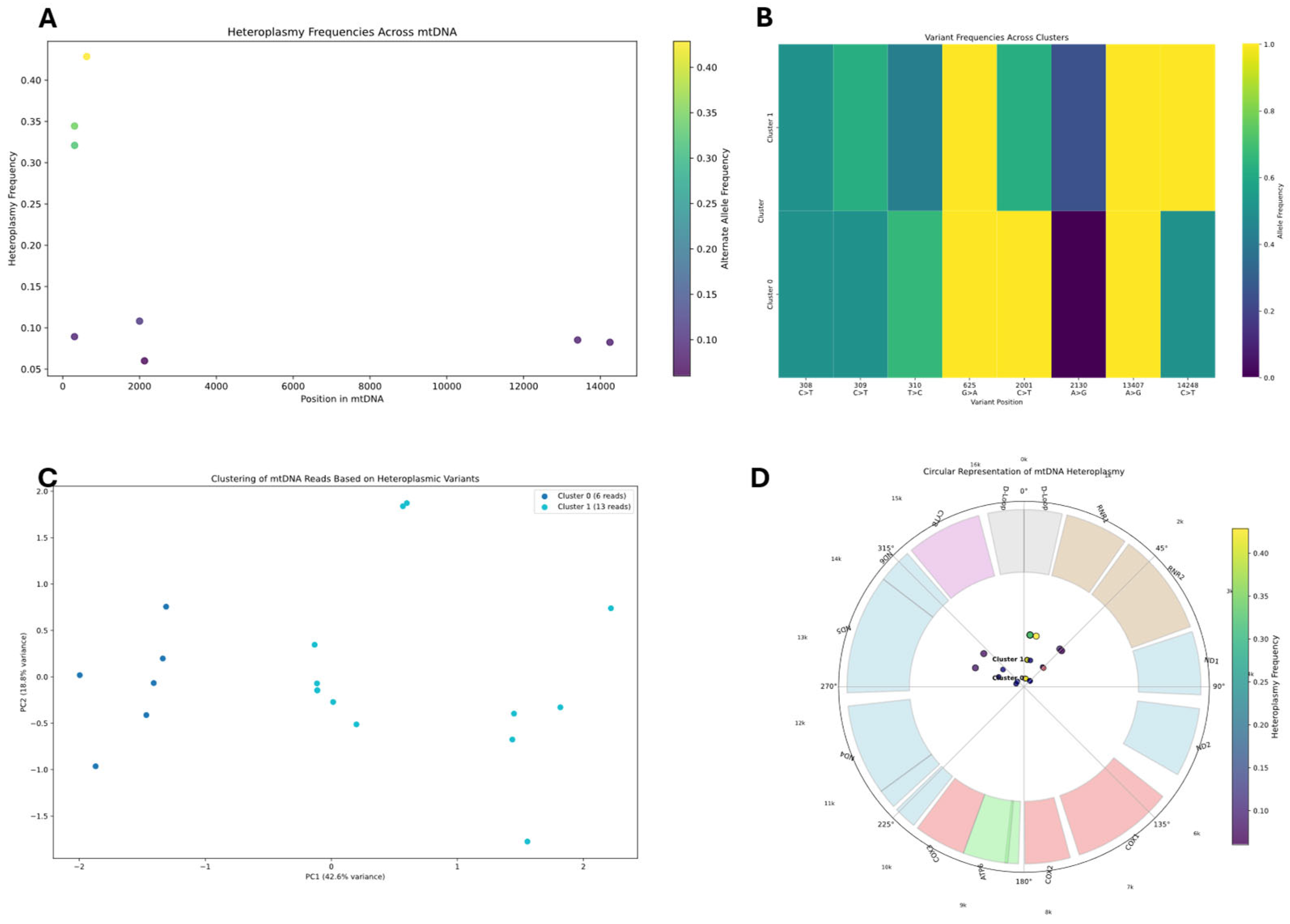
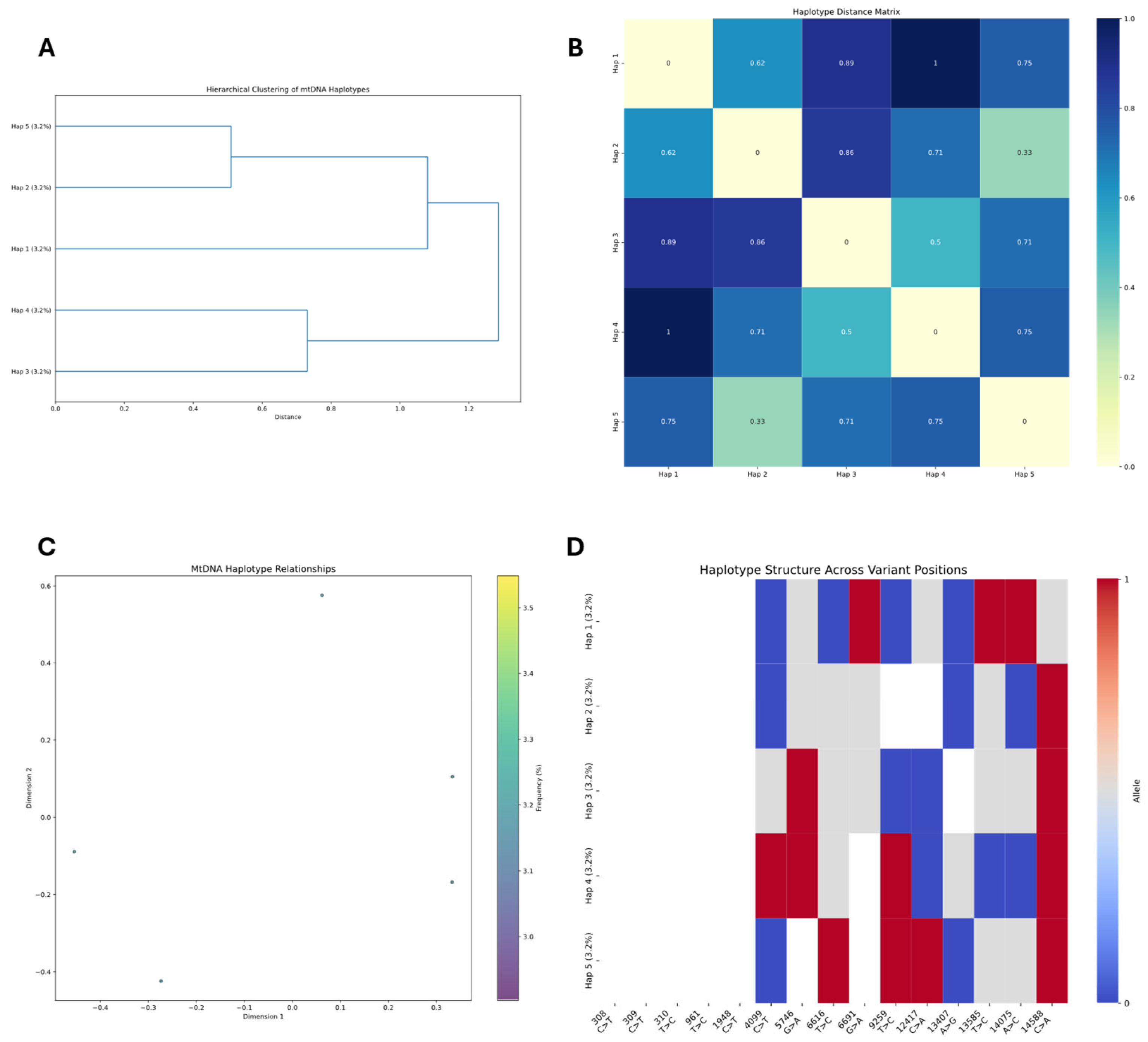
Heteroplasmic variant analysis was conducted by examining the mitochondrial genome for positions showing significant alternate allele frequencies (≥5%). The analysis pipeline processed BAM files using pysam, applying quality filters (mapping quality ≥ 20, base quality ≥ 20) to identify reliable variants. The pipeline built a read-variant matrix to analyze the distribution of heteroplasmic variants across individual reads. We employed K-means clustering with silhouette score optimization to identify distinct mitochondrial subpopulations (K-means k = 3 chosen by silhouette score = 0.71; first two PCs explaining 87% variance). Principal component analysis (PCA) was used to visualize the clustering of reads based on their variant profiles. For each identified cluster, we calculated allele frequencies at heteroplasmic sites and created consensus sequences.
Reads covering heteroplasmic sites were then extracted to reconstruct potential haplotypes, with reads grouped by their shared nucleotide patterns at variant positions. In samples with insufficient heteroplasmic sites for standard haplotype reconstruction, methylation patterns were employed as alternative clustering features using K-means and hierarchical clustering algorithms.
Integration of epigenetic modifications with haplotype data was performed to identify potential correlations between sequence variations and methylation patterns. For each identified haplotype, methylation frequencies were calculated at each position and compared across haplotypes to identify differentially methylated regions (DMRs). DMRs statistical significance was assessed through custom algorithms designed specifically for linking epigenetic modifications to sequence variations, by comparing methylation differences between haplotypes, with positions showing ≥30% methylation difference highlighted. These algorithms accounted for the circular nature of mtDNA and potential strand-specific methylation patterns. The combined approach enabled identification of haplotype-specific methylation signatures that would be undetectable by analyzing genomic variants or methylation patterns alone, providing insights into potential functional consequences of mitochondrial sequence variations. Visualization of these relationships was accomplished through heatmaps and profile plots comparing methylation patterns between haplotype clusters, providing insights into potential functional relationships between sequence variants and epigenetic modifications in the mitochondrial genome.
Additionally, several analytical data analysis strategies were implemented to determine if the mtDNA methylation phenomena observed was an artifact or a result from genuine 5meC methylation. First, to investigate the relationship between mitochondrial DNA (mtDNA) methylation and sequencing coverage obtained from adaptive sampling Nanopore long-read sequencing, we developed a custom Python analysis pipeline. The pipeline utilized aligned reads stored in BAM format as input. Initially, the mitochondrial chromosome was identified within the BAM header, and the presence of standard methylation call tags (e.g., MM, ML, YM) was automatically detected. Reads mapping to the mtDNA were processed using the pysam library (version dependent on environment). Reads were filtered based on a minimum mapping quality score (MAPQ ≥ 20) and exclusion of secondary or supplementary alignments. For each filtered read, per-base coverage information was recorded. Methylation status (methylated or unmethylated) for 5meC sites (or other relevant contexts depending on tag parsing logic) was extracted by parsing the appropriate BAM tags, converting relative read coordinates to genomic positions.
Data were aggregated across all reads to calculate the total coverage and the frequency of methylation (number of methylated reads/total reads assessed) at each genomic position. Only positions meeting a minimum coverage threshold (e.g., ≥10 reads) were retained for downstream analysis. The relationship between per-position coverage and methylation frequency was assessed using Pearson correlation, Spearman rank correlation, and linear regression analysis (scipy.stats, sklearn.linear_model). To evaluate relationships at different genomic scales, position-level data were further aggregated into non-overlapping genomic bins (e.g., 50, 100, 250, 500 bp), calculating the average coverage and average methylation frequency per bin. Correlation analyses were repeated on these binned data. Potential coverage biases were assessed by stratifying positions into quantiles based on coverage and comparing methylation frequencies, and conversely, stratifying by methylation frequency categories and comparing average coverage using bar plots, box plots, and one-way ANOVA (scipy.stats).
Methylation Pattern Analysis: Methylation status was assessed by extracting base modification information from BAM tags (e.g., MM, ML, XM, YM), typically generated by basecallers or post-processing tools like Nanopolish, Megalodon, or Guppy+modkit. For reads aligned to the mtDNA reference, methylation frequencies were calculated within each 100 bp bin.
4. Discussion
Organ-on-chip models have become increasingly relevant research instruments. Crucial for their application is their proper characterization, including ADME pharmacogenomic profiling, in order for one to fully understand their response upon challenge. While targeted sequencing achieved significantly greater sequencing depth for the ADME genes than WGS, the relative coverage variability among these target genes, as indicated by comparable enrichment ratio ranges, suggests that intrinsic factors associated with specific gene regions influence coverage uniformity irrespective of the sequencing methodology. Both methods showed there were some trade-offs. AS provides deeper and more uniform coverage across the targets (reflected in higher, more consistent normalized coverage). WGS coverage across these specific genes is inherently more variable. However, both methods appear capable of detecting gene-to-gene variations in methylation levels, and the relative methylation patterns look broadly similar, despite the differences in coverage characteristics.
These findings have important implications for pharmacogenomic research, where the accurate characterization of ADME gene methylation may influence drug metabolism phenotypes. The consistent methylation patterns detected across both sequencing approaches confirm that the observed epigenetic modifications in these genes represent genuine biological signals rather than technical artifacts. Notably, we identified differential methylation patterns in key drug-metabolizing enzyme genes that correlated with specific genetic variants, suggesting potential epigenetic regulation of drug response. The detection of these methylation patterns was consistent across both AS and WGS methodologies, though AS provided greater resolution for detecting subtle methylation differences at critical regulatory regions. These epigenetic variations may explain some of the inter-individual variability in drug metabolism that cannot be attributed to genetic polymorphisms alone. Future studies should investigate the functional consequences of these methylation patterns on gene expression and enzymatic activity, potentially offering new insights into previously unexplained pharmacokinetic variability and adverse drug reactions.
Previously, Sanger sequencing, Southern blot, and quantitative PCR have been the methods of choice for the mtDNA analysis [
13,
23,
24,
25,
26]. However, these techniques are expensive, with low sensitivity and throughput. Recently, short-read next-generation sequencing techniques have been more and more explored for mtDNA sequencing and single point mutations detection. However, short reads also possess multiple limitations and are unable to detect complex rearrangements and heteroplasmic levels [
27]. They also have limitations in resolving low-frequency variants and homopolymeric regions, which are often included within the mt genome as stretches of the same base, like AAAAA and as repetitive sequences. This can lead to errors in variant calling and gaps in the coverage [
28]. For these reasons, there is a diagnostic need for appropriate methods to investigate mtDNA.
Long-read sequencing techniques are particularly suitable for applications where a more complete and detailed understanding of the genome is needed, because of some features like the ability to sequence through repetitive and homopolymeric regions, the improved accuracy in detecting structural variants, the better assembly of complex genomes, and the accurate detection of heteromplasmy [
20,
29]. Furthermore, they sequence native molecules, which allows them to detect epigenetic modifications [
19,
30,
31] that are likely to play a crucial role in the future diagnosis of mitochondrial diseases, thus enhancing its clinical utility. Progress in flow cells and chemistry has further improved sequencing accuracy, offering better detection of low-level heteroplasmy. Precise quantification of the heteroplasmy level is extremely important because it classifies certain variants as the likely cause of the disease or as a benign polymorphism.
Despite the high copy number mitochondria per cell, mtDNA contributes only with 0.1–0.2% of the total DNA amount. This is why previous sequencing methods rely on PCR amplification [
32,
33]. PCR, however, can introduce biases, especially if there are regions with varying GC content, leading to uneven coverage across the mtDNA genome. This can also complicate the detection and quantification of mtDNA variants, especially in samples with heteroplasmy. Adaptive sampling is a method of software-controlled enrichment unique to nanopore sequencing platforms [
34]. It allows for simple enrichment by loading a BED file of a target region. By sequencing the first 400–500 bp of a DNA library, adaptive sampling software can identify reads containing target region or not and pass only these fragments that contain the target region. Adaptive sampling is a relatively new bioinformatic tool [
35] but is being more and more explored for mitochondrial DNA analysis, because all published data clearly show that it consistently enriches reference sequences. Moreover, long-read sequencing of mtDNA can be considered a consistently accurate approach for the identification of subjects based on their mtDNA haplogroup/haplotype (for forensic purposes or population genetics).
We are aware of other studies that used adaptive sampling of ONT to analyze mitochondrial DNA; however, none of them performed the analysis with mesenchymal stem cells (MSC). Mesenchymal stem cells are non-hematopoietic progenitors that are found in various tissues, most commonly in bone marrow, but also in adipose tissue, umbilical cord tissue, dental pulp, and other sites. They can differentiate into multiple lineages, which makes them very promising tool for therapeutic applications. Unfortunately, their regenerative potential decreases during ageing [
18,
36]. Therefore, therapeutic approaches have been investigated and already proposed to either eliminate, revitalize, or substitute senescent MSCs. Decline in mitochondrial function and biogenesis and the accumulation of mtDNA mutations are the primary reasons for ageing [
37]. MtDNA mutations are known to accumulate in stem cell populations [
38], accompanied by an increase in the somatic mutations [
39]. However, the isolation of good quality and a good amount of MSC remains a challenge. AdMSC are a relatively abundant source, with high proliferative capacity and easy harvesting, making them extremely suitable for different applications in regenerative medicine. Another thing is that obesity and obesity-related complications have become more serious issues, affecting people all over the world [
40]. Adipose tissue is a major regulator of energy metabolism and serves not only as a fat tissue storing, mobilizing, and distributing site, but different types of cells within it can produce heat (brown adipose tissue) and many adipokines. These are biologically active substances that exert diverse functions like immune (e.g., complement factors, haptoglobin), endocrine function (e.g., leptin, sex steroids, various growth factors), metabolic function (e.g., fatty acids, adiponectin, resistin), and cardiovascular function (e.g., angiotensinogen, PAI-1) [
41,
42,
43]. Studying the physiology and pathophysiology of conditions associated with metabolic inflammation is yet more than ever important. For this reason, AdMSC are considered as the main “players” and new studies for biomimetic models implementing AdMSC are being published more often [
44,
45].
Because of all these important considerations, a straightforward, accurate, reproducible, and PCR-free protocol for studying mitochondrial defects in adipose-derived MSC has been established. The analysis pipeline allows the alignment of reads containing large deletions, which leads to an accurate detection of the boundaries of mtDNA deletion and its heteroplasmy levels. This approach could serve as an optimal method for conducting full mtDNA genetic studies in a single experiment, offering complete mitochondrial genome sequencing and the detection of both point mutations and large deletions, along with quantification of heteroplasmy.
The custom algorithm developed for linking epigenetic modifications to sequence variations provided critical advantages for comprehensive mtDNA analysis. By simultaneously processing methylation signals and heteroplasmic variants from individual long reads, the algorithm preserved the phase relationship between epigenetic marks and sequence polymorphisms—a connection that would be lost in traditional separate analyses. This integrated approach revealed that certain heteroplasmic variants consistently co-occurred with specific methylation patterns in the D-loop, suggesting functional relationships between genetic and epigenetic variations that influence mitochondrial gene expression.
Furthermore, the algorithm’s ability to handle the circular nature of mtDNA and account for strand-specific methylation patterns enabled the identification of methylation clusters that correlated with particular haplotypes. This led to the discovery of previously uncharacterized mitochondrial epigenetic signatures associated with specific sequence variants. In samples with limited heteroplasmy, the approach successfully leveraged methylation patterns as surrogate markers for lineage discrimination, effectively increasing the resolution of mtDNA classification beyond what would be possible with sequence analysis alone. This methodology provided new insights into the complex interplay between mitochondrial genetics and epigenetics that may contribute to our understanding of mitochondrial heterogeneity in various biological contexts.
The current literature on integrated approaches to mitochondrial DNA methylation and heteroplasmy is still nascent. Most studies have analyzed mtDNA methylation and sequence variants separately, without directly examining their interplay. For example, Patil et al. mapped extensive mtDNA methylation patterns (mostly at non-CpG sites) across the mitochondrial genome [
22], and Liu et al. characterized the distribution of CpG methylation in mtDNA [
46]. However, these studies considered epigenetic and genetic variations independently, inferring any links only indirectly. The advent of long-read sequencing now allows both genetic and epigenetic features to be read concurrently from the same DNA molecule, a major technological advance in genomics [
47]. Recent reviews highlight that nanopore sequencing can directly detect nucleotide modifications along with the DNA sequence, enabling simultaneous calling of heteroplasmic variants and methylation marks from one experiment [
47]. Similar integrative methods have already been applied in the nuclear genome: Using nanopore reads, researchers have phased methylation patterns with SNP haplotypes to reveal allele-specific methylation in human chromosomes [
48].
Yet, applying these techniques to mtDNA remains challenging. Mitochondrial methylation levels are very low and sometimes controversial [
22,
49,
50], making true signals hard to distinguish from noise. Most controversy stems from CpG specifically targeted detection by most methods, including early DNA methylation detection algorithms, like Nanopolish and Megalodon, as shown by the higher DNA methylation within Guppy basecalling reads [
50]. Novel Dorado modes are better suited to detect 5meC, but perhaps other non-CpG modifications (like 6mA) should be specifically trained to enable proper and verifiable detection. Substantial evidence argues against widespread, functional CpG methylation in mammalian mtDNA. This scarcity might be linked to the potential gene silencing effects such methylation could exert within the extremely compact mitochondrial genome, combined with the lack of known canonical CpG methyltransferases targeted to mitochondria [
51]. This is in stark contrast to the nuclear genome where CpG methylation is a fundamental epigenetic mark. Consequently, earlier studies or methods focusing primarily or solely on CpG methylation might have provided an incomplete picture or underestimated other forms of modification [
50].
Additionally, specialized bioinformatics tools are needed to handle the circular mtDNA and to jointly analyze heteroplasmy with methylation. Our approach addresses these gaps by using algorithms tailored to mtDNA’s unique characteristics, allowing us to simultaneously assess sequence heteroplasmy and methylation on single molecules. This represents a methodological advance in the emerging field of mitochondrial epigenomics, laying the groundwork for deeper insights into mtDNA regulation in aging and disease.
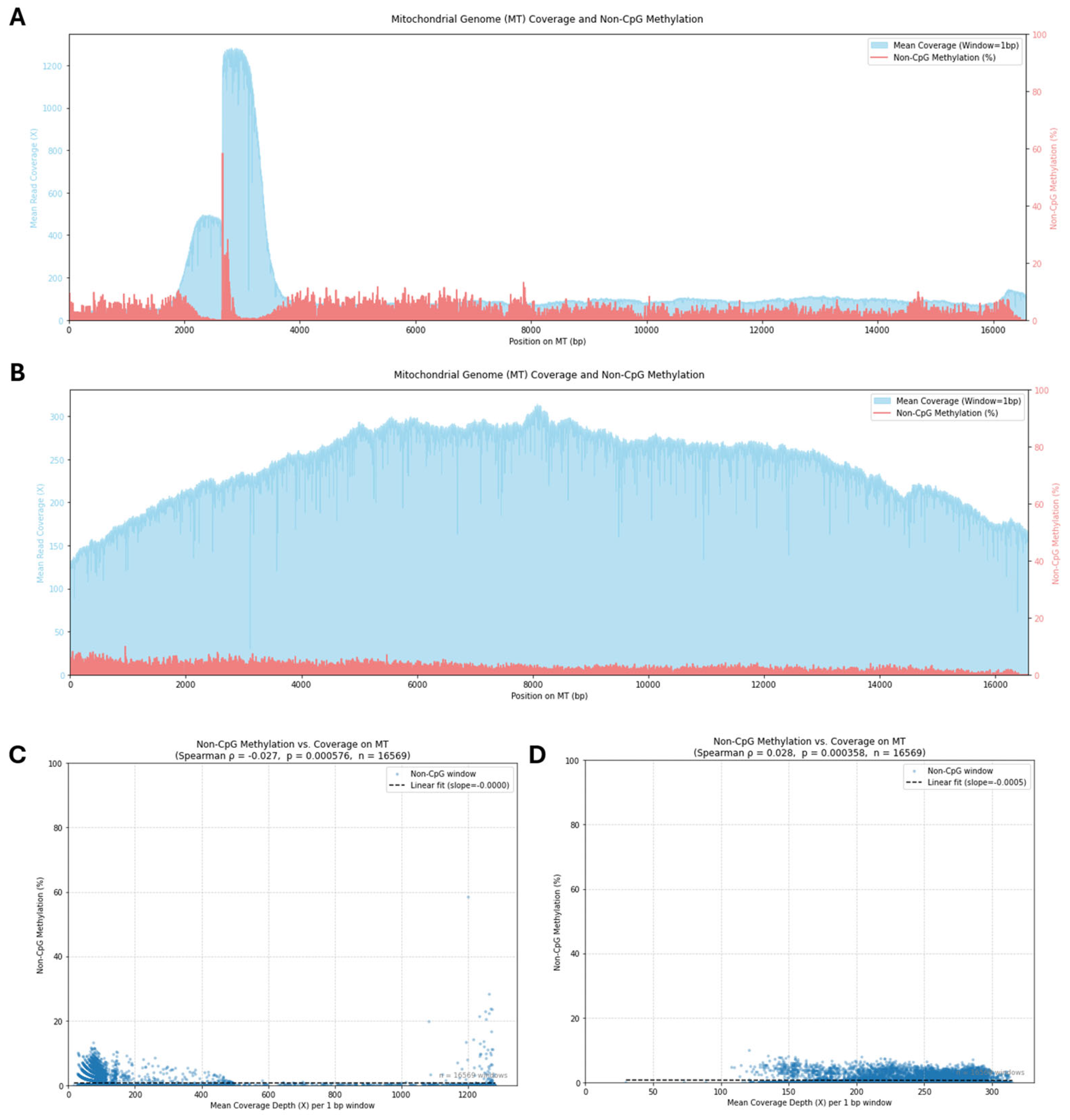
Building on our findings that nanopore adaptive sampling effectively decouples base modification detection from sequencing coverage artifacts (as suggested by the analysis in
Figure 10C,D), our data support the detection of genuine, albeit low-level, non-CpG methylation signals directly from native mtDNA molecules. Our analysis pipeline, utilizing Dorado basecalling with MM and ML tags to identify cytosine modifications (
Figure 10), revealed globally low levels of methylation, predominantly appearing independent of CpG context. This finding aligns with studies such as Patil et al. [
22] which used orthogonal methods and also concluded that non-CpG (CpH) methylation is the more prevalent, though still potentially sparse, form in human mtDNA. While our study lacks independent validation (a limitation discussed previously), the observation of consistent low-level patterns irrespective of coverage depth (
Figure 10A vs.
Figure 10B) or sequencing strategy (AS vs WGS,
Figure 10C vs.
Figure 10D) lends support to these signals reflecting features of the sequenced molecules rather than mere technical noise. Furthermore, the potential functional relevance of
maintaining low mtDNA methylation levels is underscored by studies indicating that artificial or pathological
hypermethylation can perturb mitochondrial gene expression and function [
31]. For instance, hypermethylation of genes like ND6 observed under specific pathological conditions associated with obesity models appeared detrimental [
52], suggesting that significant increases above baseline methylation might be functionally disruptive. Bicci et al. CpG considers mtDNA methylation as technical artifact of low sequencing coverage [
49]. Interestingly, AS application produces specific co-occurrence patterns between genetic variants and methylation marks, particularly in functionally significant regions like the D-loop. This integrated approach not only preserves the phase relationship between epigenetic modifications and sequence polymorphisms but also the identification of putative methylation clusters regardless of the modification that could correlate with particular haplotypes—relationships that would remain undetectable using conventional separate analyses. One important limitation of this study is that only the AdMSC cell line under the intact condition was subject to this analysis. As another study recently proved increased methylation in ND5 mitochondrial gene after challenge [
31], and we should expect methylation phenomena in mitochondria to be dynamic phenomena, regardless of how rare and low frequency they are. Their lower frequency should not be regarded as a result of lesser importance, as this could be due to signaling regulation phenomena. Beyond 5mC, it is increasingly recognized that other base modifications contribute to the mitochondrial epitranscriptome and, potentially, the epigenome. Notably, N6-methyladenosine (m6A) has been identified and characterized as a distinct epigenetic mark within mammalian mtDNA, installed by enzymes like METTL3/14 and potentially regulated by demethylases such as ALKBH1 [
31]. This presents an alternative or parallel layer of epigenetic control operating within mitochondria. The functional role of mtDNA m6A methylation is still under investigation, but evidence suggests it could be involved in modulating crucial processes such as mtDNA replication and the expression of mitochondrial genes [
31] and happened to be hyper-methylated [
52]. Our current study, focused on cytosine modifications detected by standard Dorado models, was not designed to assess m6A; exploring the landscape and interplay of different modifications using tailored approaches is a pertinent future direction.



 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}