

ProLinker–Generator: Design of a PROTAC Linker Base on a Generation Model Using Transfer and Reinforcement Learning



Abstract
1. Introduction
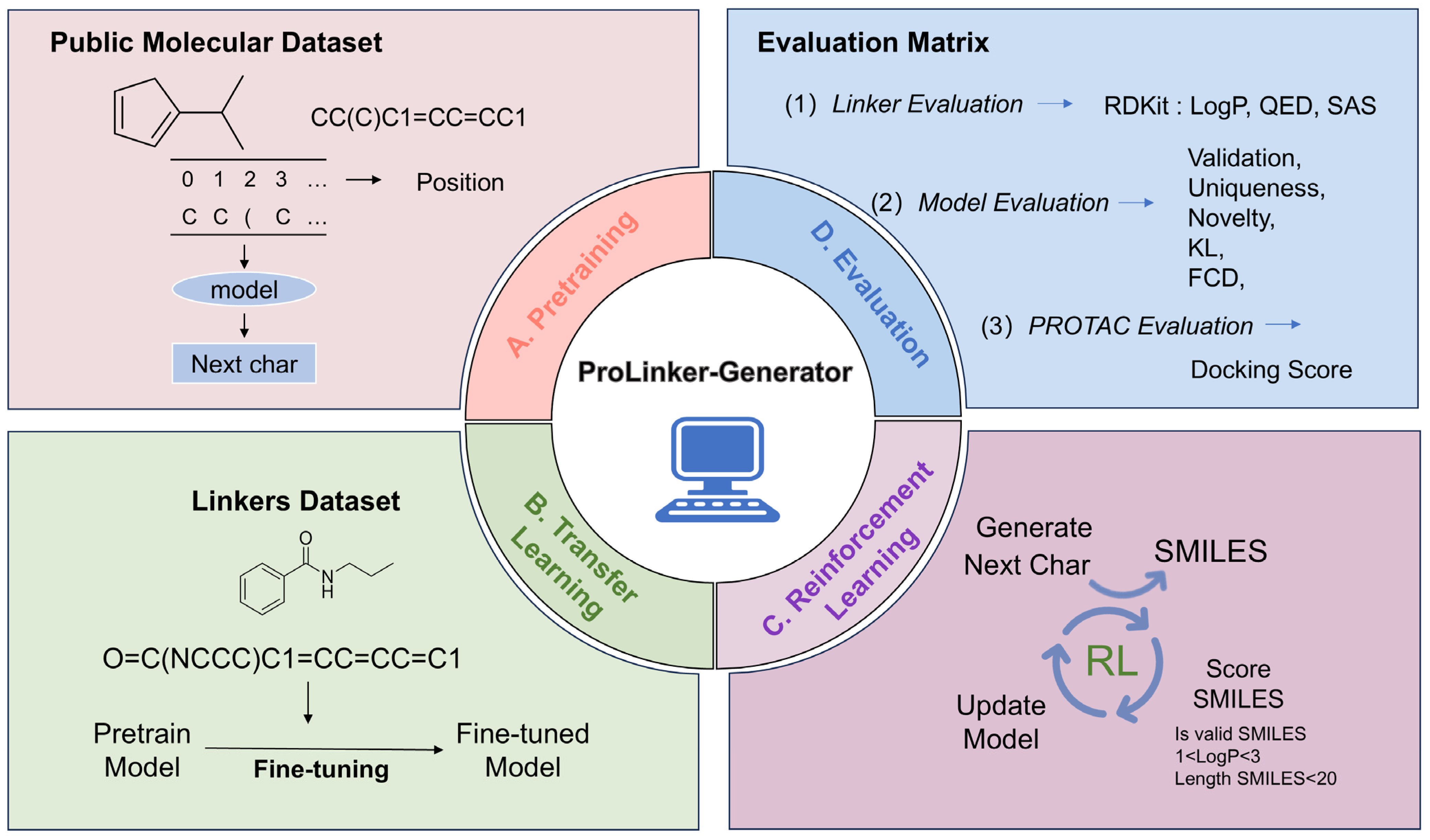
2. Method
2.1. DATA
2.2. SMILES Augmentation
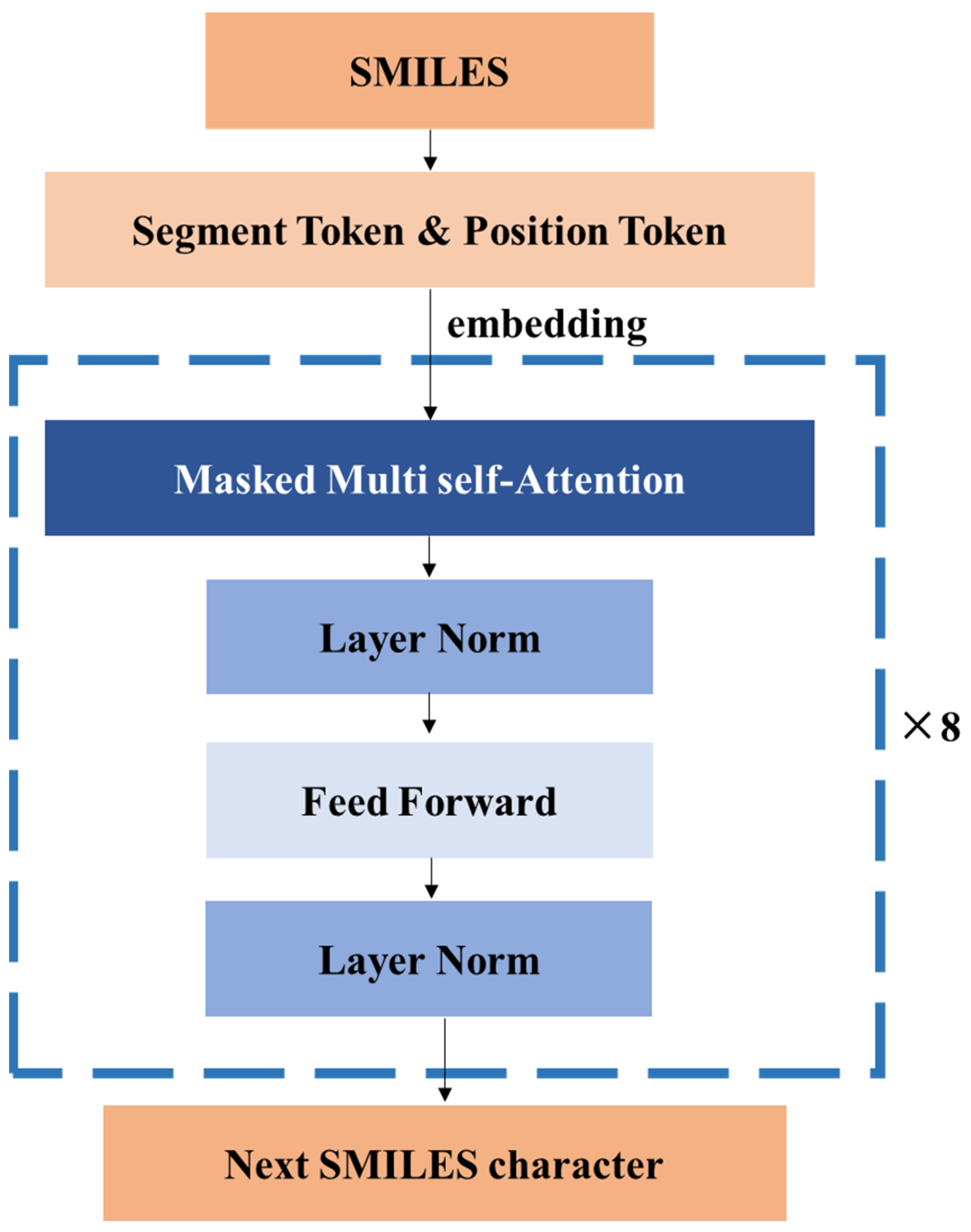
2.3. Model
2.4. Benchmark Models
2.4.1. RNN
2.4.2. LSTM
2.4.3. AAE
2.4.4. VAE
2.5. Reinforcement Learning
- If the generated SMILES is invalid (e.g., due to syntactic errors), the reward is set to 0 (Equation (4)).
- If the SMILES is valid, additional property-based scoring is applied. For LogP, a score of 1 is assigned if the value falls within the target range (1 < LogP < 3); otherwise, the score is 0 (Equation (2)).
- For molecular length, a score of 1 is given if the number of atoms is less than 20; otherwise, the score is 0 (Equation (3)).
- The final reward is the sum of these individual scores (Equation (4)).
2.6. Evaluation Metrics
3. Results
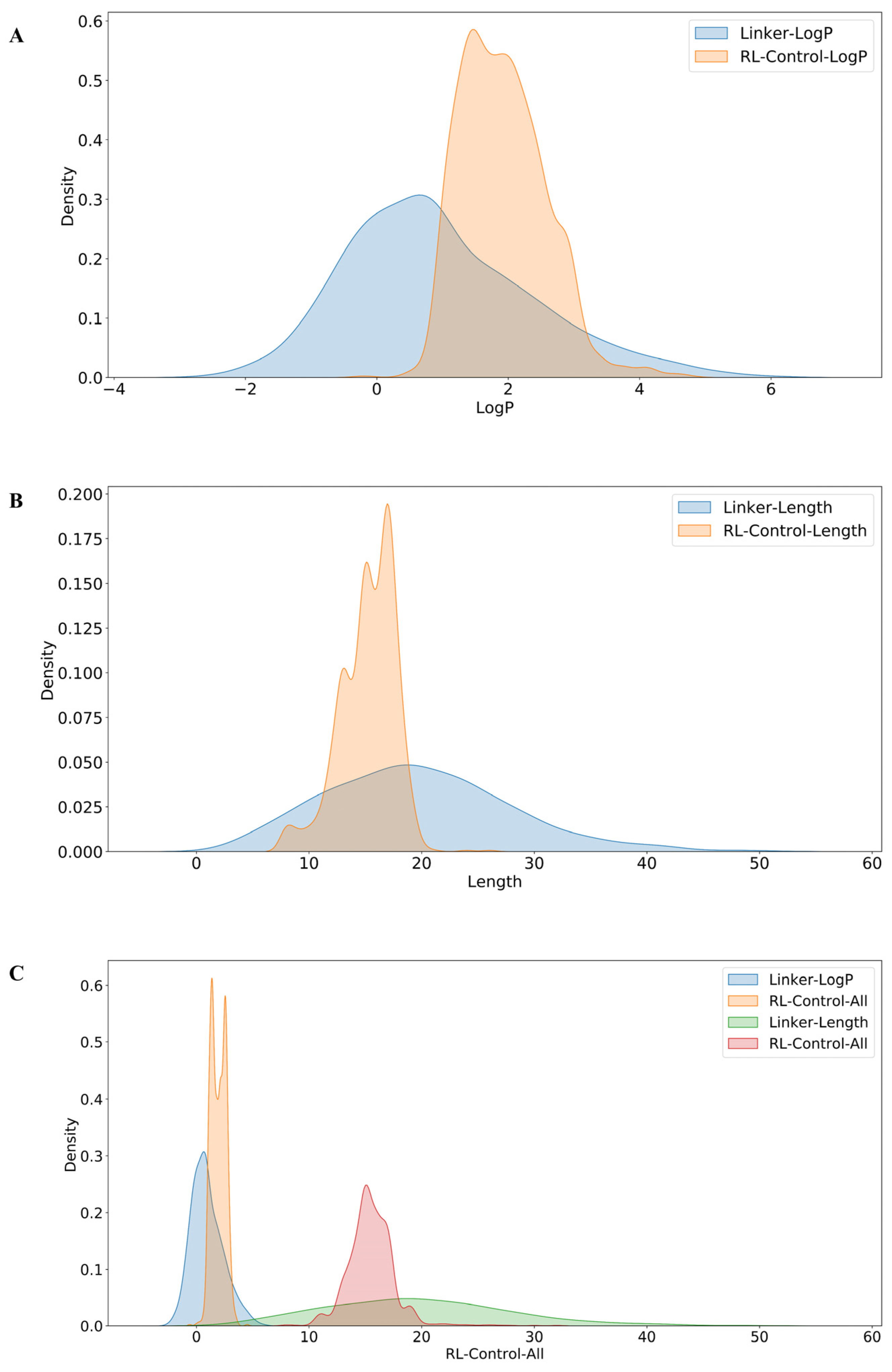
3.1. Molecular Generation Based on Reinforcement Learning
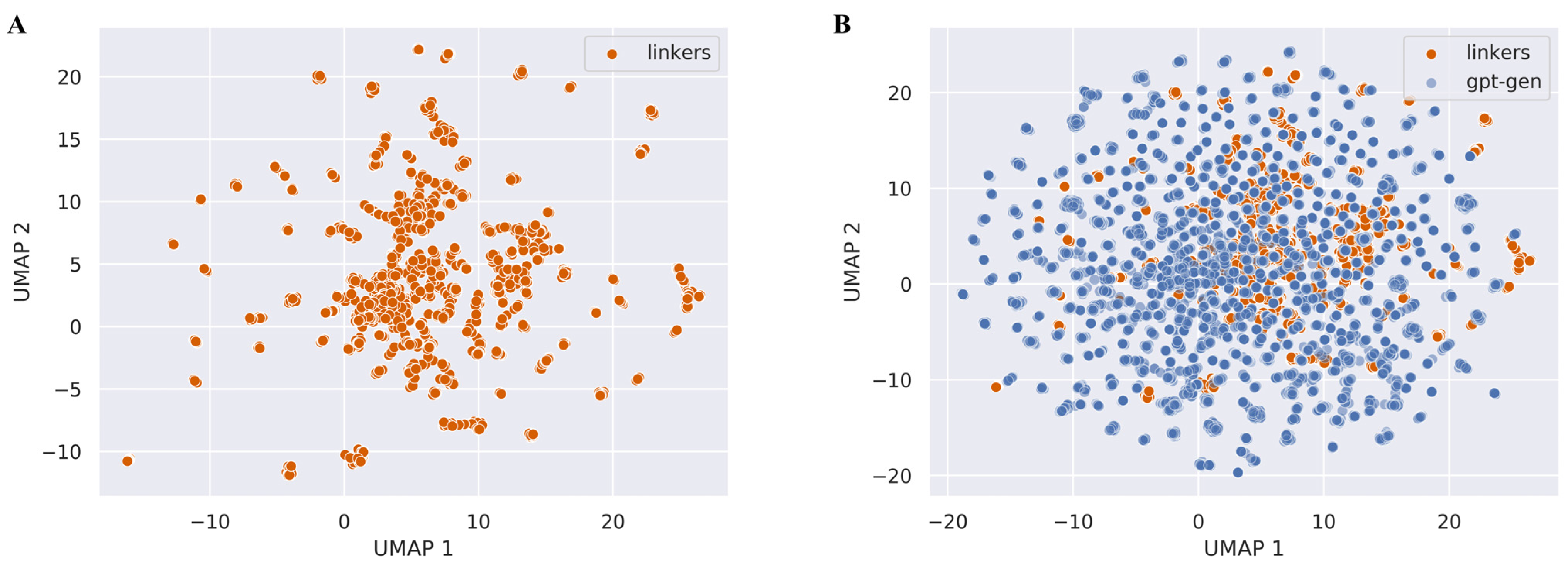
3.2. Molecular Generation Based on Transfer Learning
- (1)
- Effect of Data Augmentation
- (2)
- Effect of Different Pre-training Datasets
3.3. Linker Generation for Molecular Docking Examples
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. Protacs: Great Opportunities for Academia and Industry. Signal Transduct. Target. Ther. 2019, 4, 64. [Google Scholar] [CrossRef] [PubMed]
- Ottis, P.; Crews, C.M. Proteolysis-Targeting Chimeras: Induced Protein Degradation as a Therapeutic Strategy. ACS Chem. Biol. 2017, 12, 892–898. [Google Scholar] [CrossRef]
- Deshaies, R.J. Prime Time for Protacs. Nat. Chem. Biol. 2015, 11, 634–635. [Google Scholar] [CrossRef]
- Schneekloth, A.R.; Pucheault, M.; Tae, H.S.; Crews, C.M. Targeted Intracellular Protein Degradation Induced by a Small Molecule: En Route to Chemical Proteomics. Bioorg. Med. Chem. Lett. 2008, 18, 5904–5908. [Google Scholar] [CrossRef]
- Zhong, G.; Chang, X.; Xie, W.; Zhou, X. Targeted Protein Degradation: Advances in Drug Discovery and Clinical Practice. Signal Transduct. Target. Ther. 2024, 9, 308. [Google Scholar]
- Békés, M.; Langley, D.R.; Crews, C.M. Protac Targeted Protein Degraders: The Past Is Prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Toure, M.; Crews, C.M. Small-Molecule Protacs: New Approaches to Protein Degradation. Angew. Chem. Int. Ed. 2016, 55, 1966–1973. [Google Scholar] [CrossRef]
- Chirnomas, D.; Hornberger, K.R.; Crews, C.M. Protein Degraders Enter the Clinic—A New Approach to Cancer Therapy. Nat. Rev. Clin. Oncol. 2023, 20, 265–278. [Google Scholar] [CrossRef]
- Li, X.; Pu, W.; Zheng, Q.; Ai, M.; Chen, S.; Peng, Y. Proteolysis-Targeting Chimeras (Protacs) in Cancer Therapy. Mol. Cancer 2022, 21, 99. [Google Scholar] [CrossRef]
- Zou, Y.; Ma, D.; Wang, Y. The Protac Technology in Drug Development. Cell Biochem. Funct. 2019, 37, 21–30. [Google Scholar] [CrossRef]
- Cecchini, C.; Pannilunghi, S.; Tardy, S.; Scapozza, L. From Conception to Development: Investigating Protacs Features for Improved Cell Permeability and Successful Protein Degradation. Front. Chem. 2021, 9, 672267. [Google Scholar] [CrossRef] [PubMed]
- Gharbi, Y.; Mercado, R. A Comprehensive Review of Emerging Approaches in Machine Learning Forde Novoprotac Design. Digit. Discov. 2024, 3, 2158–2176. [Google Scholar] [CrossRef]
- Park, D.; Izaguirre, J.; Coffey, R.; Xu, H. Modeling the Effect of Cooperativity in Ternary Complex Formation and Targeted Protein Degradation Mediated by Heterobifunctional Degraders. ACS Bio. Med. Chem. Au 2022, 3, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Zorba, A.; Nguyen, C.; Xu, Y.; Starr, J.; Borzilleri, K.; Smith, J.; Zhu, H.; Farley, K.A.; Ding, W.; Schiemer, J.; et al. Delineating the Role of Cooperativity in the Design of Potent Protacs for Btk. Proc. Natl. Acad. Sci. USA 2018, 115, E7285–E7292. [Google Scholar] [CrossRef]
- Danishuddin; Jamal, M.S.; Song, K.-S.; Lee, K.-W.; Kim, J.-J.; Park, Y.-M. Revolutionizing Drug Targeting Strategies: Integrating Artificial Intelligence and Structure-Based Methods in Protac Development. Pharmaceuticals 2023, 16, 1649. [Google Scholar] [CrossRef]
- Zheng, S.; Tan, Y.; Wang, Z.; Li, C.; Zhang, Z.; Sang, X.; Chen, H.; Yang, Y. Accelerated Rational Protac Design via Deep Learning and Molecular Simulations. Nat. Mach. Intell. 2022, 4, 739–748. [Google Scholar] [CrossRef]
- Abbas, A.; Ye, F. Computational Methods and Key Considerations for in Silico Design of Proteolysis Targeting Chimera (Protacs). Int. J. Biol. Macromol. 2024, 277, 134293. [Google Scholar] [CrossRef]
- Li, B.; Ran, T.; Chen, H. 3d Based Generative Protac Linker Design with Reinforcement Learning. Brief. Bioinform. 2023, 24, bbad323. [Google Scholar] [CrossRef]
- Poongavanam, V.; Atilaw, Y.; Ye, S.; Wieske, L.H.E.; Erdelyi, M.; Ermondi, G.; Caron, G.; Kihlberg, J. Predicting the Permeability of Macrocycles from Conformational Sampling—Limitations of Molecular Flexibility. J. Pharm. Sci. 2021, 110, 301–313. [Google Scholar] [CrossRef]
- Liu, Q.; Allamanis, M.; Brockschmidt, M.; Gaunt, A. Constrained Graph Variational Autoencoders for Molecule Design. Adv. Neural Inf. Process. Syst. 2018, 31, 7806–7815. [Google Scholar]
- Lim, J.; Ryu, S.; Kim, J.W.; Kim, W.Y. Molecular Generative Model Based on Conditional Variational Autoencoder for De Novo Molecular Design. J. Cheminform. 2018, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef]
- Weng, G.; Shen, C.; Cao, D.; Gao, J.; Dong, X.; He, Q.; Yang, B.; Li, D.; Wu, J.; Hou, T. Protac-Db: An Online Database of Protacs. Nucleic Acids Res. 2021, 49, D1381–D1387. [Google Scholar] [CrossRef]
- Li, F.; Hu, Q.; Zhang, X.; Sun, R.; Liu, Z.; Wu, S.; Tian, S.; Ma, X.; Dai, Z.; Yang, X.; et al. Deepprotacs Is a Deep Learning-Based Targeted Degradation Predictor for Protacs. Nat. Commun. 2022, 13, 7133. [Google Scholar] [CrossRef]
- Bagal, V.; Aggarwal, R.; Vinod, P.K.; Priyakumar, U.D. Molgpt: Molecular Generation Using a Transformer-Decoder Model. J. Chem. Inf. Model. 2021, 62, 2064–2076. [Google Scholar] [CrossRef]
- Wang, J.; Luo, H.; Qin, R.; Wang, M.; Wan, X.; Fang, M.; Zhang, O.; Gou, Q.; Su, Q.; Shen, C.; et al. 3dsmiles-Gpt: 3d Molecular Pocket-Based Generation with Token-Only Large Language Model. Chem. Sci. 2025, 16, 637–648. [Google Scholar] [CrossRef]
- Fan, W.; He, Y.; Zhu, F. Rm-Gpt: Enhance the Comprehensive Generative Ability of Molecular Gpt Model via Localrnn and Realformer. Artif. Intell. Med. 2024, 150, 102827. [Google Scholar] [CrossRef]
- Blaschke, T.; Arús-Pous, J.; Chen, H.; Margreitter, C.; Tyrchan, C.; Engkvist, O.; Papadopoulos, K.; Patronov, A. Reinvent 2.0: An Ai Tool for De Novo Drug Design. J. Chem. Inf. Model. 2020, 60, 5918–5922. [Google Scholar] [CrossRef]
- Guo, J.; Knuth, F.; Margreitter, C.; Janet, J.P.; Papadopoulos, K.; Engkvist, O.; Patronov, A. Link-Invent: Generative Linker Design with Reinforcement Learning. Digit. Discov. 2023, 2, 392–408. [Google Scholar] [CrossRef]
- Bou, A.; Thomas, M.; Dittert, S.; Navarro, C.; Majewski, M.; Wang, Y.; Patel, S.; Tresadern, G.; Ahmad, M.; Moens, V.; et al. Acegen: Reinforcement Learning of Generative Chemical Agents for Drug Discovery. J. Chem. Inf. Model. 2024, 64, 5900–5911. [Google Scholar] [CrossRef]
- Polykovskiy, D.; Zhebrak, A.; Sanchez-Lengeling, B.; Golovanov, S.; Tatanov, O.; Belyaev, S.; Kurbanov, R.; Artamonov, A.; Aladinskiy, V.; Veselov, M.; et al. Molecular Sets (Moses): A Benchmarking Platform for Molecular Generation Models. Front. Pharmacol. 2018, 11, 565644. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The Chembl Database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. Zinc: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef]
- Wu, Z.; Ramsundar, B.; Feinberg, E.N.; Gomes, J.; Geniesse, C.; Pappu, A.S.; Leswing, K.; Pande, V. Moleculenet: A Benchmark for Molecular Machine Learning. Chem. Sci. 2018, 9, 513–530. [Google Scholar] [CrossRef]
- Wildman, S.A.; Crippen, G.M. Prediction of Physicochemical Parameters by Atomic Contributions. J. Chem. Inf. Comput. Sci. 1999, 39, 868–873. [Google Scholar] [CrossRef]
- Ertl, P.; Schuffenhauer, A. Estimation of Synthetic Accessibility Score of Drug-like Molecules Based on Molecular Complexity and Fragment Contributions. J. Cheminform. 2009, 1, 8. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the Chemical Beauty of Drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef]
- Radford, A.; Narasimhan, K.; Salimans, T.; Sutskever, I. Improving Language Understanding by Generative Pre-Training. 2018. [Google Scholar]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, L.; Polosukhin, I. Attention Is All You Need. Adv. Neural Inf. Process. Syst. 2017, 30. [Google Scholar] [CrossRef]
- Hocky, G.M.; White, A.D. Natural Language Processing Models That Automate Programming Will Transform Chemistry Research and Teaching. Digit. Discov. 2022, 1, 79–83. [Google Scholar] [CrossRef]
- Radford, A.; Wu, J.; Child, R.; Luan, D.; Amodei, D.; Sutskever, I. Language Models Are Unsupervised Multitask Learners. OpenAI Blog 2019, 1, 9. [Google Scholar]
- Shen, T.; Guo, J.; Han, Z.; Zhang, G.; Liu, Q.; Si, X.; Wang, D.; Wu, S.; Xia, J. Automoldesigner for Antibiotic Discovery: An Ai-Based Open-Source Software for Automated Design of Small-Molecule Antibiotics. J. Chem. Inf. Model. 2024, 64, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Polykovskiy, D.; Zhebrak, A.; Vetrov, D.; Ivanenkov, Y.; Aladinskiy, V.; Mamoshina, P.; Bozdaganyan, M.; Aliper, A.; Zhavoronkov, A.; Kadurin, A. Entangled Conditional Adversarial Autoencoder for De Novo Drug Discovery. Mol. Pharm. 2018, 15, 4398–4405. [Google Scholar] [CrossRef]
- Kadurin, A.; Nikolenko, S.; Khrabrov, K.; Aliper, A.; Zhavoronkov, A. Drugan: An Advanced Generative Adversarial Autoencoder Model for De Novo Generation of New Molecules with Desired Molecular Properties in Silico. Mol. Pharm. 2017, 14, 3098–3104. [Google Scholar] [CrossRef]
- Wang, J.; Hsieh, C.Y.; Wang, M.; Wang, X.; Wu, Z.; Jiang, D.; Liao, B.; Zhang, X.; Yang, B.; He, Q.; et al. Multi-Constraint Molecular Generation Based on Conditional Transformer, Knowledge Distillation and Reinforcement Learning. Nat. Mach. Intell. 2021, 3, 914–922. [Google Scholar] [CrossRef]
- Cyrus, K.; Wehenkel, M.; Choi, E.-Y.; Han, H.-J.; Lee, H.; Swanson, H.; Kim, K.-B. Impact of Linker Length on the Activity of Protacs. Mol. BioSyst. 2011, 7, 359–364. [Google Scholar] [CrossRef]
- Bemis, T.A.; La Clair, J.J.; Burkart, M.D. Unraveling the Role of Linker Design in Proteolysis Targeting Chimeras. J. Med. Chem. 2021, 64, 8042–8052. [Google Scholar] [CrossRef]
- Dong, Y.; Ma, T.; Xu, T.; Feng, Z.; Li, Y.; Song, L.; Yao, X.; Ashby, C.R., Jr.; Hao, G.F. Characteristic Roadmap of Linker Governs the Rational Design of Protacs. Acta Pharm. Sin. B 2024, 14, 4266–4295. [Google Scholar] [CrossRef]
- Bjerrum, E.J. Smiles Enumeration as Data Augmentation for Neural Network Modeling of Molecules. arXiv 2017, arXiv:1703.07076. [Google Scholar]
- Zhang, C.; Zhai, Y.; Gong, Z.; Duan, H.; She, Y.B.; Yang, Y.F.; Su, A. Transfer Learning across Different Chemical Domains: Virtual Screening of Organic Materials with Deep Learning Models Pretrained on Small Molecule and Chemical Reaction Data. J. Cheminform. 2024, 16, 89. [Google Scholar] [CrossRef]
- Entezari, M.; Wortsman, M.; Saukh, O.; Shariatnia, M.; Sedghi, H.; Schmidt, L. The Role of Pre-Training Data in Transfer Learning. Comput. Sci. 2023. [Google Scholar] [CrossRef]
- Trozzi, F.; Wang, X.; Tao, P. Umap as a Dimensionality Reduction Tool for Molecular Dynamics Simulations of Biomacromolecules: A Comparison Study. J. Phys. Chem. B 2021, 125, 5022–5034. [Google Scholar] [CrossRef] [PubMed]
- McInnes, L.; Healy, J.; Saul, N.; Großberger, L. Umap: Uniform Manifold Approximation and Projection. J. Open Source Softw. 2018, 3, 861. [Google Scholar] [CrossRef]
- Yang, Y.; Hsieh, C.-Y.; Kang, Y.; Hou, T.; Liu, H.; Yao, X. Deep Generation Model Guided by the Docking Score for Active Molecular Design. J. Chem. Inf. Model. 2023, 63, 2983–2991. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, X.; Gan, J.; Chen, S.; Xiao, Z.X.; Cao, Y. Cb-Dock2: Improved Protein-Ligand Blind Docking by Integrating Cavity Detection, Docking and Homologous Template Fitting. Nucleic Acids Res. 2022, 50, W159–W164. [Google Scholar] [CrossRef]
- Schiemer, J.; Horst, R.; Meng, Y.; Montgomery, J.I.; Xu, Y.; Feng, X.; Borzilleri, K.; Uccello, D.P.; Leverett, C.; Brown, S.; et al. Snapshots and Ensembles of Btk and Ciap1 Protein Degrader Ternary Complexes. Nat. Chem. Biol. 2021, 17, 152–160. [Google Scholar] [CrossRef]
- Tong, X.; Liu, X.; Tan, X.; Li, X.; Jiang, J.; Xiong, Z.; Xu, T.; Jiang, H.; Qiao, N.; Zheng, M. Generative Models for De Novo Drug Design. J. Med. Chem. 2021, 64, 14011–14027. [Google Scholar] [CrossRef]
- Sousa, T.; Correia, J.; Pereira, V.; Rocha, M. Generative Deep Learning for Targeted Compound Design. J. Chem. Inf. Model. 2021, 61, 5343–5361. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Validity | Uniqueness | Novelty | KL | FCD | |
|---|---|---|---|---|---|
| Length-controlled | 0.995 | 0.249 | 0.976 | 0.579 | 14.6 |
| LogP-controlled | 0.998 | 0.157 | 0.921 | 0.686 | 11.5 |
| Two properties controlled | 0.995 | 0.129 | 0.931 | 0.466 | 17.5 |
| Without RL | 0.989 | 0.404 | 0.968 | 0.879 | 7.48 |
| Valid | Unique | Novelty | FCD | KL | |
|---|---|---|---|---|---|
| Linker_Only | 0.146 | 0.973 | 1.00 | 15.63 | 0.71 |
| Linker_Aug | 0.848 | 0.587 | 0.973 | 6.96 | 0.86 |
| Linker_TL | 0.989 | 0.404 | 0.968 | 7.48 | 0.88 |
| Valid | Unique | Novelty | KL | FCD | |
|---|---|---|---|---|---|
| ChEMBL | 0.989 | 0.404 | 0.968 | 0.88 | 7.48 |
| ZINC | 0.979 | 0.333 | 0.998 | 0.86 | 7.71 |
| QM9 | 0.975 | 0.331 | 0.998 | 0.86 | 7.21 |
| Valid | Unique | Novelty | KL | FCD | |
|---|---|---|---|---|---|
| linker_SMILES | 0.989 | 0.304 | 0.968 | 0.87 | 7.03 |
| linker_SELFIES_1K | 1.0 | 0.874 | 1.0 | 0.94 | 3.606 |
| linker_SELFIES_10K | 1.0 | 0.619 | 1.0 | 0.911 | 3.576 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, Y.; Song, D.; Zhang, C.; Su, A. ProLinker–Generator: Design of a PROTAC Linker Base on a Generation Model Using Transfer and Reinforcement Learning. Appl. Sci. 2025, 15, 5616. https://doi.org/10.3390/app15105616
Luo Y, Song D, Zhang C, Su A. ProLinker–Generator: Design of a PROTAC Linker Base on a Generation Model Using Transfer and Reinforcement Learning. Applied Sciences. 2025; 15(10):5616. https://doi.org/10.3390/app15105616
Chicago/Turabian StyleLuo, Yanlin, Danyang Song, Chengwei Zhang, and An Su. 2025. "ProLinker–Generator: Design of a PROTAC Linker Base on a Generation Model Using Transfer and Reinforcement Learning" Applied Sciences 15, no. 10: 5616. https://doi.org/10.3390/app15105616
APA StyleLuo, Y., Song, D., Zhang, C., & Su, A. (2025). ProLinker–Generator: Design of a PROTAC Linker Base on a Generation Model Using Transfer and Reinforcement Learning. Applied Sciences, 15(10), 5616. https://doi.org/10.3390/app15105616