

Country-Based COVID-19 DNA Sequence Classification in Relation with International Travel Policy



Abstract
1. Introduction
2. Data and Method
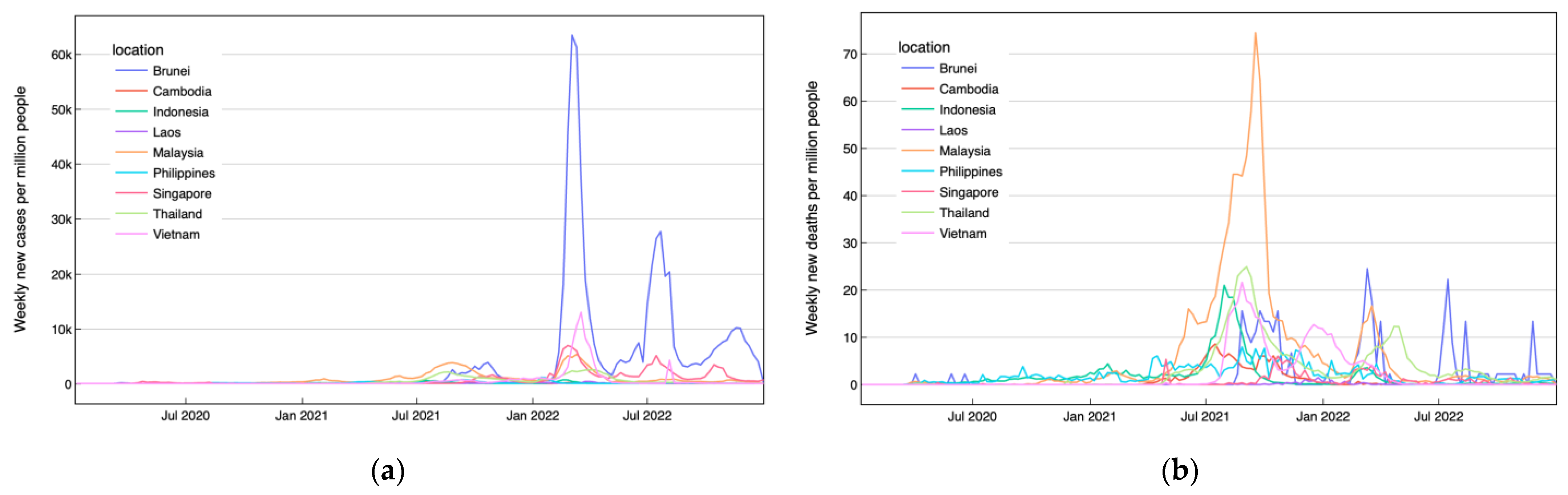
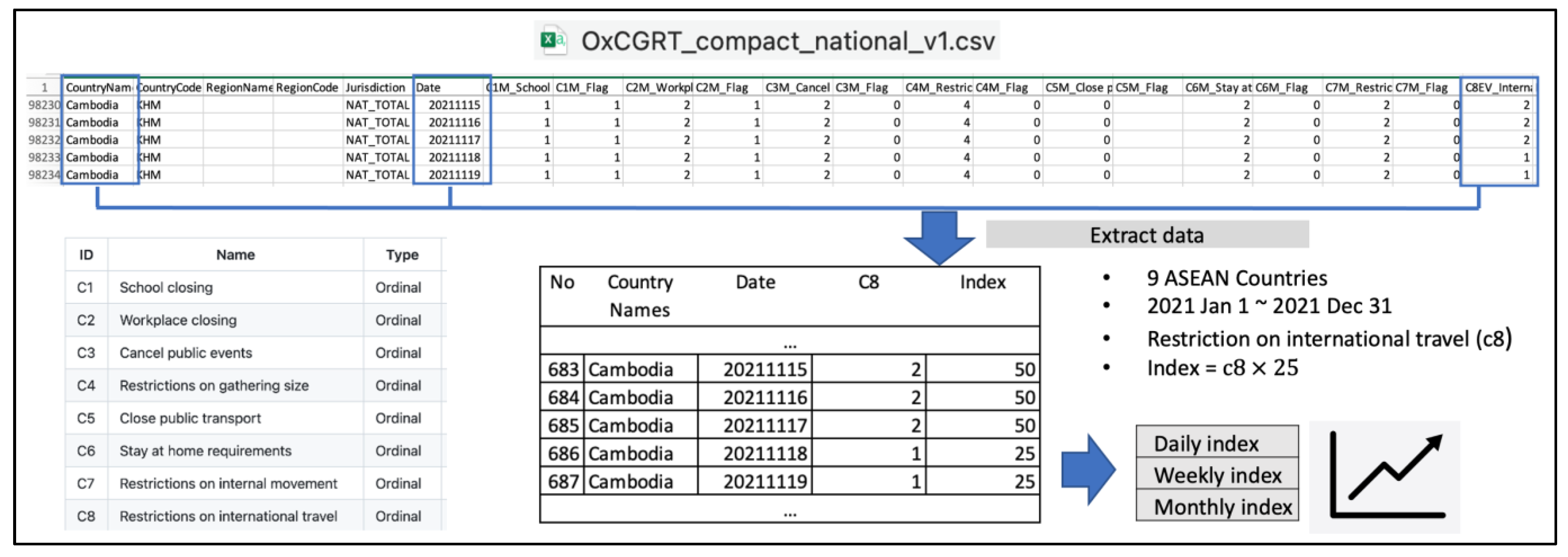
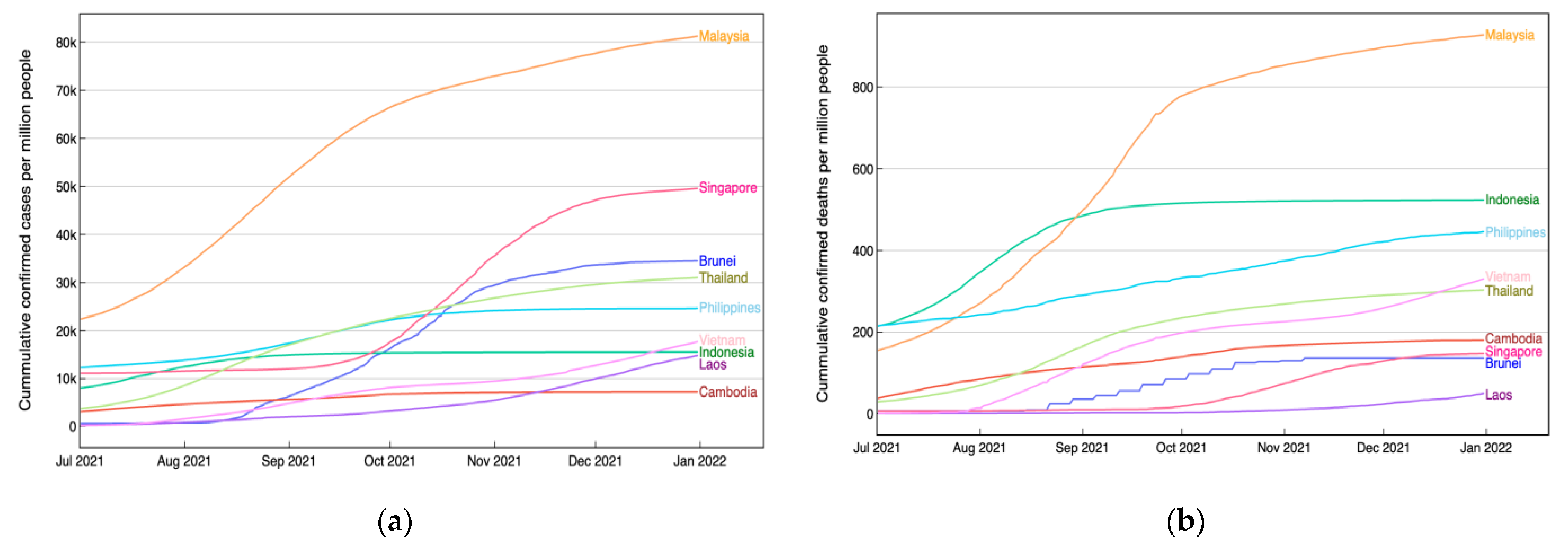
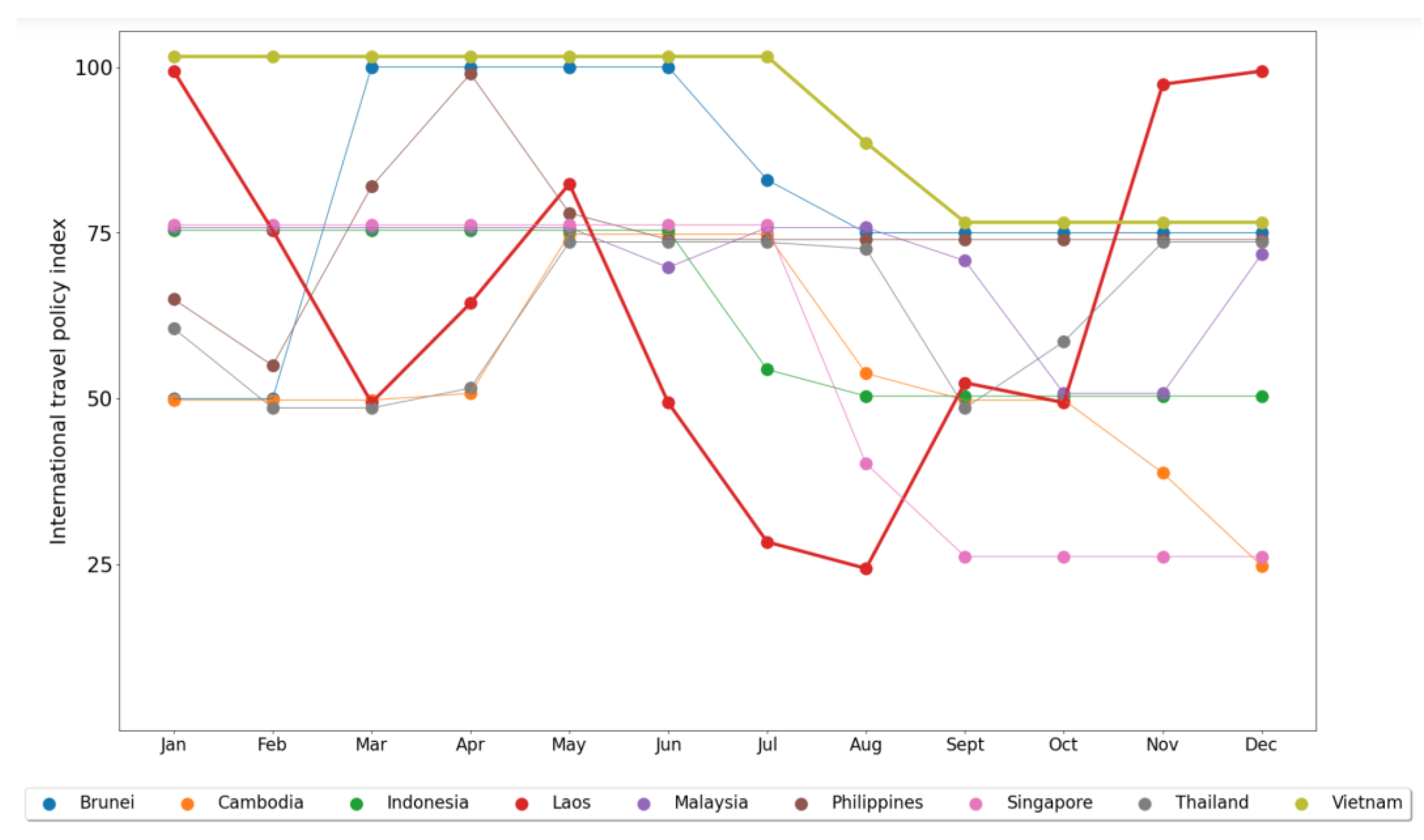
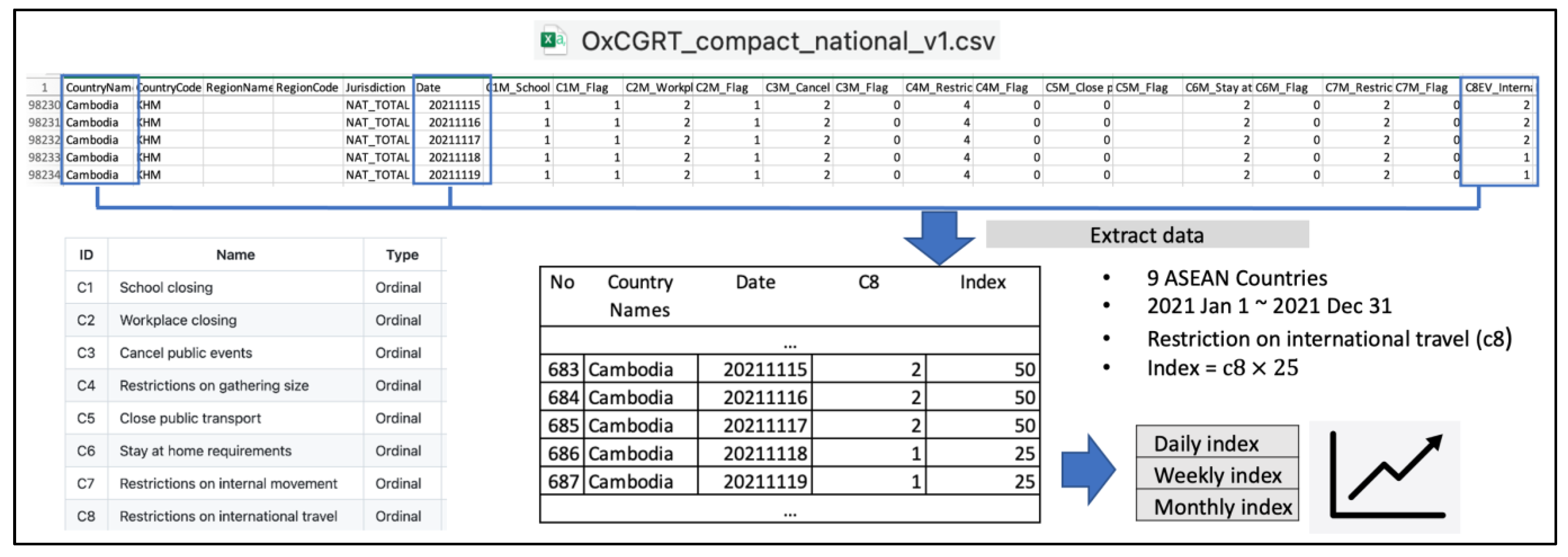
2.1. Data
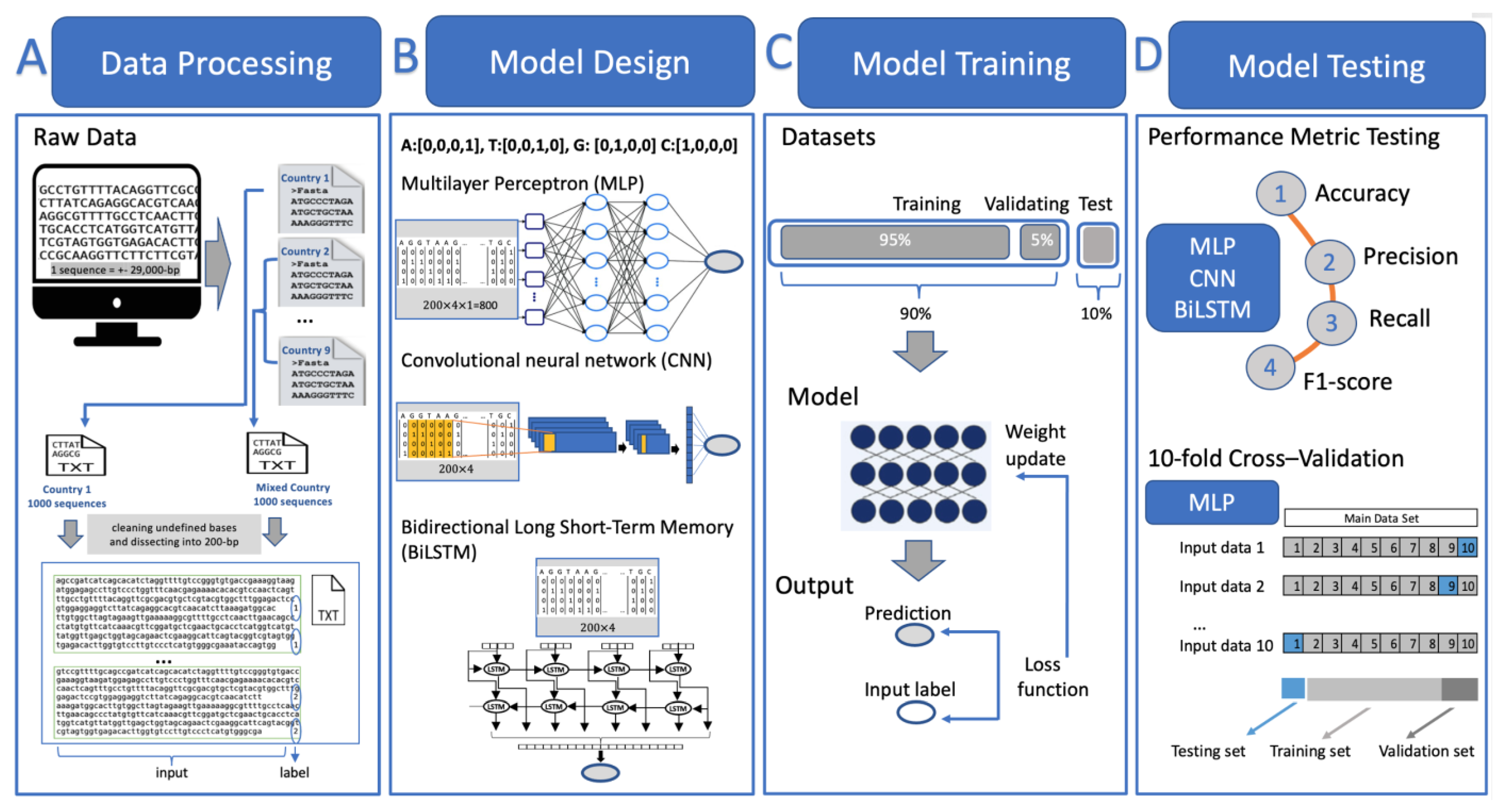
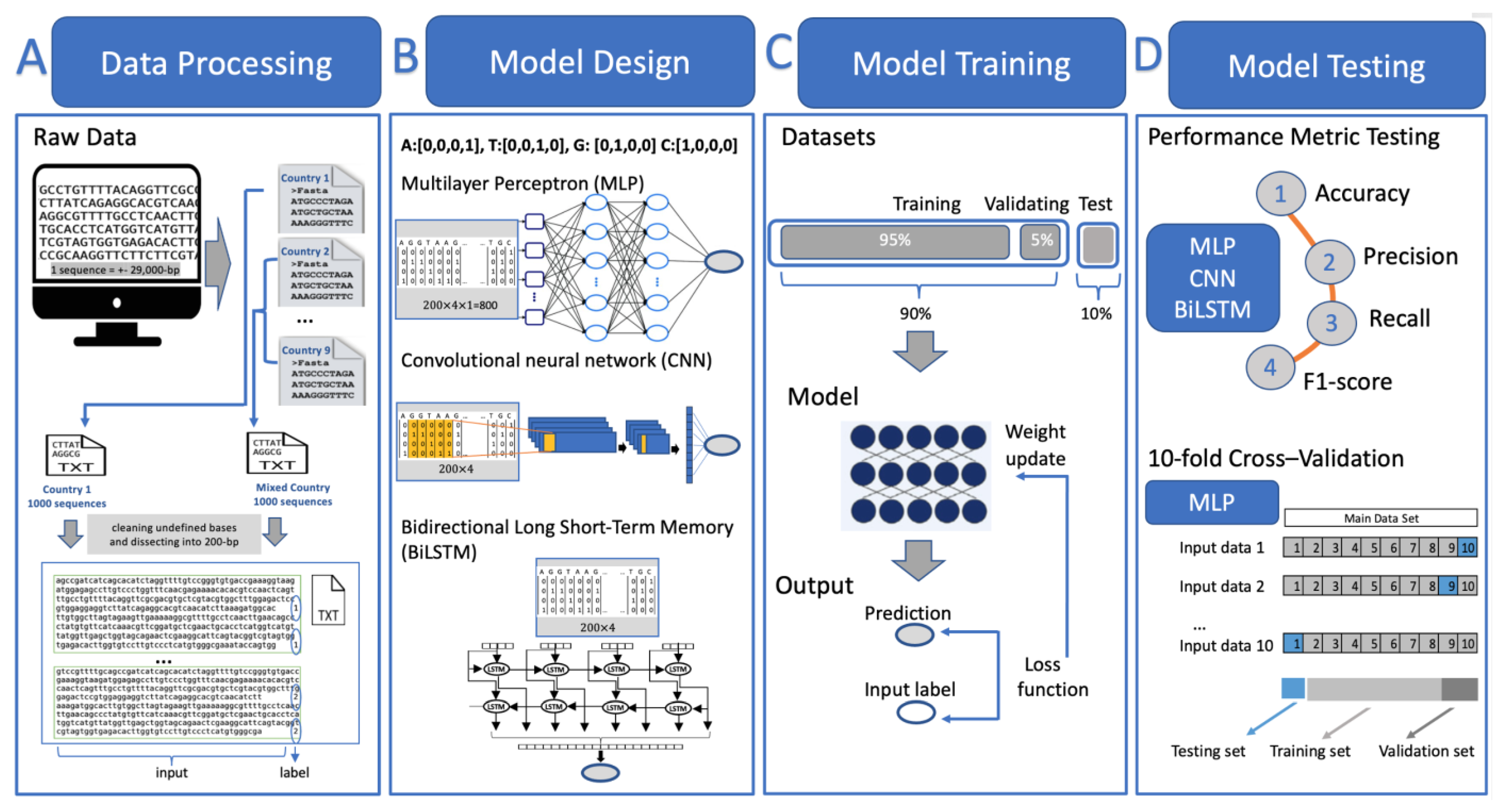
2.2. Method
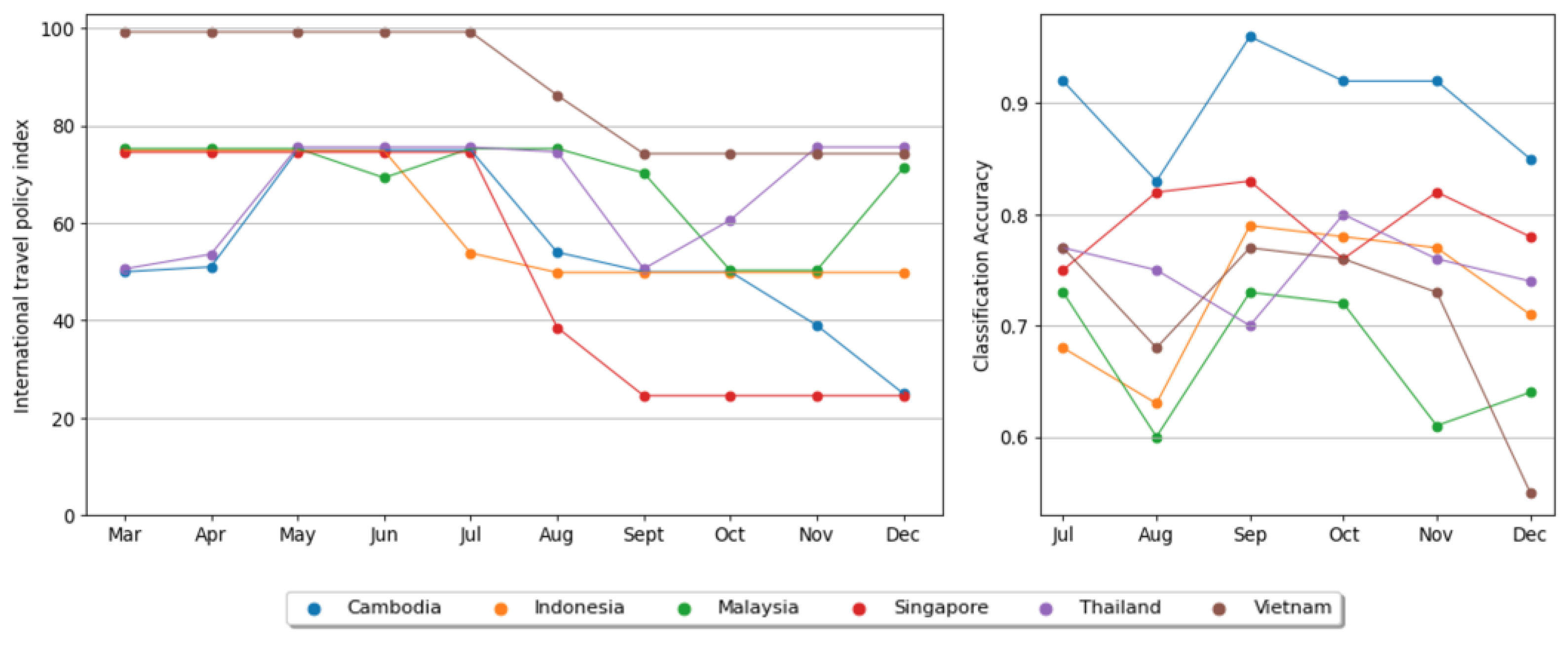
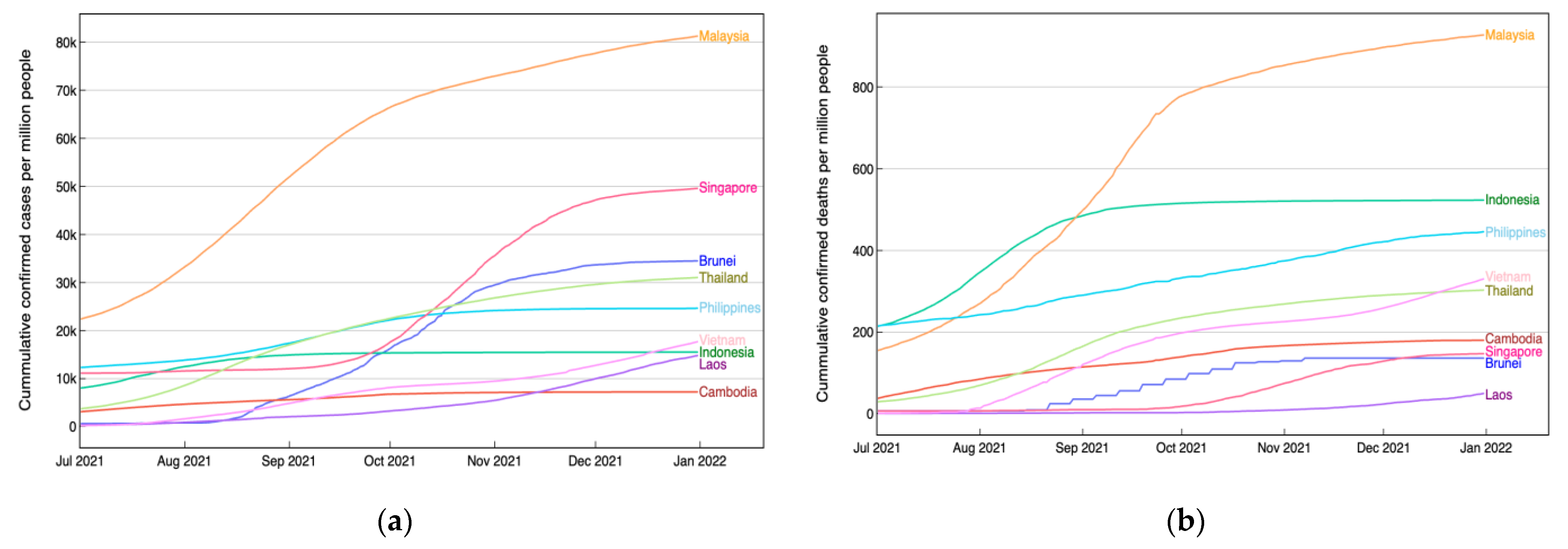
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jayakumar, P.; Brohi, S.N.; Jhanjhi, N.Z. Top 7 Lessons Learned from COVID-19 Pandemic. Authorea Prepr. 2020. preprints. [Google Scholar] [CrossRef]
- Rathnayaka, I.W.; Khanam, R.; Rahman, M.M. The Efficacy of Government Strategies to Control the COVID-19 Pandemic. Int. J. Sociol. Soc. Policy 2023, 44, 43–58. [Google Scholar] [CrossRef]
- Gunasekaran, H.; Ramalakshmi, K.; Arokiaraj, A.R.M.; Kanmani, S.D.; Venkatesan, C.; Dhas, C.S.G. Analysis of DNA Sequence Classification Using CNN and Hybrid Models. Comput. Math. Methods Med. 2021, 2021, 1835056. [Google Scholar] [CrossRef] [PubMed]
- El-Tohamy, A.; Maghwary, H.A.; Badr, N.L. A Deep Learning Approach for Viral DNA Sequence Classification Using Genetic Algorithm. Int. J. Adv. Comput. Sci. Appl. 2022, 13, 0130861. [Google Scholar] [CrossRef]
- Ahmed, I.; Jeon, G. Enabling Artificial Intelligence for Genome Sequence Analysis of COVID-19 and Alike Viruses. Interdiscip. Sci. Comput. Life Sci. 2021, 14, 504–519. [Google Scholar] [CrossRef]
- Basu, S.; Campbell, R.H. Classifying COVID-19 Variants Based on Genetic Sequences Using Deep Learning Models. bioRxiv 2021. preprint. [Google Scholar] [CrossRef]
- Ullah, A.; Malik, K.M.; Saudagar, A.K.J.; Khan, M.B.; Hasanat, M.H.A.; AlTameem, A.; Alkhathami, M.; Sajjad, M. COVID-19 Genome Sequence Analysis for New Variant Prediction and Generation. Mathematics 2022, 10, 4267. [Google Scholar] [CrossRef]
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.A.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The Evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 361–379. [Google Scholar] [CrossRef]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated Transmissibility and Impact of SARS-CoV-2 Lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef]
- Saxena, S.K.; Kumar, S.; Ansari, S.; Paweska, J.T.; Maurya, V.K.; Tripathi, A.K.; Abdel-Moneim, A.S. Transmission Dynamics and Mutational Prevalence of the Novel Severe Acute Respiratory Syndrome Coronavirus-2 Omicron Variant of Concern. J. Med. Virol. 2022, 94, 2160–2166. [Google Scholar] [CrossRef]
- Moya, A.; Holmes, E.C.; González-Candelas, F. The Population Genetics and Evolutionary Epidemiology of RNA Viruses. Nat. Rev. Microbiol. 2004, 2, 279–288. [Google Scholar] [CrossRef]
- Islam, M.R.; Hoque, M.N.; Rahman, M.S.; Alam, A.S.M.R.U.; Akther, M.; Puspo, J.A.; Akter, S.; Sultana, M.; Crandall, K.A.; Hossain, M.A. Genome-Wide Analysis of SARS-CoV-2 Virus Strains Circulating Worldwide Implicates Heterogeneity. Sci. Rep. 2020, 10, 14004. [Google Scholar] [CrossRef]
- Namazi, H.; Dawi, N.B.M. Information and complexity-based analysis of the variations of the coronavirus genome between different countries. Fractals 2020, 28, 2050134, Correction in Sci. Rep. 2021, 11, 20568. [Google Scholar] [CrossRef]
- Sarkar, R.; Mitra, S.; Chandra, P.; Saha, P.; Banerjee, A.; Dutta, S.; Chawla-Sarkar, M. Comprehensive Analysis of Genomic Diversity of SARS-CoV-2 in Different Geographic Regions of India: An Endeavour to Classify Indian SARS-CoV-2 Strains on the Basis of Co-Existing Mutations. Arch. Virol. 2021, 166, 801–812. [Google Scholar] [CrossRef]
- Toyoshima, Y.; Nemoto, K.; Matsumoto, S.; Nakamura, Y.; Kiyotani, K. SARS-CoV-2 Genomic Variations Associated with Mortality Rate of COVID-19. J. Hum. Genet. 2020, 65, 1075–1082. [Google Scholar] [CrossRef]
- Eaaswarkhanth, M.; Madhoun, A.A.; Al-Mulla, F. Could the D614G Substitution in the SARS-CoV-2 Spike (S) Protein Be Associated with Higher COVID-19 Mortality? Int. J. Infect. Dis. 2020, 96, 459–460. [Google Scholar] [CrossRef]
- Becerra-Flores, M.; Cardozo, T. SARS-CoV-2 Viral Spike G614 Mutation Exhibits Higher Case Fatality Rate. Int. J. Clin. Pract. 2020, 74, e13525. [Google Scholar] [CrossRef] [PubMed]
- Omais, S.; Kharroubi, S.A.; Zaraket, H. No Association between the SARS-CoV-2 Variants and Mortality Rates in the Eastern Mediterranean Region. medRxiv 2021. preprints. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.; Hill, V.; McCrone, J.; Price, A.; Jorgensen, D.A.; O’Toole, Á.; Southgate, J.; Johnson, R.A.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75.e11. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence That D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef] [PubMed]
- Abadi, S.A.R.; Mohammadi, A.; Koohi, S. A New Profiling Approach for DNA Sequences Based on the Nucleotides’ Physicochemical Features for Accurate Analysis of SARS-CoV-2 Genomes. BMC Genom. 2023, 24, 266. [Google Scholar] [CrossRef]
- Chu, D.-T.; Ngoc, S.-M.V.; Thi, H.V.; Thi, Y.-V.N.; Ho, T.-T.; Hoang, V.-T.; Singh, V.; Al-Tawfiq, J.A. COVID-19 in Southeast Asia: Current Status and Perspectives. Bioengineered 2022, 13, 3797–3809. [Google Scholar] [CrossRef]
- Yap, P.S.X.; Tan, T.S.; Chan, Y.F.; Tee, K.K.; Kamarulzaman, A.; Teh, C.S.J. An Overview of the Genetic Variations of the SARS-CoV-2 Genomes Isolated in Southeast Asian Countries. J. Microbiol. Biotechnol. 2020, 30, 962–966. [Google Scholar] [CrossRef]
- Muflikhah, L.; Rahman, M.A.; Widodo, A. Profiling DNA Sequence of SARS-CoV-2 Virus Using Machine Learning Algorithm. Bull. Electr. Eng. Informatics 2022, 11, 1037–1046. [Google Scholar] [CrossRef]
- GISAID—Gisaid.org. GISAID. Available online: https://gisaid.org/ (accessed on 14 October 2023).
- Mathieu, E. Coronavirus Pandemic (COVID-19). Our World in Data. Available online: https://ourworldindata.org/coronavirus (accessed on 6 May 2023).
- COVID-19 Government Response Tracker. Blavatnik School of Government. Available online: https://www.bsg.ox.ac.uk/research/covid-19-government-response-tracker (accessed on 13 December 2023).
- Amul, G.G.H.; Ang, M.; Kraybill, D.; Ong, S.E.; Yoong, J. Responses to COVID-19 in Southeast Asia: Diverse Paths and Ongoing Challenges. Asian Econ. Policy Rev. 2021, 17, 90–110. [Google Scholar] [CrossRef]
- Tegally, H.; Moir, M.; Everatt, J.; Giovanetti, M.; Scheepers, C.; Wilkinson, E.; Subramoney, K.; Makatini, Z.; Moyo, S.; Amoako, D.G.; et al. Emergence of SARS-CoV-2 Omicron Lineages BA.4 and BA.5 in South Africa. Nat. Med. 2022, 28, 1785–1790. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Ranganathan, P. Common Pitfalls in Statistical Analysis: The Use of Correlation Techniques. Perspect. Clin. Res. 2016, 7, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, B. “p < 0.05” Might Not Mean What You Think: American Statistical Association Clarifies p Values. JNCI J. Natl. Cancer Inst. 2016, 108, djw194. [Google Scholar] [CrossRef] [PubMed]
- Lü, Y.; Belitskaya-Levy, I. The Debate about p-Values. PubMed 2015, 27, 381–385. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Country Name | Number of Sequences Submitted in: | |||||
|---|---|---|---|---|---|---|
| January–June 2020 | July–December 2020 | January–June 2021 | July–December 2021 | January–June 2022 | July–December 2022 | |
| Brunei | 5 | 2 | 2 | 488 | 118 | 54 |
| Cambodia | 20 | 26 | 460 | 1487 | 578 | 86 |
| Indonesia | 204 | 581 | 4186 | 4428 | 715 | 332 |
| Laos | - | - | - | 862 | 233 | 12 |
| Malaysia | 165 | 326 | 1341 | 5777 | 2800 | 848 |
| Philippines | 29 | 337 | 4293 | 3292 | 24 | 148 |
| Singapore | 1092 | 363 | 1664 | 7171 | 969 | 303 |
| Thailand | 307 | 144 | 2161 | 6532 | 1277 | 398 |
| Vietnam | 121 | 55 | 275 | 1658 | 411 | 15 |
| Country Name | The Distribution Percentage of Variant: | |||||
|---|---|---|---|---|---|---|
| Delta | Omicron | Alpha | Beta | Gamma | Others | |
| Brunei | 100.00% | |||||
| Cambodia | 72.02% | 27.98% | ||||
| Indonesia | 97.70% | 0.07% | 0.02% | 2.21% | ||
| Laos | 99.65% | 0.23% | 0.12% | |||
| Malaysia | 98.67% | 0.09% | 0.31% | 0.93% | ||
| Philippines | 70.47% | 13.61% | 12.52% | 3.40% | ||
| Singapore | 99.78% | 0.13% | 0.03% | 0.01% | 0.06% | |
| Thailand | 94.32% | 0.05% | 5.10% | 0.38% | 0.15% | |
| Vietnam | 100.00% | |||||
| Model | Main Layer: Unit/Filter [Dropout] | Fully Connected Layer: Unit, Activation, Dropout | Output Layer | Hyperparameters: Optimizer, Learning Rate, Batch Size |
|---|---|---|---|---|
| MLP | Dense (6 layers): 400, 300, 200, 100, 50, 10 [0.3] | ReLU | Dense (2, softmax) | Adam, 0.001, 1000 |
| CNN | Conv1D (3 layers): 100, 100, 80. MaxPooling1D (3 layers): 4, 2, 2 [0.2]. | 20, ReLU, 0.5 | Dense (2, softmax) | Adam, 0.001, 1000 |
| BiLSTM | BiLSTM (1 layer): 80 [0.2] | 20, ReLU, 0.5 | Dense (2, softmax) | Adam, 0.001, 1000 |
| Country Name | MLP Model | CNN Model | BiLSTM Model | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Acc. | Prec. | Recall | F1-Score | Time | Acc. | Prec. | Recall | F1-Score | Time | Acc. | Prec. | Recall | F1-Score | Time | |
| Brunei | 0.98 | 0.98 | 0.98 | 0.98 | 00:00:58 | 0.98 | 0.98 | 0.98 | 0.98 | 00:02:09 | 0.98 | 0.98 | 0.98 | 0.98 | 0:12:17 |
| Cambodia | 0.85 | 0.87 | 0.85 | 0.85 | 00:01:18 | 0.85 | 0.87 | 0.85 | 0.85 | 00:05:16 | 0.84 | 0.86 | 0.84 | 0.83 | 0:13:29 |
| Indonesia | 0.69 | 0.71 | 0.69 | 0.69 | 00:01:35 | 0.71 | 0.72 | 0.71 | 0.70 | 00:08:54 | 0.69 | 0.7 | 0.69 | 0.69 | 0:23:30 |
| Laos | 0.84 | 0.86 | 0.84 | 0.83 | 00:01:22 | 0.86 | 0.88 | 0.86 | 0.86 | 00:06:14 | 0.85 | 0.88 | 0.85 | 0.85 | 0:10:50 |
| Malaysia | 0.66 | 0.68 | 0.66 | 0.65 | 00:01:26 | 0.70 | 0.72 | 0.7 | 0.69 | 00:11:15 | 0.66 | 0.67 | 0.66 | 0.65 | 0:18:12 |
| Philippines | 0.81 | 0.82 | 0.81 | 0.80 | 00:01:47 | 0.83 | 0.84 | 0.83 | 0.82 | 00:07:58 | 0.81 | 0.83 | 0.81 | 0.81 | 0:20:47 |
| Singapore | 0.79 | 0.79 | 0.79 | 0.79 | 00:01:34 | 0.82 | 0.82 | 0.82 | 0.82 | 00:11:22 | 0.78 | 0.8 | 0.78 | 0.77 | 0:19:29 |
| Thailand | 0.79 | 0.79 | 0.79 | 0.79 | 00:01:40 | 0.80 | 0.80 | 0.80 | 0.80 | 00:07:27 | 0.80 | 0.80 | 0.80 | 0.80 | 0:14:34 |
| Vietnam | 0.70 | 0.71 | 0.70 | 0.70 | 00:01:38 | 0.73 | 0.74 | 0.73 | 0.72 | 00:10:02 | 0.70 | 0.71 | 0.70 | 0.69 | 0:13:30 |
| Country Name | Average of Accuracy for All Folds | Standard Deviation | Execution Time |
|---|---|---|---|
| Brunei | 97.97 | 0.05 | 00:16:06 |
| Cambodia | 85.22 | 0.23 | 00:14:07 |
| Indonesia | 66.72 | 0.52 | 00:16:08 |
| Laos | 83.44 | 0.27 | 00:18:33 |
| Malaysia | 65.07 | 0.44 | 00:16:46 |
| Philippines | 78.37 | 0.58 | 00:20:27 |
| Singapore | 78.64 | 0.22 | 00:22:03 |
| Thailand | 78.38 | 0.30 | 00:18:00 |
| Vietnam | 67.01 | 0.31 | 00:21:02 |
| i Months Before | Cambodia | Indonesia | Malaysia | Singapore | Thailand | Vietnam | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| r | p-Value | r | p-Value | r | p-Value | r | p-Value | r | p-Value | r | p-Value | |
| i = 4 | 0.28 | 0.17 | −0.48 | 0.02 | −0.10 | 0.58 | 0.34 | 0.09 | 0.07 | 0.71 | 0.49 | 0.01 |
| i = 3 | 0.04 | 0.83 | −0.50 | 0.01 | −0.40 | 0.06 | −0.03 | 0.88 | 0.01 | 0.97 | 0.77 | 0.00 |
| i = 2 | 0.08 | 0.68 | −0.50 | 0.01 | 0.32 | 0.12 | 0.26 | 0.21 | −0.34 | 0.09 | 0.20 | 0.34 |
| i = 1 | −0.16 | 0.43 | −0.41 | 0.05 | 0.55 | 0.00 | 0.05 | 0.80 | −0.37 | 0.07 | 0.50 | 0.01 |
| i = 0 | 0.31 | 0.13 | −0.15 | 0.48 | 0.02 | 0.92 | −0.18 | 0.37 | 0.34 | 0.09 | 0.20 | 0.35 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khatizah, E.; Park, H.-S. Country-Based COVID-19 DNA Sequence Classification in Relation with International Travel Policy. Appl. Sci. 2024, 14, 1916. https://doi.org/10.3390/app14051916
Khatizah E, Park H-S. Country-Based COVID-19 DNA Sequence Classification in Relation with International Travel Policy. Applied Sciences. 2024; 14(5):1916. https://doi.org/10.3390/app14051916
Chicago/Turabian StyleKhatizah, Elis, and Hyun-Seok Park. 2024. "Country-Based COVID-19 DNA Sequence Classification in Relation with International Travel Policy" Applied Sciences 14, no. 5: 1916. https://doi.org/10.3390/app14051916
APA StyleKhatizah, E., & Park, H.-S. (2024). Country-Based COVID-19 DNA Sequence Classification in Relation with International Travel Policy. Applied Sciences, 14(5), 1916. https://doi.org/10.3390/app14051916