1. Introduction
The use of pesticides in modern industrial agriculture is nearly ubiquitous, as they are essential in maintaining food security for the increasingly urban world population [
1]. However, a large number of currently used pesticides have been found to be detrimental to ecosystem health and prosperity. Furthermore, several pesticides with adverse effects are known to persist in soils for a long time after use, such as some banned organochlorines [
2]. Recent studies have pointed to the large amount of pesticides present in agricultural soils, including the very commonly used herbicide glyphosate [
3,
4,
5]. Due to its permanently ionic nature, glyphosate (
Figure 1) cannot be analyzed in multiresidue methods, and is often quantified separately [
3]. To our knowledge, no method has ever been published which could determine permanently ionic pesticides and their degradation products (glyphosate, aminomethylphosphonic acid, glufosinate) along with other pesticide residues: This work presents a first attempt.
In general, methods for the analysis of pesticides in soil need to reach very low detection limits, due to the small quantities of these analytes often present in agricultural soils (commonly in the µg/kg range) [
6], and also need to have a certain robustness in order to be able to extract the analytes from soils with different physical and chemical properties, the most important of which are soil texture, pH, and organic matter content [
6]. In terms of GC-MS analysis, it is important to obtain an extract which is relatively free from matrix interferents, most notably those which are not detected by the technique, e.g., non-volatile compounds such as inorganic salts, which can compromise the instrument over many runs, causing among other things adsorption of the pesticides and eventual poor peak shape. Also, moderately volatile and/or polar matrix components can cause matrix-induced response enhancement, which results in the sample peaks being significantly larger and more symmetrical than those injected in pure solvent. Generally, matrix-matching calibration has been used to mitigate this problem, but the use of analyte protectants has also been proposed, in which moderately volatile compounds such as some sugars are added to the sample in order to “protect” the analytes from chemical adsorption, resulting in the same effect as matrix-induced response enhancement [
7], both in the samples and calibration solutions.
When designing a method which could quantify glyphosate along with other commonly used pesticides, several avenues were considered. Since the extraction of glyphosate from soil is always performed with an aqueous solution [
8], two serial extractions would always be needed, since low-polarity analytes will not readily extract onto aqueous phases. Furthermore, since glyphosate extraction is commonly performed with very basic solutions (either with KOH or aqueous NH
3), and some pesticides are known to degrade under basic pH [
9], it was thought that the aqueous extraction would have to occur last. Thus, the idea was to modify a previously existent multiresidue method which can extract organochlorines, organophosphates, triazoles, among others, and then, perform the glyphosate extraction. The most important condition would be that the first extraction did not degrade or modify the sample to an extent that the glyphosate extraction would be impaired. Thus, QuEChERS was ruled out, since it requires the addition of salts, which would have made the aqueous extraction impossible.
The original conception was to perform ultrasound-assisted extraction (UAE) with an organic solvent, remove the entirety of the extract volume, and then, add the aqueous solution for glyphosate extraction. However, UAE has several drawbacks and does not inherently provide significant concentration factors [
6]. Also, the removal of the entire extract solvent volume is operationally challenging, even after centrifugation, and could induce repeatability problems. Since a method for the multiresidue analysis of pesticides from soil using direct-immersion solid-phase microextraction (DI-SPME) was being developed [
10], it was thought that it could be adapted to provide the extraction for this combined methodology.
The employment of direct-immersion SPME for soil analysis is made possible by a new type of semi-disposable fiber called SPME LC-Tips [
11,
12]. These fibers present one major advantage over traditional SPME, namely, their much reduced cost. Whereas commonly used SPME assemblies would quickly degrade when in contact with a stirred soil slurry (via abrasion), and thus, only be usable for a few samples, resulting in extremely high per-sample costs, these new fibers, although they can be used only one to three times, still provide a cost-effective analysis which rivals commonly used methodologies such as QuEChERS [
13]. The major downside of this technique is that, unlike traditional SPME where the fiber can undergo direct desorption onto the GC inlet [
14], SPME LC-Tips require solvent desorption prior to injection onto the GC or LC. This greatly reduces the concentration factor of the extraction, because only a small percentage of the solvent extract is injected onto the chromatographic system, whereas for direct thermal desorption nearly the entire analyte mass on the fiber can be introduced onto the column. An attractive (but costly) possibility for direct desorption using LC-Tips is to use direct analysis in real time coupled with mass spectrometry (DART-MS), especially using a high-resolution mass analyzer to differentiate analytes with similar masses, where the pesticides trapped in the fiber are desorbed and ionized by a heated stream of excited and ionized helium or nitrogen, allowing their analysis with increased sensitivity and without the need for solvent retroextraction, chromatographic separation, or the increase in soil sample mass [
15]. Nevertheless, for the proposed combined method, solvent desorption is ideal, as the SPME extract can easily be combined with the glyphosate extract.
2. Materials and Methods
2.1. Soil Sampling
The soil sample was collected from Idanha-a-Nova Municipality in Portugal (39′8454º N, 7′2544º W), at a depth of 25–30 cm. The land where the soil was sampled used to grow tobacco. Once in the laboratory, samples were sieved through a 2 mm mesh and allowed to air-dry at 22 ºC. The soil had a sandy-loam texture (25% coarse sand, 40.2% fine sand, 17.7% silt, and 17.1% clay), and 3.3% organic matter (st. deviation = 0.3, n = 5). Soil pH was 7.73 ± 0.06 (4 g with 10 mL of milli-Q water, shaken for 1 h, n = 3) and 6.94 ± 0.05 (4 g with 10 mL of 1 M KCl in milli-Q water, shaken for 1 h, n = 3).
2.2. Standards and Chemicals
Individual standards for 2-methyl-4-chlorophenoxyacetic acid (MCPA), buprofezin, 2,4-dichlorophenoxyacetic acid (2,4-D), aminomethylphosphonic acid (AMPA), boscalid, chlorpyrifos, diflufenican, epoxiconazole, glyphosate, malathion, metalaxyl, metazachlor, metolachlor, penconazole, prosulfocarb tebuconazole, tefluthrin, tetraconazole, and terbuthylazine were of analytical grade, obtained from Sigma-Aldrich (Steinheim, Germany). A multiresidue standard for organochlorine pesticides (at 100 µg/mL each) was obtained from Restek (Bellefonte, PA, USA). Penconazole-d7 was obtained from Toronto Research Chemicals (Toronto, ON, Canada). Glyphosate 1,2-
13C
2-
15N was obtained from Dr. Ehrenstorfer (Augsburg, Germany). Water used in the extraction and dilutions was ultrapure, produced in a Milli-Q plus system from Millipore (Bedford, MA, USA). Absolute ethanol (EtOH) and methanol were of HPLC grade, obtained from Honeywell (Charlotte, NC, USA). Dichloromethane was GC-MS grade, purchased from Carlo-Erba (Emmendingen, Germany). The remaining materials were all purchased from Sigma-Aldrich, namely, C
18 SPME LC-Tips, trifluoroacetic anhydride and trifluoroethanol (≥99% purity), 25% aqueous ammonia solution (p. a. grade), 3-ethoxy-1,2-propanediol, gulonolactone, and D-sorbitol (analyte protectants) of purities 98%, 95%, and 99%, respectively. Stock solutions for the pesticides were prepared in methanol at 250 µg/mL, and kept refrigerated at −20 ºC, for at most one month. Dilutions for injection and soil spiking were performed in methanol. A stock solution of the three analyte protectants was prepared in a 50/50 (
v/
v) solution of ethanol–methanol at 1000 µg/mL each [
7]. The internal standard solution (penconazole-d7 and glyphosate 1,2-
13C
2-
15N) was prepared with 80/20 (
v/
v) methanol–water at 800 ng/mL each.
2.3. GC-MS/MS Analysis
The analyses were performed by gas chromatography–tandem mass spectrometry (GC-MS/MS) on a Bruker GC 456 and a Bruker Scion TQ (Triple Quadrupole) system equipped with a CTC CombiPAL automatic injector and a programmable temperature vaporizer (PTV) inlet (Bruker 1079). Data were acquired with Bruker MSWS 8.2 and analyzed with Bruker MS Data Review 8.0. Chromatographic separation was achieved on a ZB-5MS Plus capillary column (20 m × 0.18 mm i.d., 0.18 µm df). The oven temperature program began at 40 ºC, where it was held for 3 min, increased at 20 ºC/min to 140 ºC, then 4 ºC/min to 250 ºC, and finally, 20 ºC/min to 300 ºC, where it was held for 1 min. Helium of 99.9999% purity was used as the carrier gas at a constant flow rate of 0.7 mL/min. The injection volume was 5 µL, performed in PTV large-volume mode, starting at 80 ºC with a split ratio of 1:120, held for 30 s, then splitless and with a temperature increase of 200 ºC/min to 270 ºC. At 3 min, the split valve was opened at a ratio of 1:60, and after 3 more minutes reduced to 15 mL/min and held for the entire run. The mass spectrometer system was operated in multiple-reaction monitoring (MRM), with argon as collision gas at 2.4 mTorr. The transfer line was held at 290 ºC, and the ion source at 270 ºC. The solvent delay was set to 7 min. MRM transitions associated with the selected precursor and product ion pairs of the analytes can be found in the
Supplementary Material.
2.4. Final Method
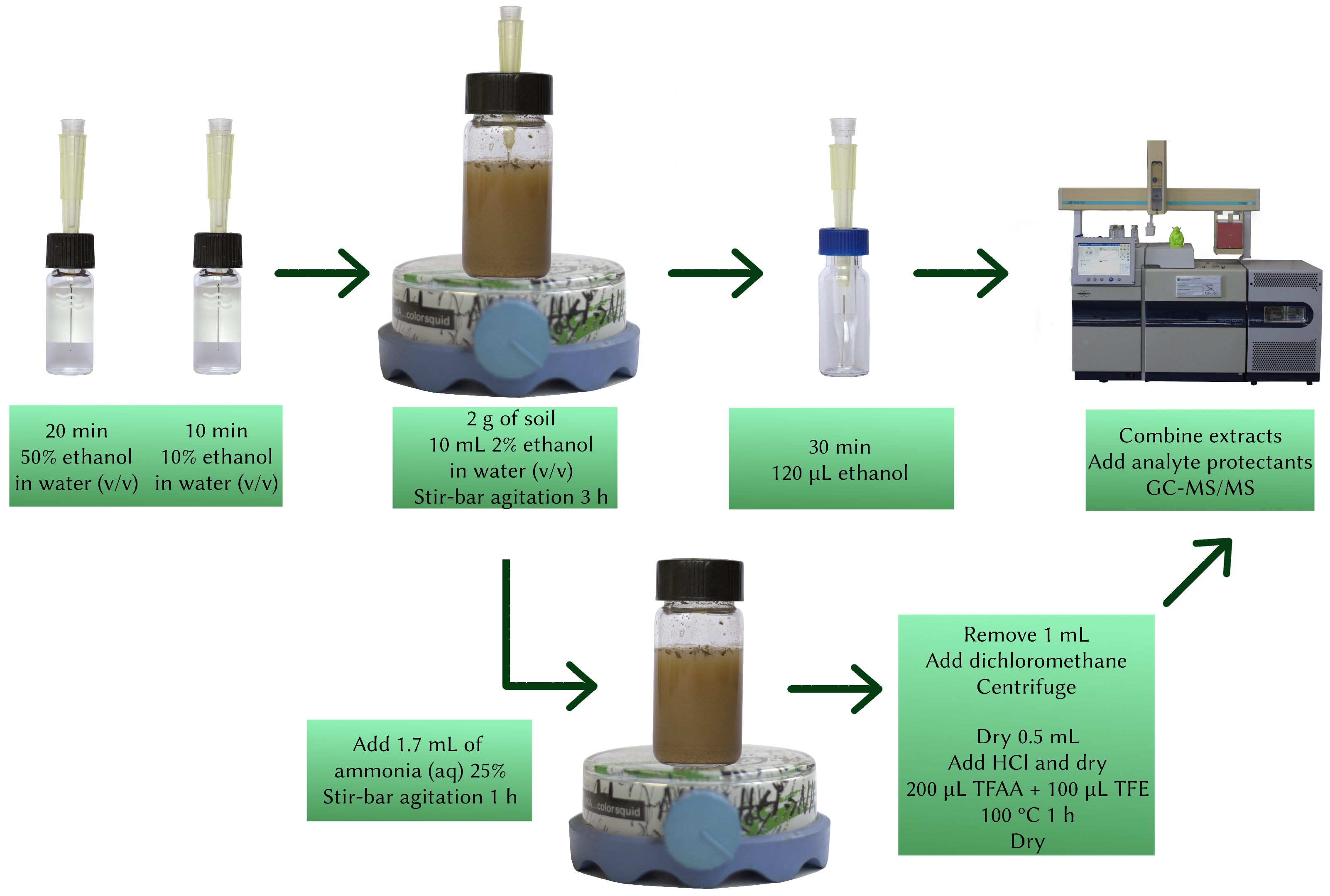
A mass of 2 g of soil sample was weighed onto the extraction vial, to which was added 50 µL of internal standard solution, and it was allowed to air-dry for 1 h. C18 LC-Tip SPME fibers were conditioned by being inserted into a 2 mL glass vial containing 50/50 (v/v) ethanol–water for 20 min, followed by re-equilibration in another 2 mL glass vial containing water with 10% ethanol (v/v) for 10 min, under constant agitation at 300 rpm.
DI-SPME extraction: A volume of 10 mL of 2% ethanol in water (v/v) was added to the extraction vial, then the conditioned fiber was inserted into this solution, which was agitated for 3 h at 1000 rpm with a magnetic stir-bar. Then, the fiber was removed and immediately inserted into a 300 µL vial containing 120 µL of ethanol, and retroextracted for 30 min at 300 rpm, after which it was removed and the extract was stored in the fridge.
Glyphosate extraction: As soon as the fiber was removed from the extraction vial, 1.7 mL of 25% aqueous ammonia solution was added to it, and the soil was stirred at 1200 rpm for 1 h, at the same time that the fiber was being retroextracted. Afterwards, 1 mL of the aqueous solution from the extraction vial was added to a 2 mL Eppendorf tube, and 50 µL of dichloromethane was added, vortexed for 10 s, and centrifuged for 5 min at 4000 rpm. Then, 500 µL of the supernatant were transferred to a 1 mL reaction vial (Thermo Scienfitic, Waltham MA, USA), and completely dried under a nitrogen stream at 60 ºC. Then, 20 µL of 18% (m/m) aqueous hydrochloric acid was added to the reaction vial, and re-dried under nitrogen at 60 ºC. Afterwards, 200 µL of trifluoroacetic acid and 100 µL of trifluoroethanol were added to the reaction vials, and they were maintained at 100 ºC for 1 h. After being allowed to cool to room temperature, the vials were opened and gently dried under nitrogen.
Final extract: The ethanolic fiber extracts were removed from the fridge and allowed to return to room temperature, after which 80 µL of the extract was added to the dry reaction vial, which was then closed, vortexed for 10 s, and sonicated for 1 min. Finally, 20 µL of the analyte protectant solution was added to the reaction vial and the mixture was removed onto an autosampler vial and analyzed by GC-MS/MS. A graphical representation of the method can be seen in
Figure 2.
2.5. Method Performance
Calibration curves were performed by spiking various 2 g samples with the appropriate amount of standard containing all the analytes to obtain seven different concentrations in the range of 0.1–100 µg per kg of soil, and then, drying the soil under a very gentle nitrogen stream, before following the full method described above. Each concentration was extracted in triplicate, except for 20 µg/kg, which was extracted five times, for the repeatability and recovery calculations. In order to determine recovery, a calibration was performed for glyphosate without internal standard, in the range of 0.1–10 ng, added directly to the derivatization vial, and then, dried under a gentle nitrogen stream.
3. Results and Discussion
3.1. Multiclass Pesticide Extraction by Immersion SPME
The pesticide selection chosen for the method development included several pesticide classes: Chloroacetamides (metazachlor and metolachor), organochlorines (aldrin, lindane and pentachloroanisole), organophosphates (malathion and chlorpyrifos), triazoles (tetraconazole, penconazole and tebuconazole), as well as one pyrethroid, thiocarbamate, and unclassified pesticides, namely, tefluthrin, prosulfocarb, and buprofezin. The aim was to gauge the applicability of the developed method towards different chemical properties.
The original multiclass extraction method which was adapted used 6% methanol in water (
v/
v) as a solvent for immersion SPME, followed by desorption of the fiber in methanol and subsequent injection [
10]. In the present work, ethanol was used instead, mainly because it has a much smaller toxicity (negligible for humans considering normal laboratory exposure levels), can be produced more easily from renewable feedstocks, and is slightly less polar, which is relevant for the extraction of organochlorines, especially. Furthermore, most salts have lower solubility in ethanol than methanol [
16], which is important if in the future this method is applied to soils with greater salt content, where the use of methanol for redissolution of the derivatized glyphosate extract might introduce salts into the chromatographic system. From the two fiber chemistries available, PDMS/DVB and C18, the latter was chosen because the method introduces the analysis of organochlorines, not contemplated in the previous work [
10].
Initially, a three factor Box–Behnken design was used as an attempt to optimize the immersion SPME extraction It modeled the percentage of ethanol in the extraction solvent (2%, 6%, and 10%,
v/
v), the extraction time (60, 90, and 120 min), and the retroextraction volume (100, 120, and 140 µL). However, the results showed that for most analytes, the extraction time was by far the most relevant factor, as it seemed that even 120 min were insufficient to attain equilibrium. Thus, this factor was far outside the values at which a response surface might show a maximum. As a result, the retroextraction volume was fixed to 120 µL (as a compromise between obtaining a good concentration factor and enough working volume), and a two-factor, three-level full-factorial experimental design was constructed (nine experiments), where the percentages of ethanol in the extraction solvent were 2, 6, and 10%, and the extraction times were 120, 150, and 180 min. Each experiment was repeated three times, for a total of 27 runs. The results (in terms of maximum average signal) are presented in
Table 1.
From the results, 180 min was selected as the extraction time. A long extraction time is not ideal, because the full method including glyphosate extraction and derivatization takes several hours, and at worst can be longer than a single 8 h shift, which would compromise its applicability in routine laboratories without the implementation of process automation. The results for just 180 min of extraction can be seen in
Figure 3.
From these results, 2% ethanol in water was chosen as the extraction solvent. Although the more nonpolar compounds were favored by the greater percentage of ethanol, most had the highest signals at 2%. Furthermore, a smaller amount of ethanol would be favorable for the subsequent glyphosate extraction, as the organic modifier would not help in the dissolution of glyphosate. Interestingly, no compound had a maximum at 6% ethanol, which seems to suggest that the ideal percentage for each pesticide lies either closer to 2% or 10%, or outside the evaluated range.
3.2. Glyphosate Extraction
The extraction of glyphosate from the soil matrix had to be carried out via some modifications to the extraction solvent for immersion SPME. After the three hours of extraction, the SPME fiber was removed from the soil slurry and retroextracted onto ethanol. Concurrently, the glyphosate extraction was performed. There were two possibilities explored: addition of potassium hydroxide (KOH) [
3] or ammonia solution [
17]. The ammonia was chosen because it could be almost entirely removed by drying the extract. KOH, on the other hand, could not be removed from the extract, and although it has a relatively low solubility in ethanol (as does KCl, which is formed after neutralization with hydrochloric acid), it is still sufficient to cause accumulation in the GC liner and eventual clogging and chemisorption. Thus, when an extraction with KOH was assayed (0.6 molar, added dry to the extraction slurry), an extra step of extraction after the derivatization was required. Furthermore, whether neutralization with hydrochloric acid was performed or not, the large amount of salts interfered with the derivatization step, especially at low concentrations of glyphosate.
The sample-to-extraction solvent ratio was slightly modified from a previous work [
17], except only one extraction was performed instead of two. Using a larger sample size (e.g., 3 or 4 g) while retaining a similar extraction solvent volume could have been employed in an attempt to improve detection and quantification limits for the multiresidue pesticides extracted with DI-SPME, but would have been operationally more challenging (especially in terms of shaking), and might have compromised repeatability and extractability in terms of the soil–solvent equilibrium, even though it would ultimately result in a larger analyte mass being extracted onto the SPME fiber. Also, this could compromise the glyphosate recovery, unless a larger extraction solvent volume was added after DI-SPME.
After the extraction, a small amount of dichloromethane had to be added to the extract because it was found that it contained non-negligible amounts of the other analytes, and their presence in the aqueous solvent could compromise repeatability. The simple centrifugation of the extract both removed suspended soil particles, thus eliminating the need for filtration, and settled the dichloromethane layer. However, for finer soils (notably clay), it may be required to centrifuge at higher speeds for longer. Neutralization of the extract with hydrochloric acid before centrifugation (in order to precipitate some soil matrix components) was not feasible because of the formation of non-volatile salts which could not be removed in the drying step.
For derivatization of the glyphosate extract, two methods were tested: silylation with N-(tert-butyldimethylsilyl)-N-methyltrifluoroacetamide (MTBSTFA) co-dissolved with acetonitrile [
18,
19], and simultaneous acylation and esterification with trifluoroacetic anhydride (TFAA) and trifluoroethanol (TFE) [
17,
20]. The silylation with MTBSTFA suffers from several drawbacks, including high reactivity of the reagent with water (more problematic than for TFAA), high cost per sample, and poor reaction yields for trace amounts of glyphosate [
21], possibly caused by low solubility of the underivatized compound and adsorption to the glass vial. Furthermore, neither MTBSTFA nor the other reaction products are sufficiently volatile to allow drying of the mixture after derivatization, which compromises the subsequent combination with the SPME extract. Thus, derivatization with TFAA + TFE was chosen.
One of the drawbacks of this approach was that the glyphosate derivative was much more volatile than the other pesticides analyzed. This was even more pronounced for gylphosate’s degradation product aminomethylphosphonic acid (AMPA), which was also tested in this method. When a starting GC oven temperature of 50 ºC was used, AMPA’s derivative showed poor peak shapes with significant variability between injections of the same extract. As a consequence, the initial oven temperature was reduced to 40 ºC to permit better focusing of this analyte’s band at the top of the column.
3.3. Combined Method and Performance
For the combined method, it was imperative to ensure proper dissolution of the derivative in the ethanol SPME extract. Sonication was employed in an attempt to ensure full dissolution, and thus, good repeatability. The full method was also tested for two phenoxy herbicides: 2-methyl-4-chlorophenoxyacetic acid (MCPA) and 2,4-dichlorophenoxyacetic acid (2,4-D). It was possible to extract these compounds along with glyphosate due to the fact that at the extraction pH (around 12) the carboxylate is dominant, with almost no neutral carboxylic acid molecules present (calculated from the phenoxyacetic acid pKa). However, the repeatability was poor at every concentration (20–100 µg/kg), with coefficients of determination of 0.87 for MCPA and 0.88 for 2,4-D. This was likely caused by the extraction itself, or by the fact that a certain amount of the analytes might have been dissolved in the dichloromethane phase. Naturally, the isotopically labeled glyphosate was very poor at correcting extraction variability, but it may be possible to analyze these compounds with this methodology by using another internal standard which is chemically similar to them, although adding more isotopically labeled compounds to a method will significantly increase its cost per sample. For AMPA, there was also significant lack of repeatability (R2 = 0.91), but this was probably also due to the poor chromatographic performance, since the volatile AMPA derivative is not compatible with the programmed temperature volatilization injection technique used.
Table 2 presents the method performance parameters. The extraction recovery for glyphosate was 92%, with a relative standard deviation of 12% (n = 3). Although for some compounds the internal standard is essential, for others (such as tebuconazole and tefluthrin) it did not increase repeatability. Glyphosate had a lower limit of detection, likely because of the greater concentration factor. Even though SPME is known for greatly concentrating samples, that effect is not present in this method, because instead of direct desorption onto the GC inlet (which transfers the whole extracted analyte mass onto the GC-MS), it employs solvent desorption onto 120 µL of ethanol, of which only 5 µL is injected. Nevertheless, in the future an increase in soil sample mass (e.g., to 4 g) might be an interesting avenue for exploration; it could lower limits of detection, although it could also increase RSDs. Another weakness of the method is that it does not take advantage of the inherent clean-up performed by SPME, since the SPME extract is then mixed with the glyphosate extract, whose only clean-up was the addition of dichloromethane. This results in a final sample with much more matrix interferents than the SPME extract alone, although these were not noticeable in the MRM chromatogram.
The method was also tested for other currently used pesticides, namely, boscalid, diflufenican, epoxiconazole, metalaxyl, and terbutylazine, but was found lacking in terms of precision (R
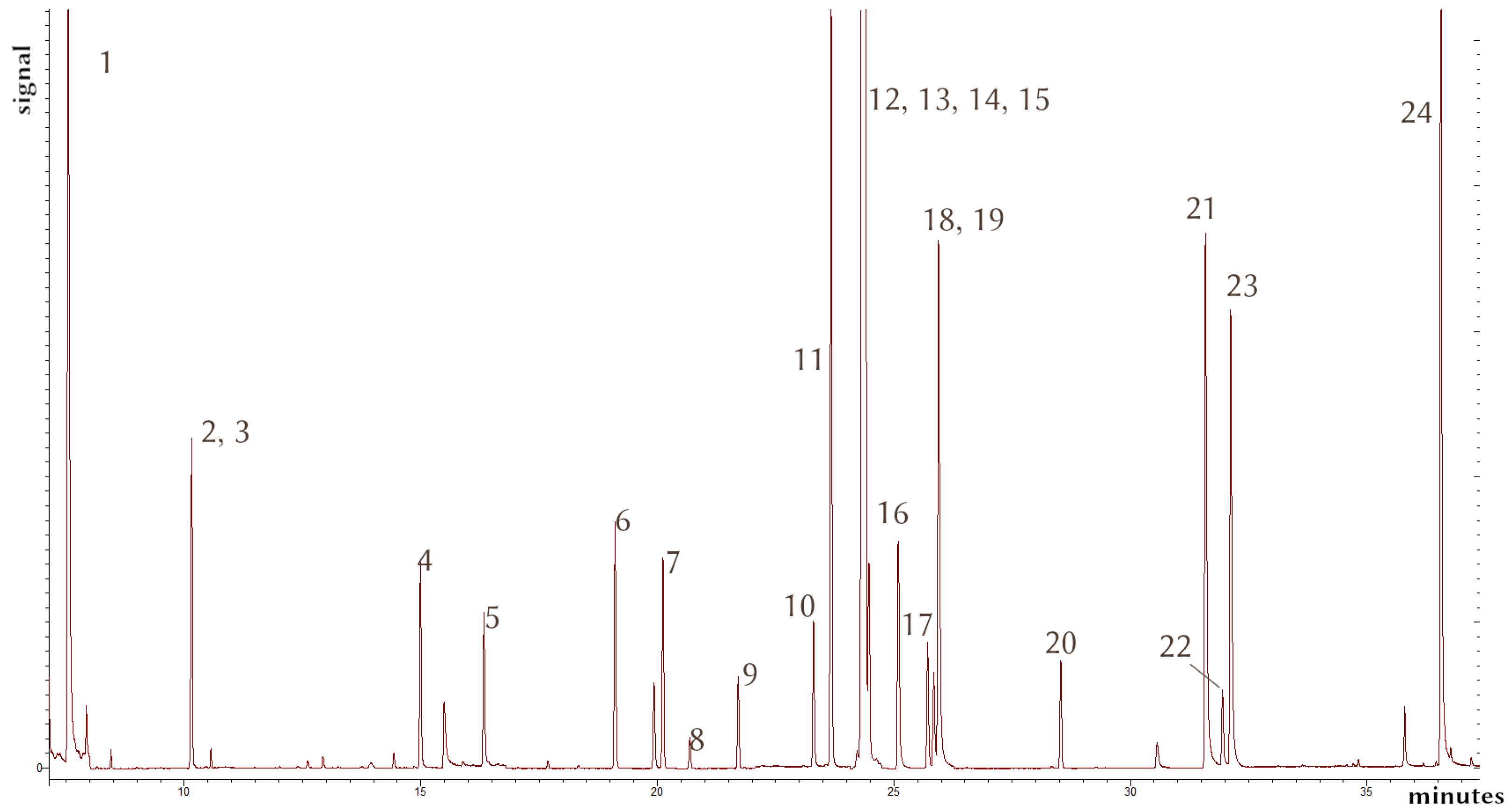
2 = 0.899–0.94; RSD as high as 40% for 20 µg/kg extractions; n = 5). In the future, an isotopically labeled internal standard which better mimics the chemical properties of these compounds should be used to mitigate such problems. Further optimization of the method in order to achieve better concentration factors from the immersion-SPME extraction would be desirable. In terms of the GC-MS/MS determination, it was possible to isolate every analytes’ signal, and thus, a combined method involving more compounds could be viable. A chromatogram from a spiked sample can be seen in
Figure 4.
The limit of quantification obtained for glyphosate was significantly lower than other published methods (e.g., Ref. [
3], LoQ of 50 µg/kg), likely due to the low extraction solvent volume used and high concentration factor generated by drying the aqueous extract, which nonetheless is a difficult and time-consuming process, often avoided. It is possible that a different soil, particularly of finer texture, might require a larger extraction solvent volume to obtain an adequate recovery for glyphosate, which would compromise the detection limit, unless an even larger concentration is performed. In terms of the other analytes, detection and quantification limits are within normal ranges for most multiresidue methods, but not significantly better [
6].

 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}