1. Background
Understanding the characteristics of the species of the fungi phylogenetic tree requires the identification of specific sequences in the proteomes, which in turn may be correlated with the environment and its conditions [
1]. The phylogenetic analysis provides a framework for research and identifying multiple similarities and conservation zones, which can be important for identifying certain protein functions or key structural regions. For example, searching for protein sequences containing a specific pattern can help to identify proteins that bind to certain ligands or have specific enzymatic activity [
2]. Searching for repetitive patterns in protein sequences can also help to identify evolutionarily related proteins, which can provide information about the evolution of proteins and their functions over time [
3]. However, a detailed analysis of proteomes is essential. Human experts can perform this task, but the analysis is challenging on a large scale.
In the biological field, hard-coded algorithms mostly traverse phylogenetic trees, and some software resources like grep and msgfdb2pepxml, among others, are described below.
Grep is a text-processing program designed for regular pattern matching within the text, allowing the search of regular expressions in a string; this string can be a proteome or any other type of text sequence [
4]. Grep requires Linux as an Operating System, command line terminal mastery and knowledge of regular expressions. It is important to note that grep operates solely within the terminal. It lacks a Graphical User Interface (GUI).
Another resource for finding regular expressions is msgfdb2pepxml; this Python library converts the output from the MS-GFDB search engine to pepXML, uses regular expressions to recognize enzyme uses and cleavage rules, and supports PSI-MS [
5]. Executing msgfdb2pepxml requires knowledge of the Python programming language and familiarity with library syntax (which also includes an understanding of regular expressions). Given its nature as a library, there is no GUI, reinforcing the importance of proficiency in understanding and manipulating the library through code.
PhyloPattern is another resource for finding regular expressions. It is a library focused on identifying regular expressions in phylogenetic trees; this library is not focused on proteomes or any other biological sequence [
6]. To execute this tool, the user must have a Prolog engine and know the syntax of this programming language. Also, as this resource is a library, there is no GUI, underscoring the importance of manipulating the library through code.
PatScan is another resource, a program focused on searching for protein or nucleotide sequences of a pattern (regular expression) [
7]. Executing this program involves compiling the source files and preparing a FASTA file for the search operation. Additionally, command-line terminal mastery for the compiling process and knowledge of regular expressions are essential.
PatMatch is another resource to identify repeats using regular expressions; this program does not automate searching for patterns within the sequences because it requires the user to write the complete sequence in which they want to search for the pattern; entering all the sequences of a genome or proteome can take a long time due to the size and number of elements [
8]. For PatMatch utilization, access to the tool in the web browser is necessary. It should be noted that this tool focuses on peptide and nucleotide sequences, not on proteomic sequences; other relevant aspects of this tool are the length limitation of a search to less than 20 residues and the fact that it can only process one sequence at a time.
However, considering the tools’ requirements and characteristics, their implementation could be challenging, especially for users without proficiency in command-line terminal mastery and knowledge of programming.
None of the described tools and resources have been dedicated to detecting FUNGI regular expressions via web scraping. Surprisingly, there is a noticeable absence of software tailored to this specific task. Also, to use some of them, a file in a particular format containing all the sequences is required; the information in this file could be subject to errors if the source is not trustworthy.
Due to the need to automate the analysis process at a large scale, software that can be easily integrated into this process, which reads and analyzes proteomes on a large scale, detects matches, and saves considerable time without downloading files, is now needed.
In this context, we present FungiRegEx v1.0, a software tool to fill the identified challenges of the described tools, taking the available information from the Joint Genome Institute (JGI) [
9] Mycocosm portal (guaranteeing that the information is trustworthy) and performing a search into the proteome databases of the multiple species with the user-defined regular expression through its web scraper module integrated into the tool; also, it integrates a Graphic User Interface (GUI) with a user-friendly interface, and as such, the user does not need to install any additional components, download additional files, or have solid programming knowledge to use it.
FungiRegEx helps in the recognition of repeated sequences, which holds substantial significance as it offers valuable insights into the functional and evolutionary roles of diverse organisms [
10,
11], driving evolution, inducing variation, and regulating gene expression [
12]. Also, FungiRegEx is focused on FUNGI pattern detection and is customizable to adapt it to the resources of the computer or server where it is executed (in case the user wants a greater or lesser number of scraper instances). Finally, this tool could be deployed on a server or a computer if the user wants to.
Notably, FungiRegEx stands out by providing a GUI, eliminating the need for additional files or programming knowledge, and performing the search of the regular expressions in multiple sequences simultaneously while saving resources and time.
2. Materials and Methods
This section presents the materials and methods used to develop FungiRegEx software.
2.1. Data Source
It is imperative to obtain proteomic information from reliable sources, meaning it has been validated, is recognized in the field with extensive coverage, and consistently updates its information. Therefore, we integrate the JGI Mycocosm database, Walnut Creek, CA, USA [
13].
2.2. Architecture of FungiRegEx
FungiRegEx front-end is based on React JS 17.0.2v, a JavaScript library that is both available and open-source and is designed for constructing interfaces [
14], and Node JS 16.17v, serving as the back-end, which is a JavaScript runtime built upon the V8 JavaScript engine [
15], as well as Chromium [
16], which is an open-source web browser. The application provides an interactive GUI, as shown in Figure 2 in the
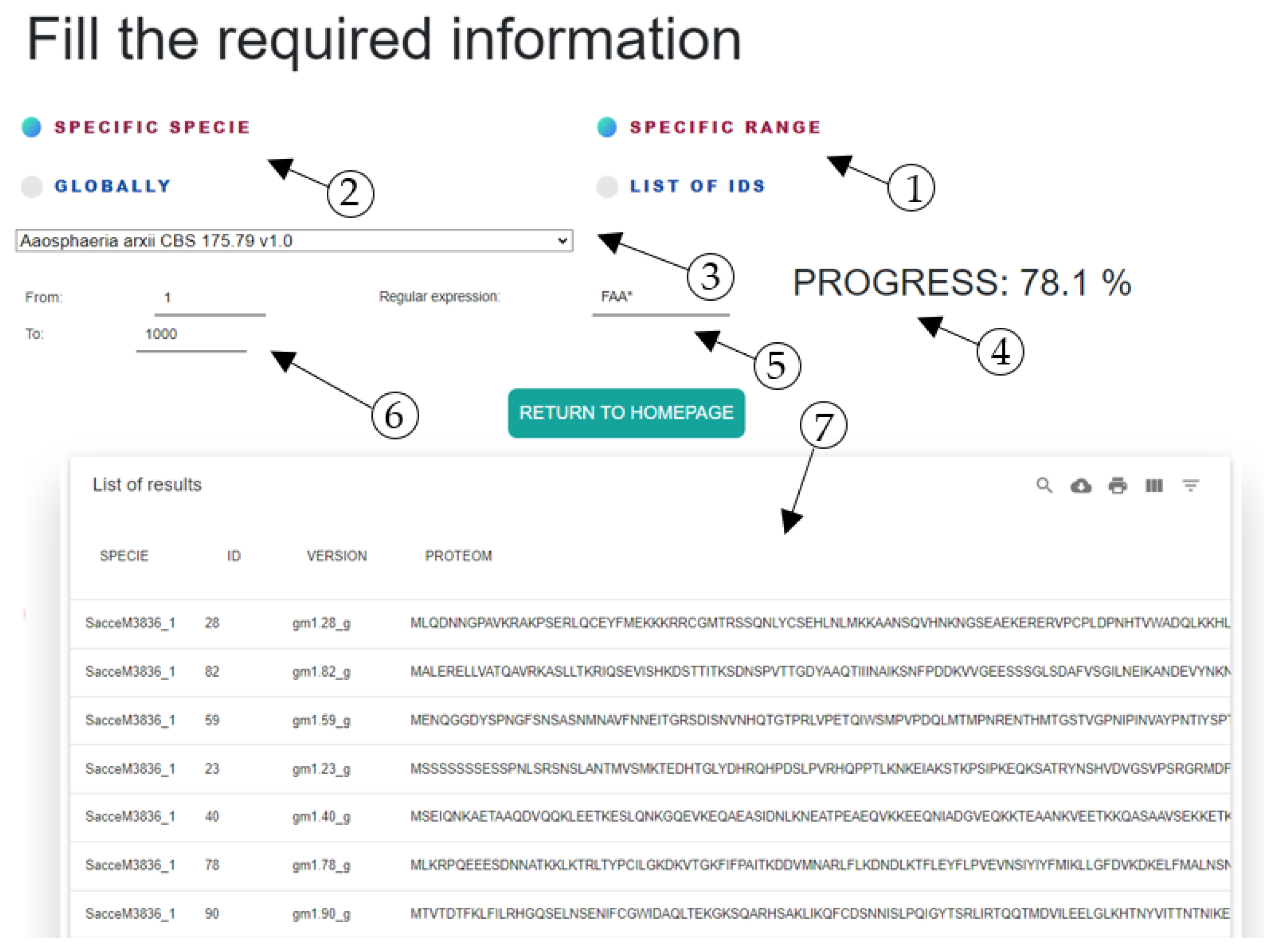
Section 3. However, it is clarified that one of the characteristics of the GUI is that it allows the table results to be downloaded in a CSV file.
Also, internally, the application launches the scrapper instances into the JGI Mycocosm database and optimizes memory consumption because the application reuses each launched instance once it has obtained the information; in case of an error, the instance is automatically restarted.
The FungiRegEx workflow for the user is as follows: first, the user selects the type of search and the species, clarifying that new taxonomic additions of a Fungi Specie will not be available in the software unless the user adds them. Second, the user inputs the range to perform the search. Third, the search starts. Fourth, as mentioned before, the results can be filtered, ordered, and downloaded in an output file in CSV format.
2.3. Implementation
FungiRegEx is distributed as a ZIP file. The source code is available for download at
https://sourceforge.net/p/fungiregex (accessed on 26 March 2024) and
https://github.com/maigolinox/fungiregex (accessed on 26 March 2024). Once FungiRegEx has been downloaded and unzipped, the user must read the documentation, which contains detailed instructions on installing it locally or on a server if required. Briefly, to run FungiRegEx, only two commands are needed in separate instances of bash: the first one to execute the front end of FungiRegEx is
npm run start:frontend and the last one to execute the back end of FungiRegEx is
npm run start:backend.
The regular expression search implemented in FungiRegEx is based on finding exact repeats of length k along the proteomic sequence. The regular expression can be whatever length the user requires. If the protein sequence of length k is diminutive, the comparative process proceeds more expeditiously. Once the regular expression is found in the protein sequence of the FUNGI organism, it is filtered to eliminate those that do not match.
2.3.1. Searching for Regular Expressions Matches
The application back-end begins its search by creating a regular expression object.
Regular expressions are patterns used to search for character combinations in text strings. Regular expressions can contain various special characters and modifiers that define the pattern to search for [
17].
The magnitude of the search range directly impacts the algorithm’s processing time; smaller ranges are preferred for optimal efficiency.
2.3.2. If Matches between Regular Expression and Protein Sequence Are Found
The search process identifies the regular expression within the amino acid chain with a length of k, as illustrated in
Figure 1, where we can assume a chain of amino acids that, in accordance with the functionality of FungiRegEx, is capable of finding the matches of the regular expression throughout the chain, in addition to counting them. As the detected repeats do not accurately reflect the true length of the repeating pattern, they need to be expanded to match the actual repeat length. This paper’s chosen approach involves reading the characters to the left or right of all repeats and storing the matches in an array.
2.3.3. If No Matches between a Regular Expression and Protein Sequence Are Found
If no matches are found, the algorithm will continue the search in another chain. The search algorithm can progress in larger intervals without overlooking any repetitions. The crucial factor in progressing with larger intervals is ensuring the search algorithm never overlooks matches.
2.3.4. Processing Speed
An approximation of the speed will take the algorithm to process the regular expression given by the next mathematical formula.
Reducing the size of search intervals can improve processing speed. However, the algorithm’s speed may decrease if the intervals become too small. This is because smaller intervals can cause the program to spend more time launching browser instances than acquiring information and performing the search for the regular expression.
The next section presents the results obtained using FungiRegEx.
3. Results
As part of the results, we developed a GUI for using FungiRegEx, shown in
Figure 2. This GUI is described in
Section 2.2.
FungiRegEx was validated and tested by searching the sequence AXSXG as a regular expression, where AXSXG is a pentapeptide of a lipase group that brings thermostability and resistance to solvents of an enzyme [
18] and that has been little described in fungi. The considerations to validate and test FungiRegEx with this sequence are as follows:
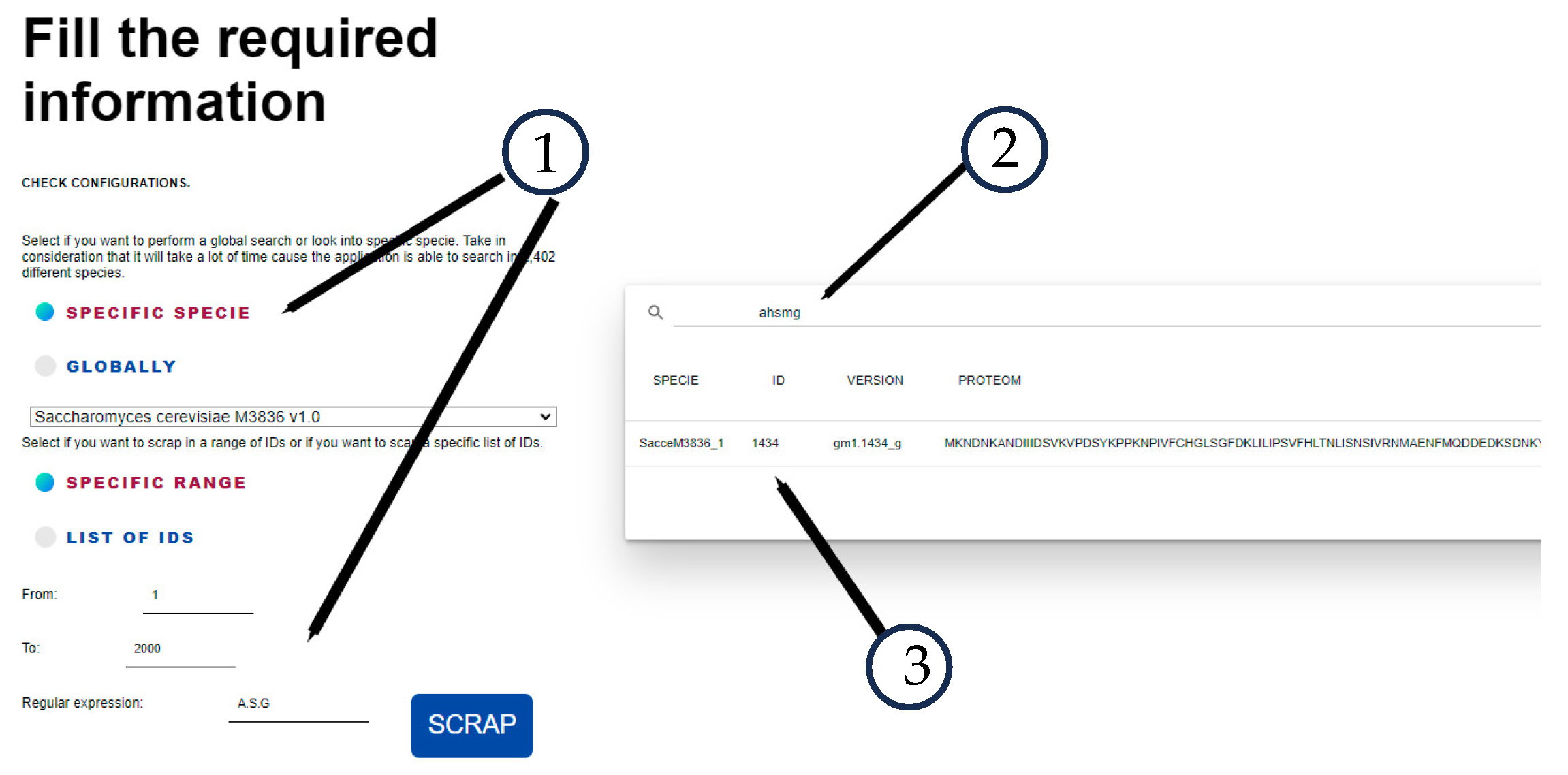
To bring some results, we perform the search with the following parameters:
- 3.
A.S.G, where “.” in regular expressions syntax means whatever character.
- 4.
Search in a specific range: 1 to 2000. This means that FungiRegEx will launch the scrapper instances to retrieve the data in the JGI Mycocosm database from 2000 proteomes.
After running the search, the tool showed that of the 2000 scraped sequences, only one with identifier 1434 has that pentapeptide only once. It also identified matches in 281 sequences with similarity.
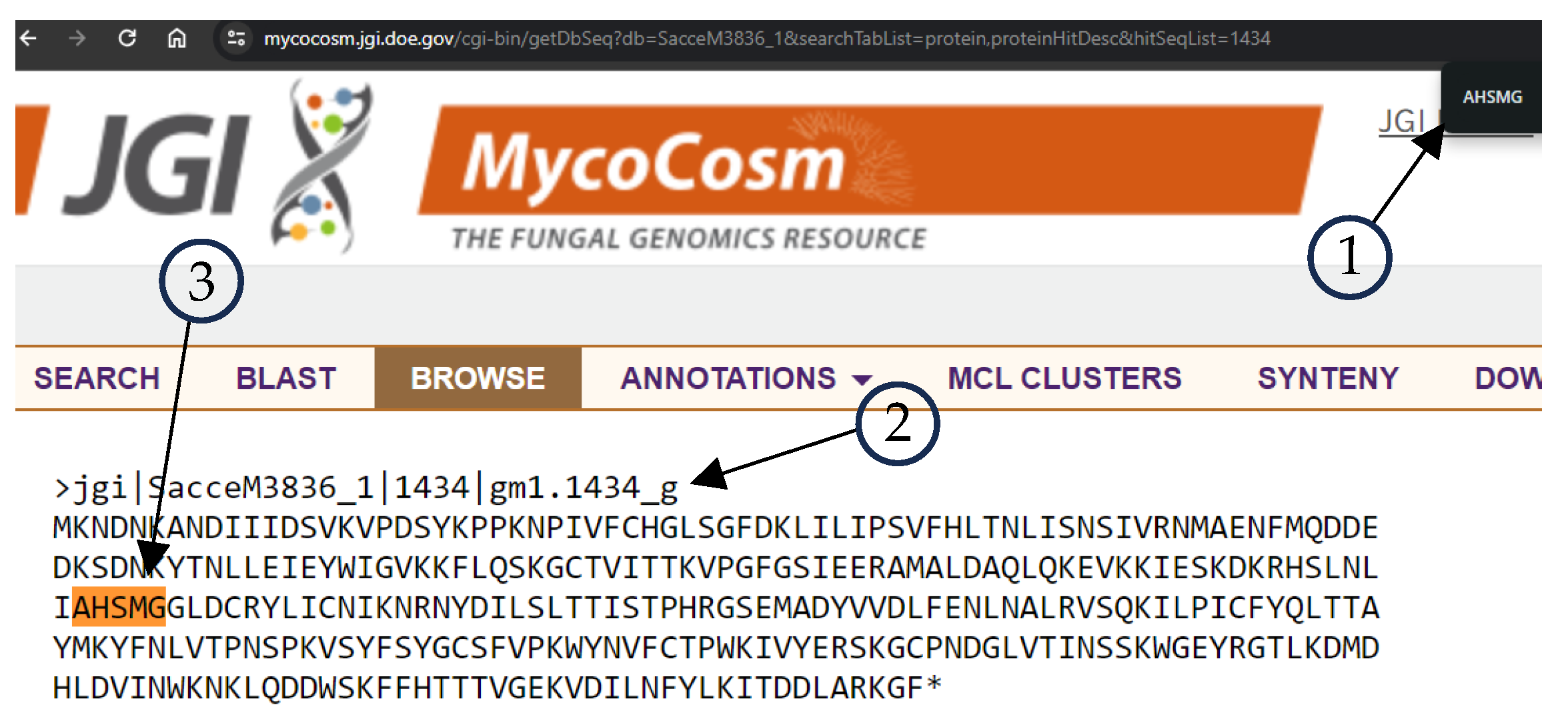
Figure 3 shows the results of the JGI Mycocosm retrieved data, where element 1 is the search tool of the browser searching for AHSMG regular expression, element 2 is the header of the sequence, specie
SacceM3836_1 id 1434, and element 3 is the coincidence of the AHSMG in the sequence.
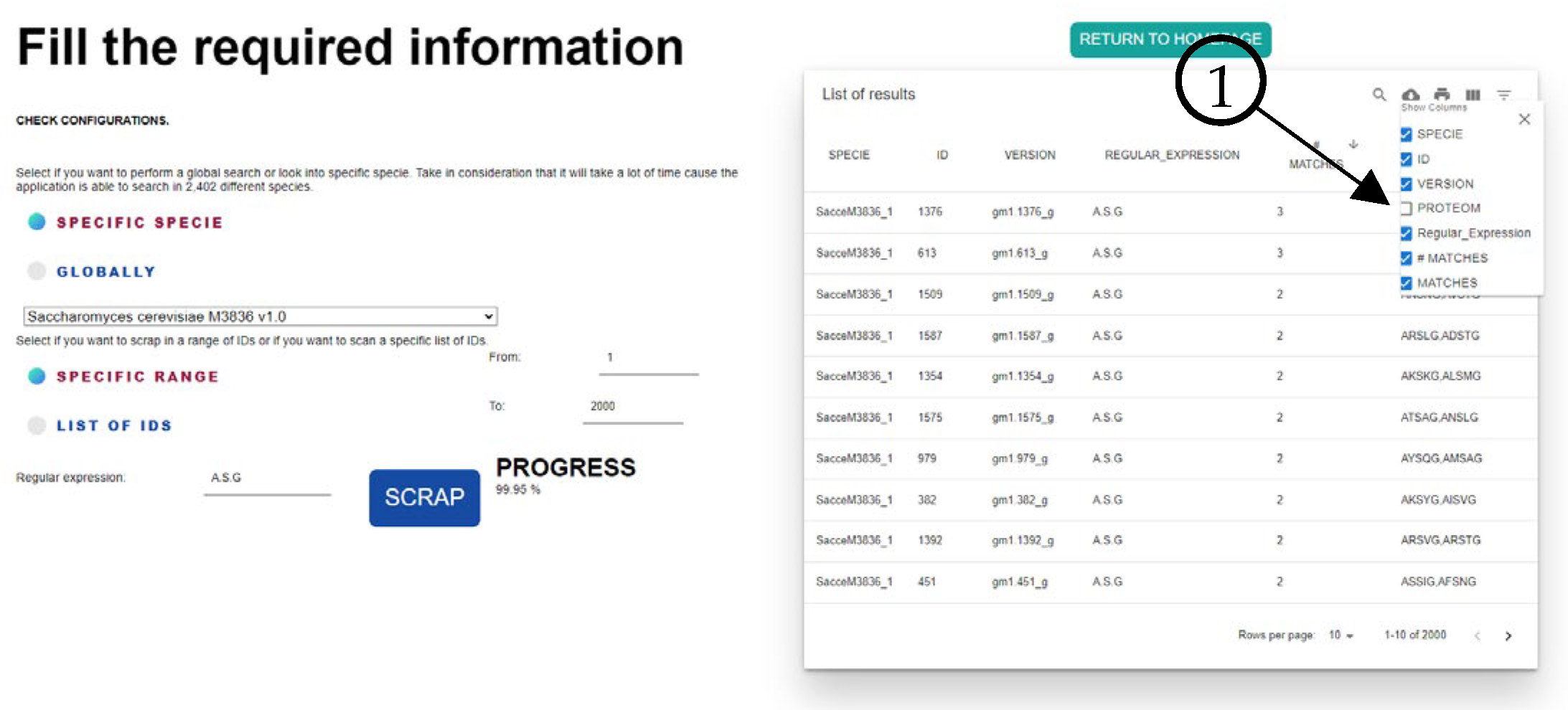
Figure 4 shows the match with the FungiRegEx tool searching for AHSMG; element 1 is the parameters to perform the search, element 2 is the table results with the regular expression, and element 3 is the sequence identifier. With this, we can appreciate that the direct result of JGI Mycocosm data and the result with FungiRegEx is the same.
As mentioned, the tool also searches the complete sequence for other similarities according to the regular expression that the user inputs. This can be seen in
Figure 5, where we hide the proteome column (Element 1) for the image size to show the results of FungiRegEx with the mentioned parameters.
In this way, the tool can find the regular expression in the user-determined search proteome, which may interest subsequent studies.
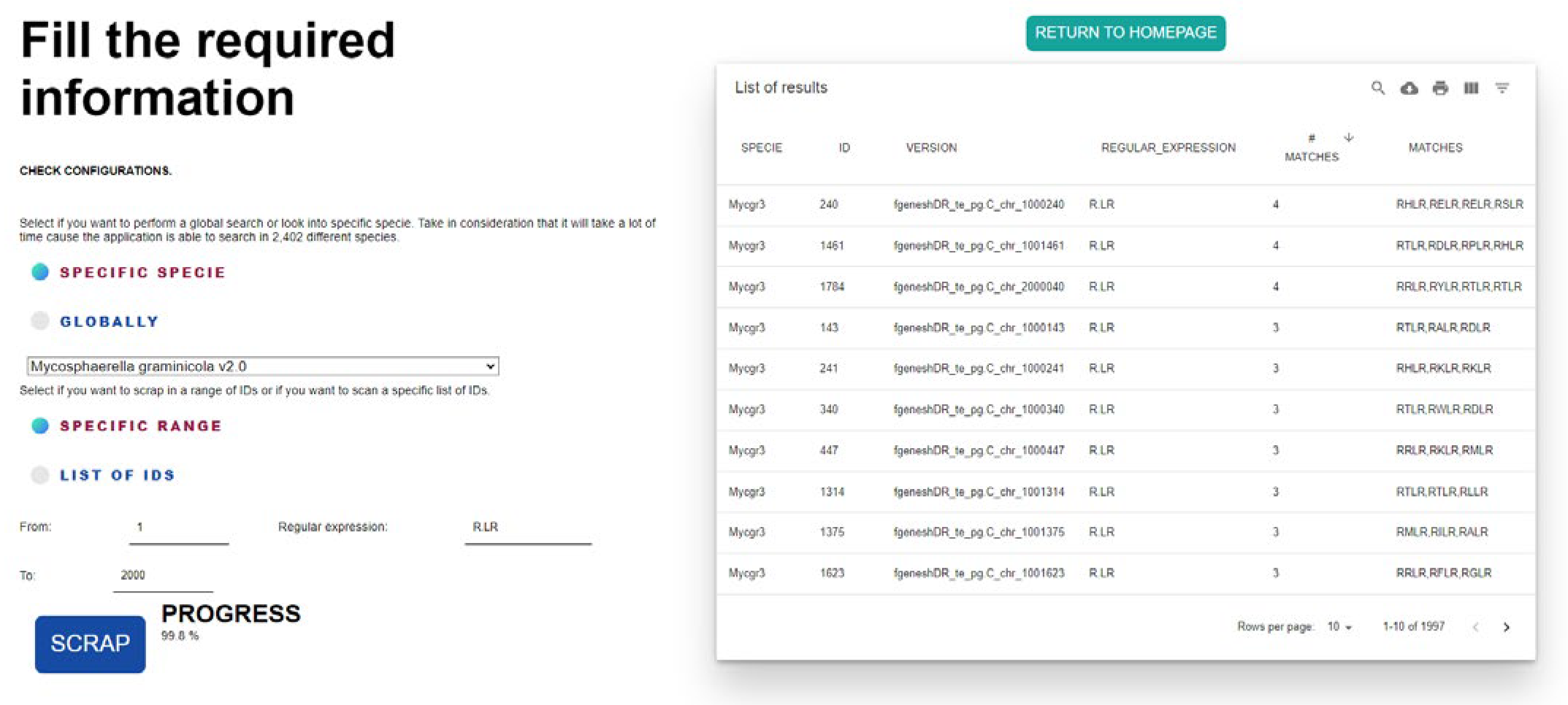
A second use case executed to validate FungiRegEx functionality involves the search for effectors. Liping Liu et al. [
23] identified different effectors, such as the RXLR, asserting that fungi, oomycetes, and bacteria release small, secreted proteins crucial for symbiotic interaction and pathogenicity. Liping Liu investigated various effectors in different species, such as Mg3LysM (Mycosphaerella graminicola LysM), secreted by Mycosphaerella graminicola [
24,
25]. For this example, the JGI Mycocosm database of Mycosphaerella graminicola v2.0 will be used with the RXLR sequence, where X represents any amino acid. In regular expression language, the regular expression is R.LR.
Figure 6 shows the results of FungiRegEx, hiding the proteome column due to the size of the proteomes to show the results in the figure.
Figure 6 shows that FungiRegEx can find the effector of interest in the proteome. In addition, it is identified that
Mycosphaerella graminicola has the effector
RXRL [
24]. Also relevant to the results is that a counter is increased each time the tool finds the regular expression in the sequence, and its value is displayed in the “# MATCHES” column.
FungiRegEx employs a straightforward sequential search method to identify regular expressions directly from the protein sequences of FUNGI organisms. Diverging from the prevalent approach of employing a suffix tree or alignment matrix as a primary data structure, the algorithm introduced in this paper operates by directly identifying regular expressions within the protein sequence. As a result, this methodology exhibits efficiency in memory usage due to launching and running the scraper instances, boasts enhanced comprehensibility and ease of implementation, and offers great speed in getting multiple sequences at the same time compared to if the process were carried out manually or using tools like PatMatch [
8] that requires the user to introduce sequences one by one. Also, FungiRegEx does not require downloading any fasta or file.
With 200 puppeteer instances, 50,000 results are obtained in approximately 139 min, as can be seen in
Figure 7, with the estimated time that indicates the monitor (depending on the resources of the computer, the computer can consult at least seven pages per second; this means that the 50,000 results can be consulted in only 12 min); it also clarifies that this depends on the available computer resources.
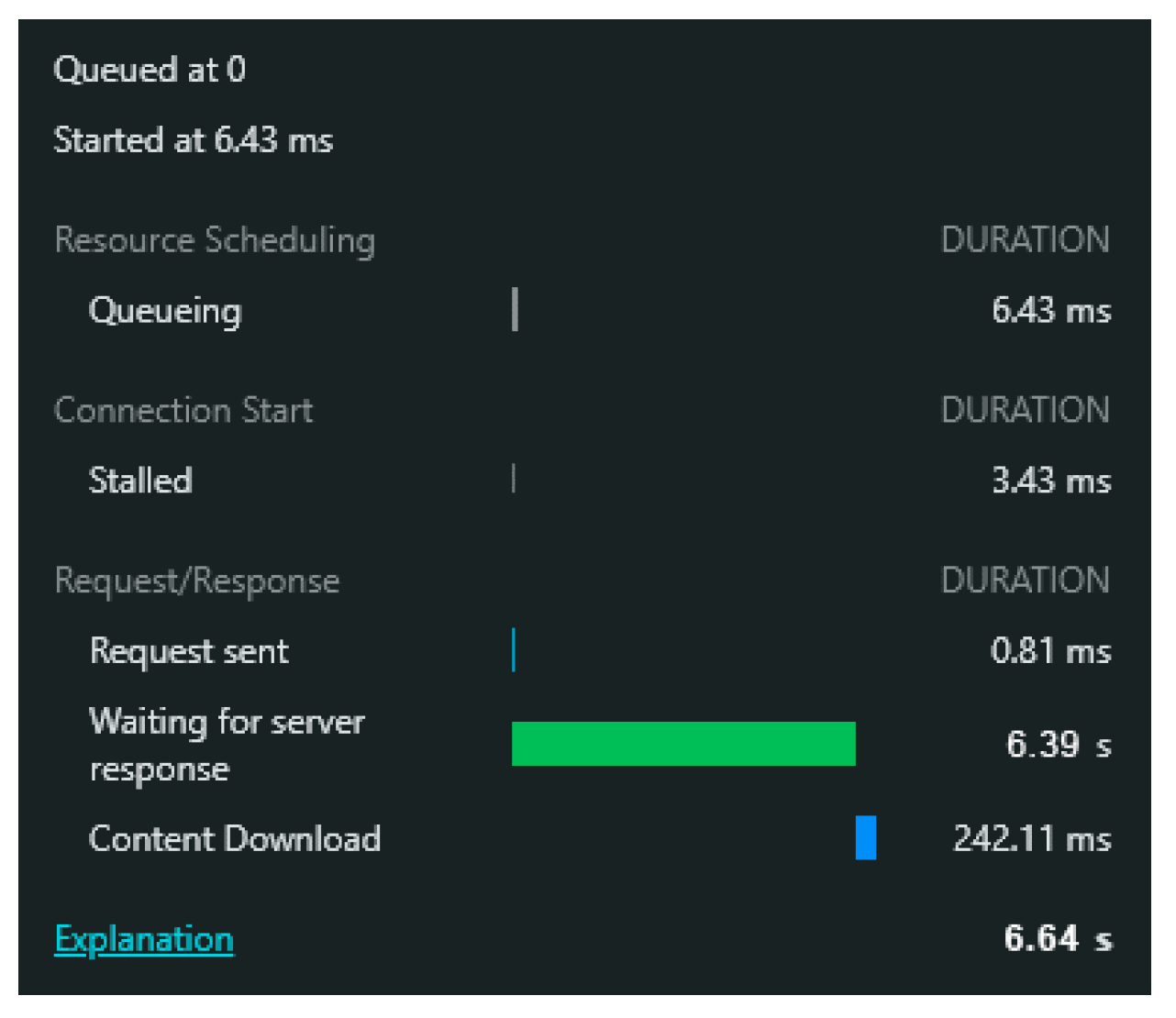
It should be mentioned that just requesting the JGI Mycocosm database and getting a response on the webpage takes around 6.64 s, as shown in
Figure 8.
FungiRegEx demonstrated efficiency in proteomic sequence analysis and could streamline the process of analyzing proteomic sequences of the available species in the JGI Mycocosm portal. It eliminates the need to download FASTA files by dynamically retrieving data from the JGI Mycocosm database, ensuring real-time access. This significantly speeds up result retrieval compared to manual methods, optimizing time and computational resource usage. It offers the adaptability to run on any computer and allows customization of scrapper instances. The user-friendly GUI facilitates the process of searching regular expressions due to not requiring coding knowledge and presents results in a customizable table, enhancing accessibility. Moreover, it operates independently of specific operating systems and offers deployment options for local or server use. Additionally, it facilitates efficient result filtering and the identification of specific sequences. The console-free user experience further enhances accessibility, while the simplified search syntax with user-defined parameters aids in targeted searches. The tool’s potential for future research is notable, as it identifies user-defined regular expressions on certain proteomes, paving the way for further exploration. Finally, it provides detailed information about matching sequences, including the exact match count.
FungiRegEx enhances the efficiency of proteomic sequence analysis and provides a user-friendly, adaptable, and customizable tool with features for interpreting and exploring results without downloading any file to perform its functions. The next section discusses the obtained results.
4. Discussion
In this section, we discuss the findings and limitations associated with FungiRegEx.
Table 1 shows the comparison between FungiRegEx’s features and those of the other tools. FungiRegEx highlights this due to its wide coverage, while the other tools cover a maximum of four features.
Below, we describe how those features are covered, highlighting the tool’s efficiency in data retrieval functionality, which allows for accelerated retrieval of results and optimized use of computational resources because each instance is reused instead of being launched again, compared to other tools that require having the data set archived to perform the search. FungiRegEx does not require downloading the file since it consults the information directly in the database. Users must manually enter characteristics of newly added fungal species to extend them beyond the registered species, potentially limiting their future applicability.
FungiRegEx introduces user-friendly features such as real-time data retrieval, customization adaptability since it allows the user to configure the parameters of the instances that can be launched depending on the computational resources available if required, and a Graphical User Interface (see
Figure 1), presenting a solution for researchers who do not have the programming knowledge or bash mastery that are necessary for the described tools in the
Section 1.
Removing FASTA file downloads through the FungiRegEx scrapper module advances real-time data recovery. Still, the dependency on Internet connectivity affects the search speed and varies depending on the quality of the user’s connection. Despite this limitation, the tool significantly speeds up result retrieval compared to manual extraction, which could revolutionize large-scale studies. The internet factors, such as speed and availability of computational resources, reveal the need to balance these factors to optimize performance, avoiding issues such as temporary IP blocking when deploying too many scraper instances. Given this last point of implementing scraper instances, it is recommended not to exceed 100 instances in parallel.
The efficient use of computational resources underlines the good functioning of FungiRegEx, coupled with the fact that users can configure the parameters for greater control of computational resources. Its adaptability to any computer and customizable parameters improve versatility despite limitations such as single-task support and possible task deletion in simultaneous use scenarios between different users.
The GUI simplifies the presentation of results, but readability problems arise in very long proteomes as the table where the results are displayed becomes very large, suggesting room for improvement in the interface.
Computer OS independence and deployment flexibility allow users to align runtime modes with their preferences and infrastructure, with a recommendation for local installation due to its single-user nature.
Regarding the last six covered features, FungiRegEx does not present any restriction regarding the length limit of the sequence to perform the regular expression search. Also, as mentioned before, this tool does not require the user to have programming knowledge or bash mastery to use it. Another relevant aspect is that this tool does not require a FASTA file; FungiRegEx can perform searches into multiple sequences at the same time. Finally, FungiRegEx allows users to store the results in CSV format if required.
The tool’s user-defined search parameters improve applicability, although users must specify proteome lengths to perform accurate searches.
The ability to filter user-defined regular expressions in results facilitates ongoing and future research by allowing the exploration of specific sequences within the returned results, provided that users already possess the regular expressions of interest they wish to search for.
Finally, it is important to note that the results can be stored in CSV format. This allows you to export the results and open the file in a text processor such as Microsoft Word or Excel, among others.
5. Conclusions
FungiRegEx represents a significant advancement in proteomic sequence analysis, offering researchers a streamlined and user-friendly approach. Handling datasets without downloading FASTA files accelerates research processes and facilitates broader investigations into fungal proteomes. Its real-time data retrieval capability from the JGI Mycocosm website enhances accessibility, although it depends on internet speed. FungiRegEx effectively utilizes computational resources and offers customization options, making it adaptable to various research needs and computational environments.
Despite the limitations mentioned in the
Section 4, FungiRegEx demonstrates clear advantages in efficiency, speed, and ease of use compared to manual methods, thus representing a valuable tool for proteomic research. Addressing these limitations could further enhance its utility and impact on the scientific community.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}