
AMP-GSM: Prediction of Antimicrobial Peptides via a Grouping–Scoring–Modeling Approach



Abstract
1. Introduction
2. Materials and Methods
2.1. Datasets
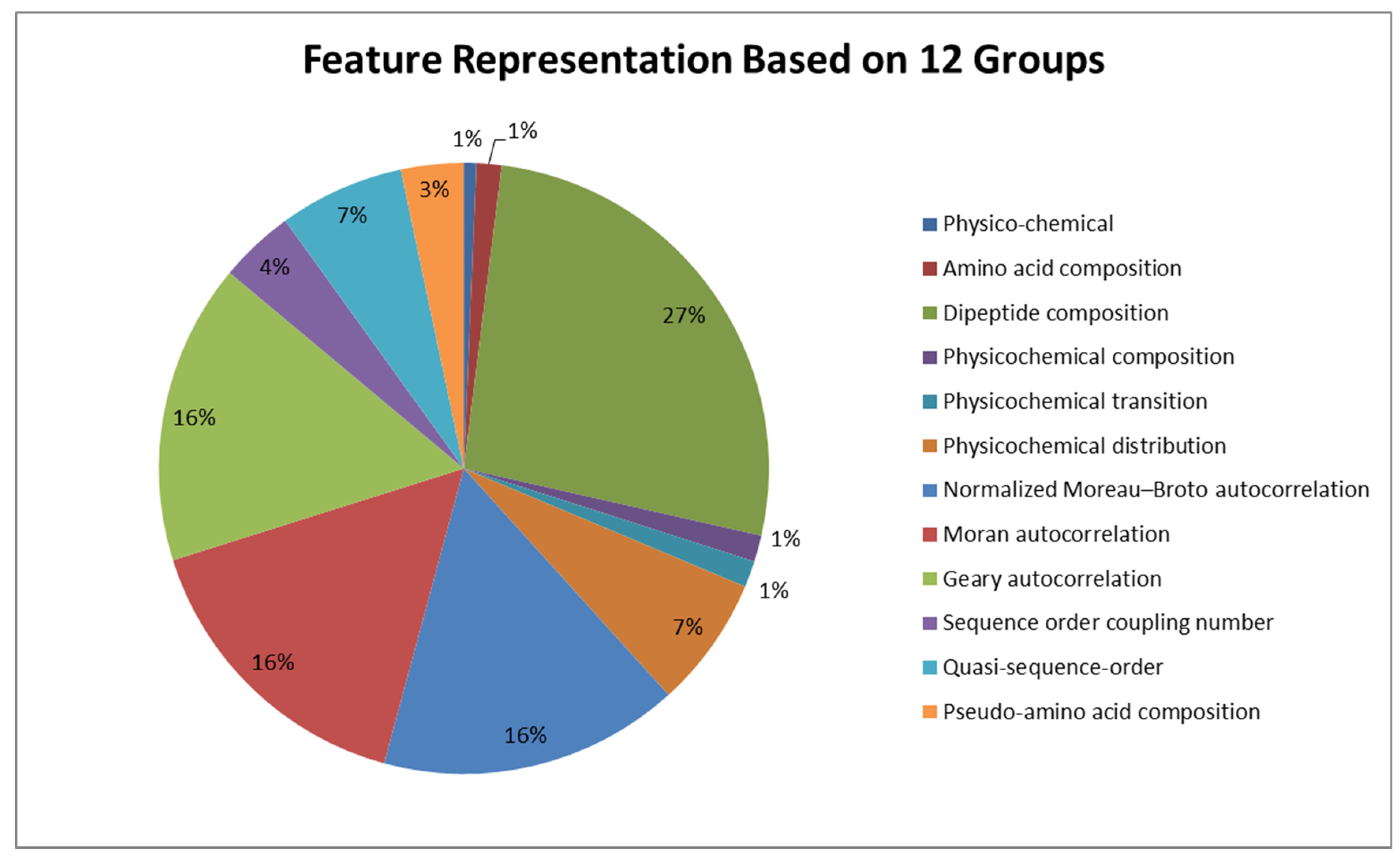
2.2. Generation of Sequence-Based, Structure-Based, and Linguistic-Based Features
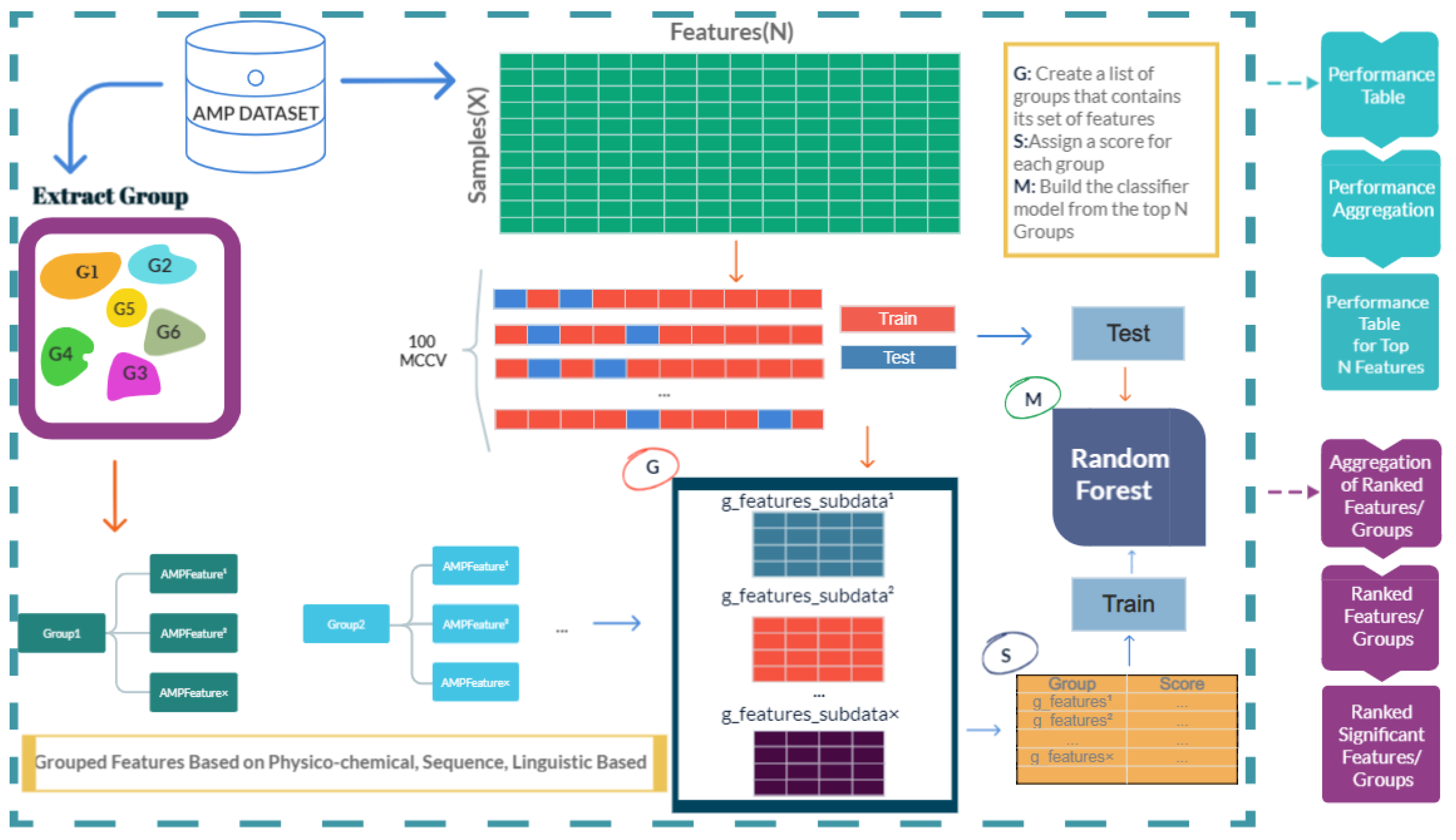
2.3. Proposed Model
2.3.1. Grouping Peptides Based on Physico-Chemical, Sequence-Based, Structure-Based, and Linguistic-Based Features
2.3.2. Scoring the Groups
2.3.3. Modeling Component
2.4. Feature Selection Methods
2.5. Performance Metrics
3. Results
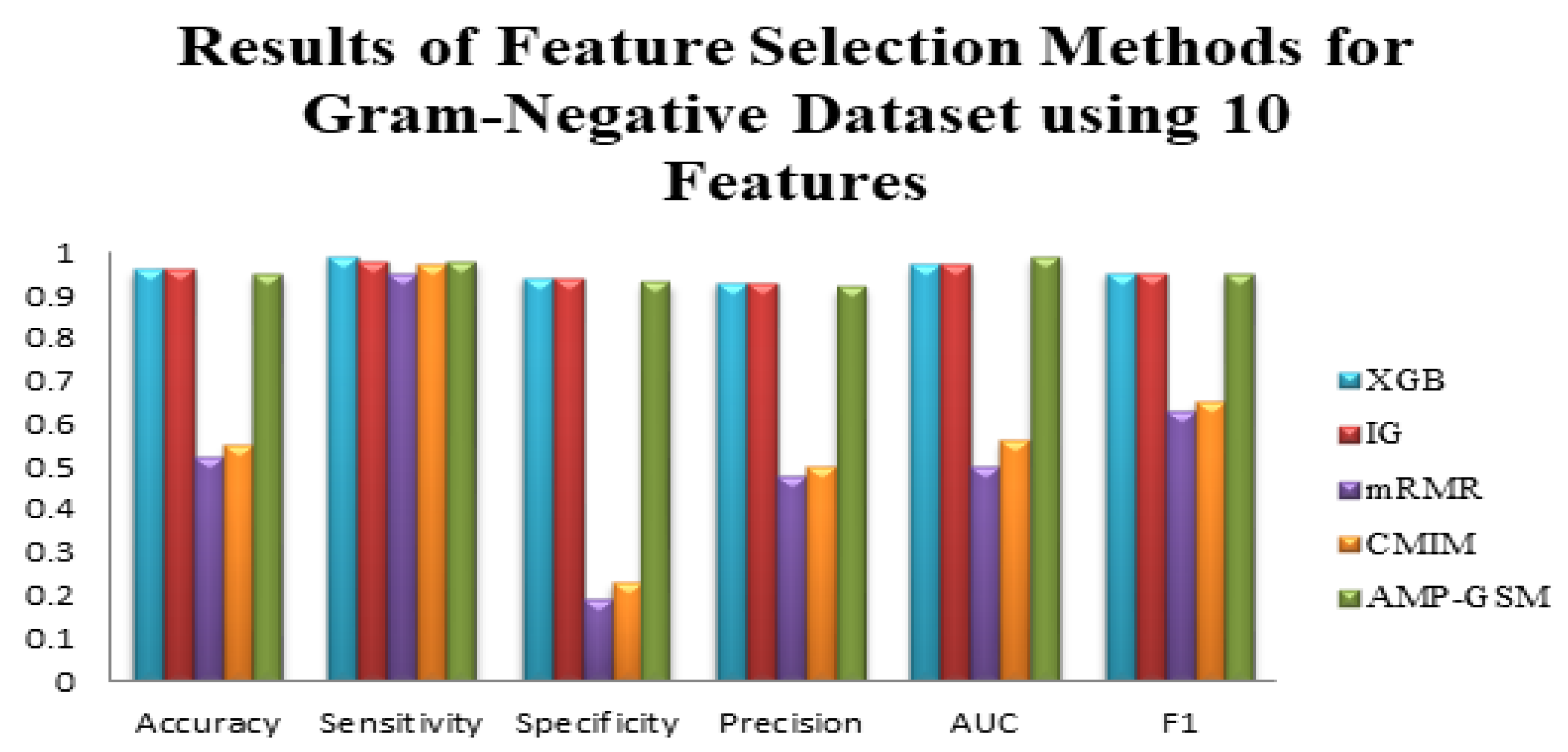
3.1. Performance Evaluation of AMP-GSM on the Gram-Negative Dataset in Dataset 1
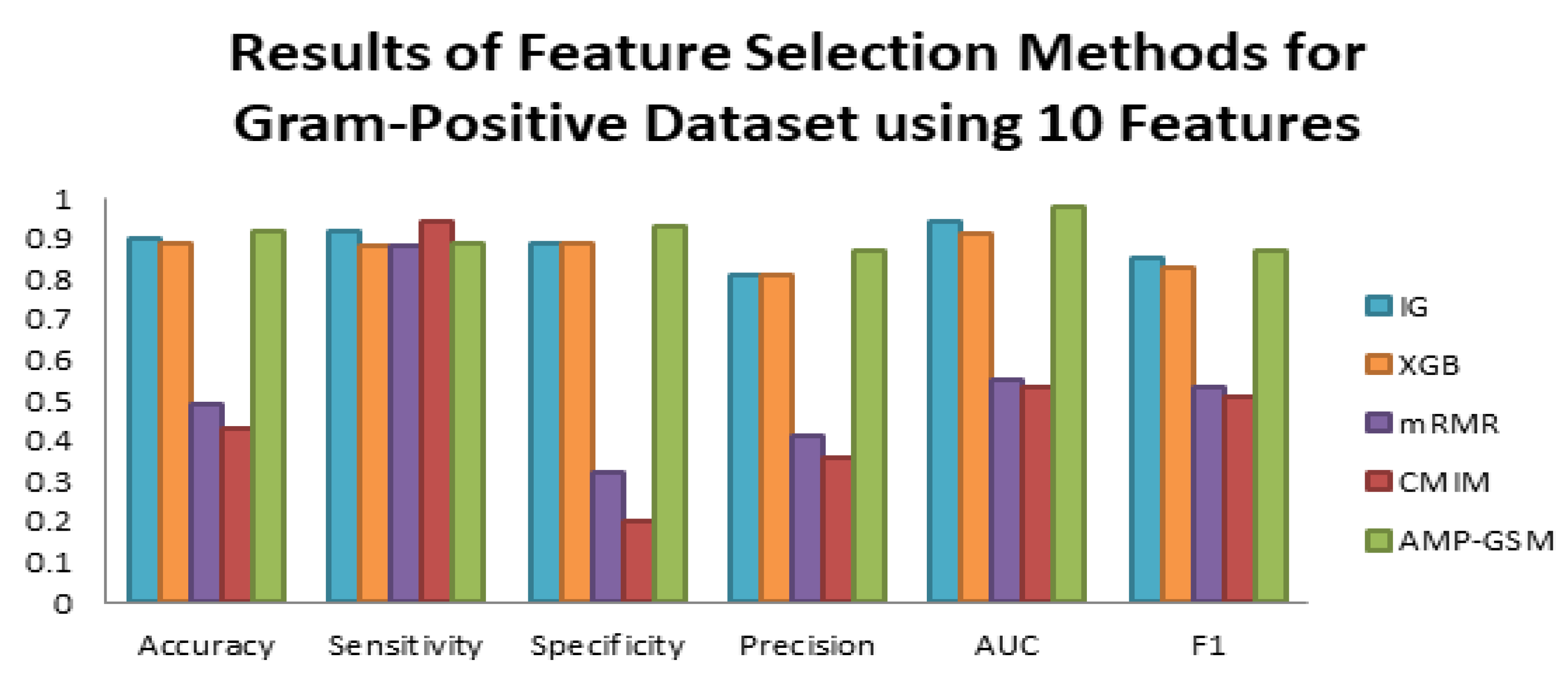
3.2. Performance Evaluation of AMP-GSM on the Gram-Positive Dataset in Dataset 1
3.3. Ranking of the Groups
3.4. Comparative Evaluation of the Proposed Method with Other Feature Selection Methods and Classifiers
3.5. Testing AMP-GSM on Different Benchmark Datasets, Comparative Evaluation with Existing Approaches
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Büyükkiraz, M.E.; Kesmen, Z. Antimicrobial peptides (AMPs): A promising class of antimicrobial compounds. J. Appl. Microbiol. 2021, 132, 1573–1596. [Google Scholar] [CrossRef]
- Vishnepolsky, B.; Grigolava, M.; Zaalishvili, G.; Karapetian, M.; Pirtskhalava, M. DBAASP Special prediction as a tool for the prediction of antimicrobial potency against particular target species. In Proceedings of the 4th International Electronic Conference on Medicinal Chemistry, Sciforum Online, 1–30 November 2018; p. 5608. [Google Scholar] [CrossRef]
- Thomas, S.; Karnik, S.; Barai, R.; Jayaraman, V.K.; Idicula-Thomas, S. CAMP: A useful resource for research on antimicrobial peptides. Nucleic Acids Res. 2009, 38, D774–D780. [Google Scholar] [CrossRef]
- Piotto, S.P.; Sessa, L.; Concilio, S.; Iannelli, P. YADAMP: Yet another database of antimicrobial peptides. Int. J. Antimicrob. Agents 2012, 39, 346–351. [Google Scholar] [CrossRef]
- Zhao, X.; Wu, H.; Lu, H.; Li, G.; Huang, Q. LAMP: A Database Linking Antimicrobial Peptides. PLoS ONE 2013, 8, e66557. [Google Scholar] [CrossRef]
- Fan, L.; Sun, J.; Zhou, M.; Zhou, J.; Lao, X.; Zheng, H.; Xu, H. DRAMP: A comprehensive data repository of antimicrobial peptides. Sci. Rep. 2016, 6, 24482. [Google Scholar] [CrossRef]
- Lee, E.Y.; Fulan, B.M.; Wong, G.C.L.; Ferguson, A.L. Mapping membrane activity in undiscovered peptide sequence space using machine learning. Proc. Natl. Acad. Sci. USA 2016, 113, 13588–13593. [Google Scholar] [CrossRef]
- Torrent, M.; Andreu, D.; Nogués, M.V.; Boix, E. Connecting Peptide Physicochemical and Antimicrobial Properties by a Rational Prediction Model. PLoS ONE 2011, 6, e16968. [Google Scholar] [CrossRef]
- Meher, P.K.; Sahu, T.K.; Saini, V.; Rao, A.R. Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou’s general PseAAC. Sci. Rep. 2017, 7, 42362. [Google Scholar] [CrossRef]
- Lata, S.; Mishra, N.K.; Raghava, G.P. AntiBP2: Improved version of antibacterial peptide prediction. BMC Bioinform. 2010, 11, S19. [Google Scholar] [CrossRef] [PubMed]
- Veltri, D.; Kamath, U.; Shehu, A. Improving Recognition of Antimicrobial Peptides and Target Selectivity through Machine Learning and Genetic Programming. IEEE/ACM Trans. Comput. Biol. Bioinform. 2015, 14, 300–313. [Google Scholar] [CrossRef]
- Randou, E.G.; Veltri, D.; Shehu, A. Binary Response Models for Recognition of Antimicrobial Peptides. In Proceedings of the International Conference on Bioinformatics, Computational Biology and Biomedical Informatics, Wshington, DC, USA, 22–25 September 2013; pp. 76–85. [Google Scholar] [CrossRef]
- Lertampaiporn, S.; Vorapreeda, T.; Hongsthong, A.; Thammarongtham, C. Ensemble-AMPPred: Robust AMP Prediction and Recognition Using the Ensemble Learning Method with a New Hybrid Feature for Differentiating AMPs. Genes 2021, 12, 137. [Google Scholar] [CrossRef]
- Vishnepolsky, B.; Grigolava, M.; Managadze, G.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M.; Pirtskhalava, M. Comparative analysis of machine learning algorithms on the microbial strain-specific AMP prediction. Brief. Bioinform. 2022, 23, 233. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.-R.; Jhong, J.-H.; Wang, Z.; Chen, S.; Wan, Y.; Horng, J.-T.; Lee, T.-Y. Characterization and Identification of Natural Antimicrobial Peptides on Different Organisms. Int. J. Mol. Sci. 2020, 21, 986. [Google Scholar] [CrossRef]
- Kavousi, K.; Bagheri, M.; Behrouzi, S.; Vafadar, S.; Atanaki, F.F.; Lotfabadi, B.T.; Ariaeenejad, S.; Shockravi, A.; Moosavi-Movahedi, A.A. IAMPE: NMR-Assisted Computational Prediction of Antimicrobial Peptides. J. Chem. Inf. Model. 2020, 60, 4691–4701. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, F.; Leier, A.; Xiang, D.; Shen, H.-H.; Lago, T.T.M.; Li, J.; Yu, D.-J.; Song, J. Comprehensive assessment of machine learning-based methods for predicting antimicrobial peptides. Brief. Bioinform. 2021, 22, 83. [Google Scholar] [CrossRef]
- Dee, W. LMPred: Predicting antimicrobial peptides using pre-trained language models and deep learning. Bioinform. Adv. 2022, 2, 021. [Google Scholar] [CrossRef]
- Ma, Y.; Guo, Z.; Xia, B.; Zhang, Y.; Liu, X.; Yu, Y.; Na Tang, N.; Tong, X.; Wang, M.; Ye, X.; et al. Identification of antimicrobial peptides from the human gut microbiome using deep learning. Nat. Biotechnol. 2022, 40, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Dai, R.; Yan, W.; Zhang, W.; Bin, Y.; Xia, E.; Xia, J. Identifying multi-functional bioactive peptide functions using multi-label deep learning. Brief. Bioinform. 2021, 23, 414. [Google Scholar] [CrossRef]
- Ahmad, A.; Akbar, S.; Khan, S.; Hayat, M.; Ali, F.; Ahmed, A.; Tahir, M. Deep-AntiFP: Prediction of antifungal peptides using distanct multi-informative features incorporating with deep neural networks. Chemom. Intell. Lab. Syst. 2020, 208, 104214. [Google Scholar] [CrossRef]
- Hussain, W. sAMP-PFPDeep: Improving accuracy of short antimicrobial peptides prediction using three different sequence encodings and deep neural networks. Brief. Bioinform. 2021, 23, 487. [Google Scholar] [CrossRef]
- Su, X.; Xu, J.; Yin, Y.; Quan, X.; Zhang, H. Antimicrobial peptide identification using multi-scale convolutional network. BMC Bioinform. 2019, 20, 730. [Google Scholar] [CrossRef] [PubMed]
- Dua, M.; Barbara, D.; Shehu, A. Exploring Deep Neural Network Architectures: A Case Study on Improving Antimicrobial Peptide Recognition. In Proceedings of the 12th International Conference on Bioinformatics and Computational Biology, San Francisco, CA, USA, 23–25 March 2020; pp. 182–191. [Google Scholar]
- Szymczak, P.; Możejko, M.; Grzegorzek, T.; Jurczak, R.; Bauer, M.; Neubauer, D.; Sikora, K.; Michalski, M.; Sroka, J.; Setny, P.; et al. Discovering highly potent antimicrobial peptides with deep generative model HydrAMP. Nat. Commun. 2023, 14, 1453. [Google Scholar] [CrossRef] [PubMed]
- Boone, K.; Camarda, K.; Spencer, P.; Tamerler, C. Antimicrobial peptide similarity and classification through rough set theory using physicochemical boundaries. BMC Bioinform. 2018, 19, 469. [Google Scholar] [CrossRef] [PubMed]
- Khaledian, E.; Broschat, S.L. Sequence-Based Discovery of Antibacterial Peptides Using Ensemble Gradient Boosting. In Proceedings of the 1st International Electronic Conference on Microbiology, Sciforum Online, 2–30 November 2020; p. 6. [Google Scholar] [CrossRef]
- Timmons, P.B.; Hewage, C.M. HAPPENN is a novel tool for hemolytic activity prediction for therapeutic peptides which employs neural networks. Sci. Rep. 2020, 10, 10869. [Google Scholar] [CrossRef]
- Spänig, S.; Heider, D. Encodings and models for antimicrobial peptide classification for multi-resistant pathogens. BioData Min. 2019, 12, 7. [Google Scholar] [CrossRef]
- Loose, C.; Jensen, K.; Rigoutsos, I.; Stephanopoulos, G. A linguistic model for the rational design of antimicrobial peptides. Nature 2006, 443, 867–869. [Google Scholar] [CrossRef]
- Khabbaz, H.; Karimi-Jafari, M.H.; Saboury, A.A.; BabaAli, B. Prediction of antimicrobial peptides toxicity based on their physico-chemical properties using machine learning techniques. BMC Bioinform. 2021, 22, 549. [Google Scholar] [CrossRef]
- Söylemez, G.; Yousef, M.; Kesmen, Z.; Büyükkiraz, M.E.; Bakir-Gungor, B. Prediction of Linear Cationic Antimicrobial Peptides Active against Gram-Negative and Gram-Positive Bacteria Based on Machine Learning Models. Appl. Sci. 2022, 12, 3631. [Google Scholar] [CrossRef]
- Veltri, D.; Kamath, U.; Shehu, A. Deep learning improves antimicrobial peptide recognition. Bioinformatics 2018, 34, 2740–2747. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Shin, T.H.; Kim, M.O.; Lee, G. AIPpred: Sequence-Based Prediction of Anti-inflammatory Peptides Using Random Forest. Front. Pharmacol. 2018, 9, 276. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, P.; Kim, Y.; Haste-Andersen, P.; Beaver, J.; Bourne, P.E.; Bui, H.-H.; Buus, S.; Frankild, S.; Greenbaum, J.; et al. Immune epitope database analysis resource (IEDB-AR). Nucleic Acids Res. 2008, 36, W513–W518. [Google Scholar] [CrossRef] [PubMed]
- Fleri, W.; Paul, S.; Dhanda, S.K.; Mahajan, S.; Xu, X.; Peters, B.; Sette, A. The Immune Epitope Database and Analysis Resource in Epitope Discovery and Synthetic Vaccine Design. Front. Immunol. 2017, 8, 278. Available online: https://www.frontiersin.org/articles/10.3389/fimmu.2017.00278 (accessed on 18 March 2023). [CrossRef]
- Dong, J.; Yao, Z.-J.; Zhang, L.; Luo, F.; Lin, Q.; Lu, A.-P.; Chen, A.F.; Cao, D.-S. PyBioMed: A python library for various molecular representations of chemicals, proteins and DNAs and their interactions. J. Cheminform. 2018, 10, 16. [Google Scholar] [CrossRef]
- Yousef, M.; Jung, S.; Showe, L.C.; Showe, M.K. Recursive Cluster Elimination (RCE) for classification and feature selection from gene expression data. BMC Bioinform. 2007, 8, 144. [Google Scholar] [CrossRef]
- Yousef, M.; Bakir-Gungor, B.; Jabeer, A.; Goy, G.; Qureshi, R.; Showe, L.C. Recursive Cluster Elimination based Rank Function (SVM-RCE-R) implemented in KNIME. F1000Research 2021, 9, 1255. [Google Scholar] [CrossRef]
- Yousef, M.; Jabeer, A.; Bakir-Gungor, B. SVM-RCE-R-OPT: Optimization of Scoring Function for SVM-RCE-R. In Database and Expert Systems Applications—DEXA 2021 Workshops; Kotsis, G., Tjoa, A.M., Khalil, I., Moser, B., Mashkoor, A., Sametinger, J., Fensel, A., Martinez-Gil, J., Fischer, L., Czech, G., et al., Eds.; Springer: Cham, Switzerland, 2021; Volume 1479, pp. 215–224. [Google Scholar] [CrossRef]
- Yousef, M.; Abdallah, L.; Allmer, J. maTE: Discovering expressed interactions between microRNAs and their targets. Bioinformatics 2019, 35, 4020–4028. [Google Scholar] [CrossRef]
- Yousef, M.; Ülgen, E.; Sezerman, O.U. CogNet: Classification of gene expression data based on ranked active-subnetwork-oriented KEGG pathway enrichment analysis. PeerJ Comput. Sci. 2021, 7, e336. [Google Scholar] [CrossRef]
- Yousef, M.; Goy, G.; Mitra, R.; Eischen, C.M.; Jabeer, A.; Bakir-Gungor, B. miRcorrNet: Machine learning-based integration of miRNA and mRNA expression profiles, combined with feature grouping and ranking. PeerJ 2021, 9, e11458. [Google Scholar] [CrossRef] [PubMed]
- Yousef, M.; Goy, G.; Bakir-Gungor, B. miRModuleNet: Detecting miRNA-mRNA Regulatory Modules. Front. Genet. 2022, 13, 767455. [Google Scholar] [CrossRef] [PubMed]
- Yousef, M.; Ozdemir, F.; Jaber, A.; Allmer, J.; Bakir-Gungor, B. PriPath: Identifying dysregulated pathways from differential gene expression via grouping, scoring, and modeling with an embedded feature selection approach. BMC Bioinform. 2023, 24, 60. [Google Scholar] [CrossRef] [PubMed]
- Yazici, M.U.; Marron, J.S.; Bakir-Gungor, B.; Zou, F.; Yousef, M. Invention of 3Mint for feature grouping and scoring in multi-omics. Front. Genet. 2023, 14, 1093326. Available online: https://www.frontiersin.org/articles/10.3389/fgene.2023.1093326 (accessed on 18 March 2023). [CrossRef]
- Yousef, M.; Sayıcı, A.; Bakir-Gungor, B. Integrating Gene Ontology Based Grouping and Ranking into the Machine Learning Algorithm for Gene Expression Data Analysis. In Database and Expert Systems Applications—DEXA 2021 Workshops; Springer: Cham, Switzerland, 2021; pp. 205–214. [Google Scholar] [CrossRef]
- Yousef, M.; Kumar, A.; Bakir-Gungor, B. Application of Biological Domain Knowledge Based Feature Selection on Gene Expression Data. Entropy 2020, 23, 2. [Google Scholar] [CrossRef]
- Xu, Q.-S.; Liang, Y.-Z. Monte Carlo cross validation. Chemom. Intell. Lab. Syst. 2001, 56, 1–11. [Google Scholar] [CrossRef]
- Kolde, R.; Laur, S.; Adler, P.; Vilo, J. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics 2012, 28, 573–580. [Google Scholar] [CrossRef]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Thiel, K.; Wiswedel, B. KNIME—The Konstanz information miner. ACM SIGKDD Explor. Newsl. 2009, 11, 26–31. [Google Scholar] [CrossRef]
- Brown, G.; Pocock, A.; Zhao, M.-J.; Lujan, M. Conditional Likelihood Maximisation: A Unifying Framework for Information Theoretic Feature Selection. J. Mach. Learn. Res. 2012, 13, 27–66. [Google Scholar]
- Peng, H.; Long, F.; Ding, C. Feature selection based on mutual information criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 2005, 27, 1226–1238. [Google Scholar] [CrossRef] [PubMed]
- Kent, J.T. Information gain and a general measure of correlation. Biometrika 1983, 70, 163–173. [Google Scholar] [CrossRef]
- Chen, T.; He, T. xgboost: eXtreme Gradient Boosting. R Package Vers. 0.4-2 2015, 4, 1–4. [Google Scholar]
- Liang, J.; Hou, L.; Luan, Z.; Huang, W. Feature Selection with Conditional Mutual Information Considering Feature Interaction. Symmetry 2019, 11, 858. [Google Scholar] [CrossRef]
- Ding, C.; Peng, H. Minimum Redundancy Feature Selection from Microarray Gene Expression Data. J. Bioinform. Comput. Biol. 2005, 03, 185–205. [Google Scholar] [CrossRef]
- Wang, P.; Hu, L.; Liu, G.; Jiang, N.; Chen, X.; Xu, J.; Zheng, W.; Li, L.; Tan, M.; Chen, Z.; et al. Prediction of Antimicrobial Peptides Based on Sequence Alignment and Feature Selection Methods. PLoS ONE 2011, 6, e18476. [Google Scholar] [CrossRef] [PubMed]
- Teimouri, H.; Medvedeva, A.; Kolomeisky, A.B. Bacteria-Specific Feature Selection for Enhanced Antimicrobial Peptide Activity Predictions Using Machine-Learning Methods. J. Chem. Inf. Model. 2023, 63, 1723–1733. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.; Karnik, S.; Nilawe, P.; Jayaraman, V.K.; Idicula-Thomas, S. ClassAMP: A Prediction Tool for Classification of Antimicrobial Peptides. IEEE/ACM Trans. Comput. Biol. Bioinform. 2012, 9, 1535–1538. [Google Scholar] [CrossRef] [PubMed]
- Tornesello, A.L.; Borrelli, A.; Buonaguro, L.; Buonaguro, F.M.; Tornesello, M.L. Antimicrobial Peptides as Anticancer Agents: Functional Properties and Biological Activities. Molecules 2020, 25, 2850. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Feature Set | Number of Features |
|---|---|---|
| Physico-chemical | Sequence Length, Normalized Hydrophobic Moment, Normalized Hydrophobicity, Net Charge, Isoelectric Point, Penetration Depth… | 10 |
| Amino acid composition | A, C, E, D, G, F, I, H, K, M… | 20 |
| Dipeptide composition | GW, GV, GT, GS, GR, GQ, ME, MD, MG, MF, MA, MC, MM, ML, MN… | 400 |
| Physico-chemical composition | _NormalizedVDWVC2, _PolarizabilityC2, _PolarizabilityC3, _ChargeC1… | 21 |
| Physico-chemical transition | _SecondaryStrT13, _SecondaryStrT12, _HydrophobicityT23, _NormalizedVDWVT23, _ChargeT12… | 21 |
| Physico-chemical distribution | _NormalizedVDWVD1075, _PolarityD1075, _SecondaryStrD2075, _SolventAccessibilityD1100… | 105 |
| Normalized Moreau–Broto autocorrelation | MoreauBrotoAuto_ResidueASA28, MoreauBrotoAuto_ResidueVol30… | 240 |
| Moran autocorrelation | MoranAuto_FreeEnergy8, MoranAuto_FreeEnergy9, MoranAuto_Steric8 … | 240 |
| Geary autocorrelation | GearyAuto_Mutability23, GearyAuto_Mutability21, GearyAuto_FreeEnergy24, … | 240 |
| Sequence order coupling number | QSO26, QSO27, QSO_ex50, QSO_ex24, QSO_ex18, QSO_ex19… | 60 |
| Quasi-sequence-order | Taugrant23, taugrant24, tausw8, tausw9, tausw6, tausw7… | 100 |
| Pseudo-amino acid composition | PAAC34, PAAC35, APAAC20, PAAC38, PAAC39… | 50 |
| #Groups | #Features (Mean) | Acc (Mean) | Sn (Mean) | Sp (Mean) | F-Measure (Mean) | AUC (Mean) | Pr (Mean) |
|---|---|---|---|---|---|---|---|
| 10 | 1039.99 | 0.92 | 0.91 | 0.93 | 0.91 | 0.98 | 0.92 |
| 9 | 807.19 | 0.93 | 0.93 | 0.92 | 0.92 | 0.98 | 0.91 |
| 8 | 714.01 | 0.94 | 0.94 | 0.93 | 0.93 | 0.98 | 0.92 |
| 7 | 571.08 | 0.92 | 0.93 | 0.92 | 0.91 | 0.98 | 0.90 |
| 6 | 439.45 | 0.92 | 0.93 | 0.91 | 0.91 | 0.98 | 0.90 |
| 5 | 306.47 | 0.92 | 0.94 | 0.90 | 0.91 | 0.98 | 0.89 |
| 4 | 190.91 | 0.92 | 0.94 | 0.91 | 0.91 | 0.98 | 0.90 |
| 3 | 121.25 | 0.93 | 0.95 | 0.91 | 0.92 | 0.98 | 0.90 |
| 2 | 60.6 | 0.93 | 0.95 | 0.92 | 0.93 | 0.99 | 0.91 |
| 1 | 10 | 0.95 | 0.98 | 0.93 | 0.95 | 0.99 | 0.92 |
| #Groups | #Features (Mean) | Acc (Mean) | Sn (Mean) | Sp (Mean) | F-Measure (Mean) | AUC (Mean) | Pr (Mean) |
|---|---|---|---|---|---|---|---|
| 10 | 1026.75 | 0.88 | 0.69 | 0.96 | 0.77 | 0.95 | 0.91 |
| 9 | 795.75 | 0.88 | 0.72 | 0.96 | 0.79 | 0.95 | 0.90 |
| 8 | 657.26 | 0.87 | 0.70 | 0.95 | 0.77 | 0.95 | 0.89 |
| 7 | 526.35 | 0.88 | 0.73 | 0.94 | 0.78 | 0.95 | 0.88 |
| 6 | 351.87 | 0.88 | 0.75 | 0.94 | 0.80 | 0.95 | 0.87 |
| 5 | 226.48 | 0.89 | 0.78 | 0.94 | 0.82 | 0.96 | 0.88 |
| 4 | 160.75 | 0.89 | 0.77 | 0.94 | 0.81 | 0.95 | 0.87 |
| 3 | 103.51 | 0.90 | 0.80 | 0.95 | 0.83 | 0.96 | 0.89 |
| 2 | 44.28 | 0.91 | 0.82 | 0.95 | 0.85 | 0.96 | 0.90 |
| 1 | 10 | 0.92 | 0.89 | 0.93 | 0.87 | 0.98 | 0.87 |
| #Groups | #Features (Mean) | Acc (Mean) | Sn (Mean) | Sp (Mean) | F-Measure (Mean) | AUC (Mean) | Pr (Mean) |
|---|---|---|---|---|---|---|---|
| 3 | 169.73 | 0.88 | 0.87 | 0.89 | 0.86 | 0.96 | 0.87 |
| 2 | 100.65 | 0.89 | 0.89 | 0.89 | 0.87 | 0.95 | 0.86 |
| 1 | 38.27 | 0.87 | 0.86 | 0.88 | 0.85 | 0.93 | 0.85 |
| #Groups | #Features (Mean) | Acc (Mean) | Sn (Mean) | Sp (Mean) | F-Measure (Mean) | AUC (Mean) | Pr (Mean) |
|---|---|---|---|---|---|---|---|
| 3 | 135.41 | 0.85 | 0.69 | 0.92 | 0.74 | 0.92 | 0.81 |
| 2 | 85.27 | 0.84 | 0.67 | 0.91 | 0.72 | 0.91 | 0.79 |
| 1 | 37.67 | 0.82 | 0.63 | 0.91 | 0.68 | 0.90 | 0.78 |
| Results for the Gram-Negative Dataset (10 Features, 100-Fold MCCV) | |||||||
|---|---|---|---|---|---|---|---|
| ML Method | FS Method | Accuracy | Sensitivity (Recall) | Specificity | Precision | AUC | F1 |
| LogitBoost | XGB | 0.96 ± 0.03 | 0.99 ± 0.03 | 0.94 ± 0.06 | 0.93 ± 0.07 | 0.97 ± 0.02 | 0.95 ± 0.04 |
| LogitBoost | IG | 0.96 ± 0.04 | 0.98 ± 0.03 | 0.94 ± 0.06 | 0.93 ± 0.07 | 0.97 ± 0.02 | 0.95 ± 0.04 |
| Adaboost | MRMR | 0.52 ± 0.09 | 0.95 ± 0.09 | 0.19 ± 0.22 | 0.48 ± 0.07 | 0.50 ± 0.13 | 0.63 ± 0.04 |
| RF | CMIM | 0.55 ± 0.11 | 0.97 ± 0.08 | 0.23 ± 0.23 | 0.50 ± 0.09 | 0.56 ± 0.14 | 0.65 ± 0.05 |
| RF | AMP-GSM | 0.95 ± 0.04 | 0.98 ± 0.03 | 0.93 ± 0.07 | 0.92 ± 0.08 | 0.99 ± 0.006 | 0.95 ± 0.05 |
| Results for the Gram-Positive Dataset (10 Features, 100-Fold MCCV) | |||||||
|---|---|---|---|---|---|---|---|
| ML Method | FS Method | Accuracy | Sensitivity (Recall) | Specificity | Precision | AUC | F1 |
| RF | IG | 0.90 ± 0.04 | 0.92 ± 0.09 | 0.89 ± 0.07 | 0.81 ± 0.11 | 0.94 ± 0.03 | 0.85 ± 0.06 |
| RF | XGB | 0.89 ± 0.05 | 0.88 ± 0.10 | 0.89 ± 0.08 | 0.81 ± 0.12 | 0.91 ± 0.05 | 0.83 ± 0.07 |
| RF | MRMR | 0.49 ± 0.17 | 0.88 ± 0.15 | 0.32 ± 0.31 | 0.41 ± 0.14 | 0.55 ± 0.14 | 0.53 ± 0.07 |
| RF | CMIM | 0.43 ± 0.14 | 0.94 ± 0.11 | 0.20 ± 0.24 | 0.36 ± 0.08 | 0.53 ± 0.11 | 0.51 ± 0.05 |
| RF | AMP-GSM | 0.92 ± 0.04 | 0.89 ± 0.10 | 0.93 ± 0.05 | 0.87 ± 0.09 | 0.98 ± 0.02 | 0.87 ± 0.06 |
| Gram-Negative Dataset | |||
|---|---|---|---|
| FS/CLSF * Method | Features Identified by FS Methods | Features Identified by AMP-GSM | Common Features between the Top Features of the FS Method and AMP-GSM |
| XGB/Logitboost | Net Charge MoranAuto_AvFlexibility8 Tilt Angle Normalized Hydrophobic Moment MoranAuto_Hydrophobicity15 MoranAuto_ResidueVol5 _ChargeC1 QSO_ex29 Isoelectric Point tausw2 | SequenceLength Normalized Hydrophobic Moment Normalized Hydrophobicity Net Charge Isoelectric Point Penetration Depth Tilt Angle Disordered Conformation Propensity Linear Moment Propensity to in vitro Aggregation | Net Charge Tilt Angle Normalized Hydrophobic Moment Isoelectric Point |
| Gram-Positive Dataset | |||
| IG/RF | Isoelectric Point Net Charge Disordered Conformation Propensity Normalized Hydrophobicity _ChargeC1 _PolarityC3 tausw9 taugrant6 _PolarityT13 tausw6 | SequenceLength Normalized Hydrophobic Moment Normalized Hydrophobicity Net Charge Isoelectric Point Penetration Depth Tilt Angle Disordered Conformation Propensity Linear Moment Propensity to in vitro Aggregation | Normalized Hydrophobicity Net Charge Isoelectric Point Disordered Conformation Propensity |
| Method | Evaluation | Sensitivity (%) | Specificity (%) | Accuracy (%) | AUC (%) | F1 Measure |
|---|---|---|---|---|---|---|
| DNN Model | 10-fold CV | 88.81 (±3.53) | 94.21 (±2.68) | 91.51 (±0.89) | 96.58 (±0.66) | - |
| CMIM-DT | 10-fold MCCV | 51.34 ± 0.17 | 51.40 ± 0.17 | 51.37 ± 0.03 | 51.37 ± 0.03 | 50.02 ± 0.09 |
| IG-RF | 10-fold MCCV | 88.70 ± 0.02 | 91.40 ± 0.02 | 90.05 ± 0.01 | 96.36 ± 0.007 | 89.91 ± 0.01 |
| mRMR-RF | 10-fold MCCV | 33.70 ± 0.18 | 67.41 ± 0.21 | 50.56 ± 0.03 | 50.80 ± 0.05 | 37.80 ± 0.14 |
| AMP-GSM Model | 10-fold MCCV | 91.01 (±0.23) | 92.97(±0.03) | 91.71 (±0.13) | 97.07 (±0.06) | 91.59 (±0.15) |
| Method | Evaluation Set | Accuracy | Sensitivity | Specificity | AUC |
|---|---|---|---|---|---|
| AIPpred | 5-Fold CV | 0.73 | 0.75 | 0.71 | 0.80 |
| ERT | 5-Fold CV | 0.73 | 0.73 | 0.72 | 0.79 |
| SVM | 5-Fold CV | 0.65 | 0.64 | 0.67 | 0.70 |
| k-NN | 5-Fold CV | 0.64 | 0.51 | 0.77 | 0.69 |
| CMIM-LogitBoost | 5-Fold MCCV | 0.54 | 0.67 | 0.40 | 0.55 |
| IG-AdaBoost | 5-Fold MCCV | 0.69 | 0.66 | 0.72 | 0.73 |
| mRMR-LogitBoost | 5-Fold MCCV | 0.50 | 0.79 | 0.20 | 0.50 |
| AMP-GSM Model | 5-Fold MCCV | 0.99 | 1 | 0.99 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Söylemez, Ü.G.; Yousef, M.; Bakir-Gungor, B. AMP-GSM: Prediction of Antimicrobial Peptides via a Grouping–Scoring–Modeling Approach. Appl. Sci. 2023, 13, 5106. https://doi.org/10.3390/app13085106
Söylemez ÜG, Yousef M, Bakir-Gungor B. AMP-GSM: Prediction of Antimicrobial Peptides via a Grouping–Scoring–Modeling Approach. Applied Sciences. 2023; 13(8):5106. https://doi.org/10.3390/app13085106
Chicago/Turabian StyleSöylemez, Ümmü Gülsüm, Malik Yousef, and Burcu Bakir-Gungor. 2023. "AMP-GSM: Prediction of Antimicrobial Peptides via a Grouping–Scoring–Modeling Approach" Applied Sciences 13, no. 8: 5106. https://doi.org/10.3390/app13085106
APA StyleSöylemez, Ü. G., Yousef, M., & Bakir-Gungor, B. (2023). AMP-GSM: Prediction of Antimicrobial Peptides via a Grouping–Scoring–Modeling Approach. Applied Sciences, 13(8), 5106. https://doi.org/10.3390/app13085106