Abstract
The C-3 functionalization of 1H-indazole could produce a lot of highly valuable pharmaceutical precursors, which could be used for the treatment of cancer and many other inflammatory diseases. This work was focused on the C-3 functionalization of 1H-indazole through Suzuki–Miyaura cross-coupling of 3-iodo-1H-indazole with organoboronic acids, catalyzed by various palladium catalysts immobilized over imidazolium ionic liquids, as well as catalyst recycling. A series of reaction parameters, including the substrate, catalyst, and ionic liquid, were fully investigated. It is significant to note that the yields of the present Suzuki–Miyaura cross-coupling were mainly determined by the catalyst and the solvent used, more than the chemical structure of the substrate. Furthermore, ferrocene-based divalent palladium complexes showed better catalytic outputs compared to simple palladium salts. Moreover, using two imidazolium ionic liquids, BMImX (BMIm+ = 1-n-butyl-3-methylimidazolium, X− = BF4−, PF6−) not only improved the yields of cross-coupled products, but also avoided the formation of Pd(0) black, as compared to the non-ionic liquid facilitated reactions, and simultaneously making catalyst recycling more effective. On average, BMImBF4 performed better than BMImPF6. Additionally, scientific calculations revealed that 1,1′-bis(diphenylphosphino)ferrocene-palladium(II)dichloride dichloromethane complex (PdCl2(dppf)) showed a lower energy barrier in the formation of intermediates than [1,1′-bis(di-tert-butylphosphino)ferrocene]dichloropalladium(II) (PdCl2(dtbpf)), leading to higher catalytic outputs. This work may contribute to the development of 1H-indazole-derived new pharmaceuticals.
1. Introduction
Indazoles, as a series of nitrogen-containing bicyclic compounds, are composed of an electron-rich pyrazole, along with a fused benzene ring [1]. Indazole has a ten-π electron aromatic heterocyclic structure, including two tautomers, 1H-indazole and 2H-indazole (Figure 1a,b) [1,2]. It was previously reported that 1H-indazole seems much more thermodynamically stable and abundant than 2H-tautomer in both the gas phase and aqueous solution; however, some 2H-indazole derivatives are also found in nature, although their appearance is rare [2]. In practice, both 1H- and 2H-indazole derivatives have shown great potential for pharmaceutical applications (Figure 1c–e).
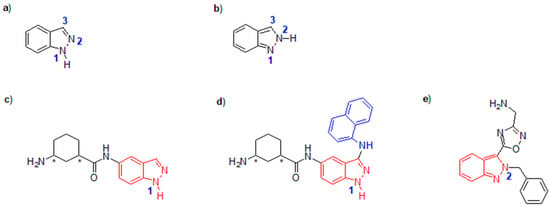
Figure 1.
The tautomers of indazole, as well as 1H- and 2H-indazoles-derived potential pharmaceuticals: (a) 1H-indazole; (b) 2H-indazole; (c) SR-17398 (anticancer), ULK1 inhibitor IC50 = 22.4 μmol L−1, * mixture of 4 stereoisomers; (d) SR-17398 derivative (anticancer), ULK1 inhibitor IC50 = 45 nmol L−1, * mixture of 4 stereoisomers; (e) Neuroprotective sodium channel modulator.
Overall, the pharmaceutical value of 1H-indazoles mainly lies in their antitumor activities [3], while 2H-indazole and its derivatives exhibit more versatile bioactivities, such as anticancer, antiplatelet, antiproliferative, anti-tubercular, antimalarial, antimicrobial, antiviral, and anti-inflammatory activities [4]. For example, cancer has been proven to be a disease marked by uncontrollable growth of abnormal cells in the human body, which can be initiated in any organ of the body and further transferred to distant organs [3]. In recent years, it has been proposed that one of the most probable pathways leading to this disease is the mutation of various types of genes and kinases (such as BRAF or KIT, etc.), causing diverse cellular anomalies and leading to the occurrence of cancer [3]. In practice, in addition to the traditional treatments for cancer using surgery, radiation therapy, chemotherapy, immunotherapy, or hormonal therapy, inhibition of kinase activity through blocking its improper phosphorylation represents a highly promising method for curing cancer [5]. 1H-indazole compounds have been selected as potential kinase inhibitors for treatment of cancer, mainly because their nitrogen-containing and other steric and electronic properties can deform the configuration of target protein kinase through formations of hydrogen bond, π-π stacking, or coordination linkage [6].
On the other hand, although the natural abundance of 2H-indazole is lower than that of 1H-indazole, the pharmaceutical potentials of 2H-indazole are still very attractive, probably owing to its unique nitrogen-containing heterocyclic structure, which can bind to protein kinase, affecting the progress of many diseases [2]. Therefore, on the basis of these findings, synthesizing both functional 1H- and 2H-indazole derivatives could contribute to the development of new pharmaceuticals.
The present developments in the synthetic functionalization of both 1H- and 2H-indazole can be generally divided into three patterns, including N- and C-functionalization, as well as fused cyclization [7]. N-functionalization means the catalytic coupling of the N-H group of indazole with electrophiles, leading to production of alkylation and arylation [8]. The C-functionalization of indazole refers to the alkylation [9], arylation [10], alkoxylation [11], amination [12], halogenation [13], and phosphonylation [14] of C-3 of indazole in a catalytic or photocatalytic manner.
Lastly, some specially functionalized indazole derivatives, such as 3-aminoindazole, can be reacted with enaminone and bromodifluoroacetic acid through three-component cyclization, leading to a fused-ring compound [15]. In light of radical capture experiments, the mechanism was illustrated as addition-elimination mechanism (SNV) mode [15]. Generally speaking, chemical functionalization of indazole usually demands a catalyst, along with an appropriate solvent, temperature, and reaction time, meaning that future large-scale production would involve many economic and environmental issues.
The C-functionalization carried out on C-3 position of 1H-indazole played a key role in promoting the pharmaceutical activity of indazole derivatives. For example, the abnormal ignition (phosphorylation) of unc-51-like kinase 1 (ULK1), a ubiquitously expressed protein activated by stress-induced autophagy under many conditions, decreases the efficiency of the current chemotherapeutics for cancer, and meanwhile increases the growth of cancer [16]. Therefore, using small molecule inhibitors of ULK1 such as SR-17398 (Figure 1c) could block the phosphorylation of ULK1 and simultaneously enhance the effect of chemotherapy [17]. The backbone of SR-17398 is indazole, whose nitrogen-containing heterocyclic structure can deform the configuration of ULK1, finally blocking the activation of ULK1 [18]. However, if the C-3 position of indazole on SR-17398 is functionalized by introducing a 1-naphthylamino group (Figure 1d), the half inhibitory concentration (IC50) is greatly decreased, and the C-3 position of indazole showed an influence on the inhibitory deformation of ULK1 [18]. On the other hand, C-3 functionalized 2H-indazole also showed high activity for the treatment of neurological diseases (Figure 1e) [19]. Functionalization of C-3 of indazoles allows developing new and powerful pharmaceuticals.
In practice, direct functionalization of C-3 on 1H-indazole suffers from several drawbacks. First, direct C-3 alkylation of 1H-indazole through the Minisci reaction (addition of nucleophilic carbon radical to protonated electron-deficient aromatic heterocycle) produced a very low yield, which can be ascribed to the unique electronic structure of the five-membered nitrogen-containing heterocycle [20]. Next, direct C-3 arylation of 1H-indazole required an uncommon catalyst and challenging reaction conditions [4]. These results delay progress to large-scale production. All in all, in view of the present progress in C-3 functionalization, exploring new synthetic methods deserves more attention.
The Suzuki–Miyaura cross-coupling reaction represents a palladium (Pd)-catalyzed C-C bond formation, already standing as a powerful methodology for the construction of sophisticated molecules for the last two decades [21,22]. First of all, taking into account the mechanism of Pd-catalyzed Suzuki–Miyaura coupling of aryl halide with organoboronic acid, a Pd(0) to Pd(II) cycle is widely accepted as the most reasonable pathway [22]. In detail, the oxidative addition of aryl halide to Pd(0) generates a Pd(II) intermediate, while the transmetalation of the Pd(II) intermediate, followed by the reductive elimination, produces the coupled product and regenerates the active Pd(0) [22,23].
Next, Pd-catalyzed Suzuki–Miyaura coupling has shown several significant advantages, particularly regarding its broad functional group tolerance and mild reaction conditions [24,25]. For instance, the suitable electrophiles cover sterically congested molecules, inert aryl, and vinyl chlorides, as well as sulfonate derivatives, while the nucleophiles also include thermally unstable polyfluorophenyl and 2-heteroaryl boron reagents [25]. Meanwhile, the organoboron compounds (electrophile) exhibit high stabilities towards air and moisture [24]. Therefore, although 1H-indazole has a sophisticated nitrogen-containing heterocycle, it is promising to develop Pd-catalyzed Suzuki–Miyaura coupling reaction for C-3 functionalization of 1H-indazole.
On the other hand, the recovery and recycling of Pd catalysts has not been well addressed. Herein, depending on the catalyst precursor, the known Pd catalytic systems can be divided into homogeneous and heterogeneous systems. The former series mainly refers to soluble Pd salts or complexes having various ligands, including phosphine and N-heterocyclic carbenes [25], while the latter usually signifies the Pd nanoparticles, clusters, or single atoms immobilized over solid supports such as silica, polymers, or activated carbon [26]. No matter which kind of catalyst is used, the active Pd species usually shows a tendency to be reduced and subsequently agglomerated to inactive Pd(0) black during catalysis [26,27]. In order to decrease this unfavorable inclination, ionic nitrogen compounds such as ammonium, amidinium, and pyridinium salts were ever introduced to stabilize the active Pd (Pd(0) or Pd2+) species, where the effects of stabilization, fixation, modulation of catalyst morphology, and reaction medium, as well as phase-transfer property on catalysis, were highly anticipated [26]. However, this endeavor was not completely effective, and quite a few ionic nitrogen compounds showed inhibitory rather than accelerating or stabilizing effects on the Pd-catalyzed reactions [26]. Obviously, there is still a lot of room to explore efficient methods to immobilize expensive Pd catalysts for recycling.
Traditionally, immobilization of Pd salts or complexes into solid supports has created heterogeneous catalysts for the Suzuki–Miyaura coupling reaction, and sometimes solid supports can serve as base generated in situ in the catalytic reaction [26]. However, the immobilization present cannot avoid leaching and the degradation of the Pd catalyst, probably because the dispersion of solvent (water and organic molecules) into the pores of solid support, which destroys the chemical or physical linkage between the Pd component and support [26]. Therefore, in order to securely fix Pd, new support materials, instead of the common solid supports (silicate, aluminum oxide or other known solid acids), deserve further exploration.
The catalysis carried out in ionic liquids (ILs) has experienced a tremendous growth over the past two decades [28]. There are numerous examples of versatile catalytic reactions that have been effectively accomplished in this kind of media [28]. Theoretically, room temperature ionic liquids (usually denoted as RTILs or ILs) are composed of ions with melting points lower than 100 °C, which are nonvolatile, nonflammable, chemically stable, easy to recover, and eco-friendly for many chemical reactions, arousing much interest in their usage as either a catalyst or solvent [28,29].
Furthermore, the great enthusiasm for exploring ILs in catalysis has not only been propelled by the eco-friendly properties of ILs, but also owing to the unique polar or ionic nature of ILs, which the common molecular solvents do not show, subsequently immobilizing catalytic components tightly and lastingly [28,30]. Therefore, the leaching of catalytically active components from ILs would become slower and minimal, product extraction and purification become more convenient, and the catalyst activity could be retained for a long time [31]. Meanwhile, certain catalytic reactions that cannot be performed in common organic solvents seem more possible in ILs [32].
Due to the demand for developing new 1H-indazole-derived pharmaceuticals, the present low efficiency of C-3 functionalization of 1H-indazole, as well as the high potential of Pd-catalyzed Suzuki–Miyaura coupling in ILs, this work aimed to develop the C-3 functionalization of 1H-indazole reactions using Pd-catalyzed Suzuki–Miyaura coupling. In order to improve the recovery and recycling of the Pd catalyst, a series of imidazolium ILs BMImX (BMIm+ = 1-n-butyl-3-methylimidazolium; X− = PF6−, BF4−) were introduced as a support material, which not only had controllable hydrophilicity, owing to variation of the anions, but also contained a nitrogen atom, showing coordination effects for stabilizing Pd ions. Lastly, on the basis of theoretical calculations, the catalytic mechanism was summarized, and comparison of Pd catalysts was carried out. In general, this work demonstrates a new and recyclable strategy for C-3 functionalization of 1H-indazole, contributing to the development of indazole-based pharmaceuticals.
2. Experimental
2.1. Starting Materials
1H-indazole (98%), di-tert-butyl dicarbonate ((Boc)2O, 99%), 4-dimethylaminopyridine (DMAP, 99%), 2-furanboronic acid (97%; compound 4, Scheme 1), 4-methoxycarbonylphenylboronic acid (97%; compound 7, Scheme 1), 3-methoxycarbonylphenylboronic acid (97%; compound 10, Scheme 1), lithium hydroxide monohydrate (LiOH·H2O, 98%) were bought from Shanghai Macklin Biochemical Technology Co., Ltd. (Shanghai, China) and used without purification.
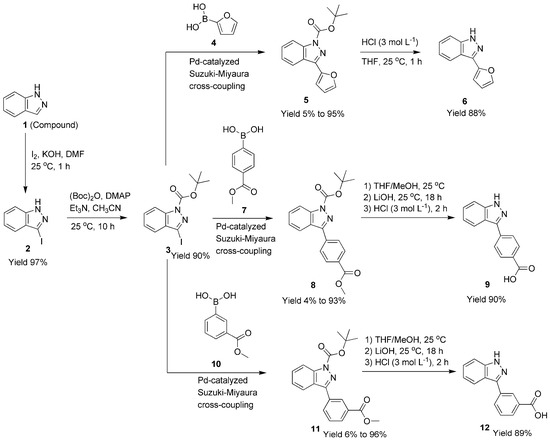
Scheme 1.
C-3 functionalization of 1H-indazole through a catalytic Suzuki–Miyaura cross-coupling reaction.
Tetrakis(triphenylphosphine)palladium(0) (Pd(PPh3)4, 99%), palladium(II) acetate (Pd(OAc)2, 99% purity, Pd 47%), palladium(II) chloride (PdCl2, 99% purity, Pd 59%), 1,1′-bis(diphenylphosphino)ferrocene-palladium(II)dichloride dichloromethane complex (PdCl2(dppf), 98% purity, Pd >13%), and [1,1′-bis(di-tert-butylphosphino)ferrocene]dichloropalladium(II) (PdCl2(dtbpf), 98%) were all commercially available from Sigma-Aldrich (St. Louis, MO, USA). Two imidazolium ILs, BMImX (BMIm+ = 1-n-butyl-3-methylimidazolium; X− = BF4−, PF6−; both 98% purities) were bought from Alfa. Deuterium reagents used in NMR, including CDCl3, CD3OD, and DMSO-d6, were purchased from Shanghai Macklin Biochemical Technology Co., Ltd. and used without purification. Other organic solvents were provided by local distributors and used after purification in our laboratory.
2.2. Analytical Instruments
The melting points of compounds 2, 3, 5, 6, 8, 9, 11, and 12 (Scheme 1) were tested using WRR-Y melting point apparatus, Shanghai INESA Physico-Optical Instrument Co., Ltd. (Shanghai, China). 1H NMR was tested on a Bruker ADVANCE III instrument (400 MHz), 13C NMR was measured on a Bruker ADVANCE III instrument (101 MHz). ESI-HRMS was obtained on micrOTOF-Q II, Bruker Daltonics equipment (Bremen, Germany). The ESI-HRMS of compounds 9 and 12 was tested using time of flight secondary ion mass spectrometry (TOF-SIMS), M6, IONTOF equipment (Münster, Germany). The C, H, and N elemental analyses were performed on an Elementar VarioEL III instrument, Elementar equipment (Langenselbold, Germany). The FT-IR spectra of the synthesized samples were taken in potassium bromide pellets using a Bruker Tensor 27 spectrometer, BRUKER equipment (Bremen, Germany).
2.3. Synthesis of 3-Iodo-1H-indazole (Compound 2)
As shown in Scheme 1, 3-iodo-1H-indazole (compound 2) was prepared by iodination of 1H-indazole (compound 1), according to a previous method developed by Bocchi and Palli [33]. In practice, iodine (16.0 g, 0.064 mol) and potassium hydroxide pellets (6.72 g, 0.12 mol) were combined with a DMF solution (60 mL) of indazole (3.77 g, 0.032 mol) at 25 °C under magnetic stirring for 1 h. The reaction mixture was then poured into aqueous NaHSO3 (10%, 200 mL) and extracted with diethyl ether (2 × 150 mL). The combined organic layers were washed with distilled water (2 × 150 mL) and brine (2 × 150 mL) and dried over anhydrous MgSO4, and the organic solvent was evaporated under reduced pressure, to give a light yellow solid (7.28 g, yield of 97%). Mp 142.7–144.3 °C (lit. mp 141 °C [34]). 1H NMR (400 MHz, Methanol-d4) δH, ppm: 7.50 (1H, dt, J = 1.7, 7.4 Hz, ArH), 7.44 (1H, d, J = 1.7 Hz, ArH), 7.43 (1H, d, J = 1.7 Hz, ArH), 7.19 (1H, m, ArH) (Figure S1, Section S1, Supplementary Materials). 13C NMR (101 MHz, CD3OD) δC, ppm: 140.75, 127.55, 127.19, 121.32, 120.60, 110.07, 92.02 (Figure S2, Section S2, Supplementary Materials). Anal. Calcd for C7H5N2I: C, 34.42; H, 2.04; N, 11.47. Found: C, 34.54; H, 1.64; N, 11.37. FT-IR (KBr) σ, cm−1: 3153 (vs, N-H), 1620 (s, C=N), 1319 (s, C-N), 424 (m, C-I).
2.4. Synthesis of Tert-butyl-3-iodo-1H-indazole-1-carboxylate (Compound 3)
As shown in Scheme 1, 3-iodo-1H-indazole (compound 2, 1.22 g, 5 mmol), (Boc)2O (1.2 g, 5.5 mmol), DMAP (30 mg, 0.24 mmol), and triethylamine (1.1 mL, 0.76 g, 7.5 mmol) were combined with CH3CN (10 mL) in a round-bottom flask (250 mL) with a condenser and sealed with a balloon. After vigorous stirring for 10 h at 25 °C, the dark orange solution was evaporated to dryness under reduced pressure, to completely remove the solvent and triethylamine. The residue was re-dissolved in diethyl ether (150 mL), then washed with brine (2 × 50 mL), dried over anhydrous MgSO4, and then concentrated under reduced pressure. Crude product was purified using flash chromatography (SiO2, 100–200 mesh; petroleum ether/ethyl acetate, 5/1, v/v; adding a few drops of triethylamine in eluent, 10 drops of triethylamine vs. 200 mL eluent) to give pure compound 3 (yellow oil, solidified overnight; 1.54 g, 90% yield). Mp 117.2–119.5 °C. 1H NMR (400 MHz, CDCl3) δH, ppm: 1.72 (9H, s, tert-butyl), 7.38 (1H, t, J = 8.0 Hz, ArH), 7.50 (1H, d, J = 8.0 Hz, ArH), 7.59 (1H, t, J = 7.6 Hz, ArH), 8.11 (1H, d, J = 8.4 Hz, ArH) (Figure S3, Section S3, Supplementary Materials). 13C NMR (101 MHz, CDCl3) δC, ppm: 148.44, 139.67, 130.26, 130.07, 124.29, 122.07, 114.64, 103.05, 85.59, 28.22 (Figure S4, Section S4, Supplementary Materials). ESI-HRMS (positive, m/z): 366.9901 (Calcd. for [M+Na]+ 367.1374) and 711.0117 (Calcd. for [2M+Na]+ 711.2851) (Figure S5, Section S5, Supplementary Materials). Anal. Calcd for C12H13N2O2I: C, 41.86; H, 3.77; N, 8.13. Found: C, 41.85; H, 3.22; N, 8.25. FT-IR (KBr) σ, cm−1: 2983 (s, methyl on tert-butyl), 1726 (vs, C=O), 1610 (m, C=N), 1381 (vs, C-N), 1234 (vs, C-O) (Figure S6, Section S6, Supplementary Materials).
2.5. Synthesis of Tert-butyl-3-(2-furyl)-1H-indazole-1-carboxylate (Compound 5)
As shown in Scheme 1, compound 5 was obtained through a Pd-catalyzed Suzuki–Miyaura coupling reaction, which is comprehensively described in Section 2.11. Yellow oils (yields of 5–95%, Table 1 and Figure 2). 1H NMR (400 MHz, CDCl3) δH, ppm: 1.77 (9H, s, tert-butyl), 7.41 (1H, d, J = 8.0 Hz, ArH), 7.49–7.51 (3H, m, ArH), 7.54–7.59 (2H, m, H on furyl), 8.21 (1H, t, J = 9.2 Hz, H on furyl) (Figure S7, Section S7, Supplementary Materials). 13C NMR (101 MHz, CDCl3) δC, ppm: 149.35, 147.77, 143.61, 141.63, 140.61, 129.19, 124.14, 123.41, 122.12, 114.81, 111.81, 110.16, 85.16, 28.25 (Figure S8, Section S8, Supplementary Materials). ESI-HRMS (positive, m/z): 307.1028 (Calcd. for [M+Na]+ 307.2989) (Figure S9, Section S9, Supplementary Materials). Anal. Calcd for C16H16N2O3: C, 67.60; H, 5.63; N, 9.86. Found: C, 68.21; H, 6.22; N, 9.25. FT-IR (KBr) σ, cm−1: 3156 (m, furyl), 2972 (m, methyl on tert-butyl), 1738 (vs, C=O), 1614 (m, C=N), 1510 (m, furyl), 1422 (m, furyl), 1374 (vs, C-N), 1243 (vs, C-O) (Figure S10, Section S10, Supplementary Materials).

Table 1.
Catalytic Suzuki–Miyaura cross-coupling of compounds 3 with 4.
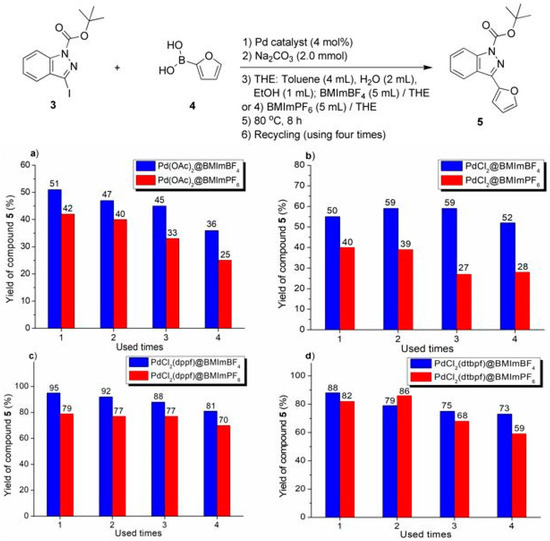
Figure 2.
Recycling experiments on Suzuki–Miyaura cross-coupling of compounds 3 with 4 catalyzed by Pd catalyst, immobilized over imidazolium ionic liquid: (a) Pd(OAc)2 over ILs; (b) PdCl2 over ILs; (c) PdCl2(dppf) over ILs; (d) PdCl2(dtbpf) over ILs.
2.6. Synthesis of 3-(2-Furyl)-1H-indazole (Compound 6)
As shown in Scheme 1, compound 5 (0.586 g, 2 mmol) was dissolved in THF (5 mL) under continuous stirring at 25 °C, the solution was then acidified through dropwise addition of HCl solution (3 mol/L) until the pH value reached 2. After stirring for 1 h, the mixture was diluted with CH2Cl2 (100 mL), the organic layer was separated and washed with brine (100 mL), dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified using flash chromatography (SiO2, 100–200 mesh; petroleum ether/ethyl acetate 5/1, v/v, adding a few drops of triethylamine in eluent, 10 drops of triethylamine vs. 200 mL eluent) to give compound 6 (yellow solid, 0.33 g, yield of 88%). Mp 163.6–164.2 °C (lit. mp 165–166 °C [35]). 1H NMR (400 MHz, CDCl3) δH, ppm: 8.00 (1H, d, J = 8.2 Hz, ArH), 7.58 (1H, d, J = 1.8 Hz, ArH), 7.43 (1H, d, J = 8.4 Hz, H on furyl), 7.31 (1H, t, J = 7.6 Hz, ArH), 7.11 (1H, t, J = 7.6 Hz, ArH), 6.87 (1H, d, J = 3.4 Hz, H on furyl), 6.50 (1H, dd, J = 1.8, 3.4 Hz, H on furyl) (Figure S11, Section S11, Supplementary Materials). 13C NMR (101 MHz, CDCl3) δC, ppm: 148.96, 142.29, 141.29, 136.92, 126.93, 121.21, 120.93, 119.64, 111.15, 109.93, 106.64 (Figure S12, Section S12, Supplementary Materials). ESI-HRMS (positive, m/z): 207.0504 (Calcd. for [M+Na]+ 207.1834). Anal. Calcd for C11H8N2O: C, 71.74; H, 4.34; N, 15.21. Found: C, 72.01; H, 4.29; N, 15.28. FT-IR (KBr) σ, cm−1: 3626 (br m, N-H), 3154 (m, furyl ring), 1617 (m, C=N), 1387 (m, C-N), 1234 (m, C-O).
2.7. Synthesis of Tert-butyl-3-(4-(methoxycarbonyl)phenyl)-1H-indazole-1-carboxylate (Compound 8)
As shown in Scheme 1, compound 8 was obtained through a Pd-catalyzed Suzuki–Miyaura coupling reaction, which is described comprehensively in Section 2.11. Mp 73.8–75.1 °C (white solids, yields of 4–93%, Table 2 and Figure 3). 1H NMR (400 MHz, CDCl3) δH, ppm: 1.75 (9H, s, tert-butyl), 3.96 (3H, s, CH3), 7.38–7.42 (1H, m, ArH), 7.58 (1H, t, J = 8.0 Hz, ArH), 7.99 (1H, d, J = 8.0 Hz, ArH), 8.09 (2H, d, J = 8.0 Hz, ArH), 8.20 (3H, t, J = 8.4 Hz, ArH) (Figure S13, Section S13, Supplementary Materials). 13C NMR (101 MHz, CDCl3) δC, ppm: 166.84, 149.33, 148.69, 141.16, 136.51, 130.68, 130.14, 129.05, 128.25, 124.25, 124.10, 121.27, 115.11, 85.27, 52.36, 28.26 (Figure S14, Section S14, Supplementary Materials). ESI-HRMS (positive, m/z): 353.1492 (Calcd. for [M+H]+ 353.3909) and 727.2764 (Calcd. for [2M+Na]+ 727.7557) (Figure S15, Section S15, Supplementary Materials). Anal. Calcd for C20H20N2O4: C, 68.18; H, 5.68; N, 7.95. Found: C, 69.01; H, 5.21; N, 7.20. FT-IR (KBr) σ, cm−1: 2929 (m, methyl on tert-butyl), 1695 (vs, C=O), 1647 (m, C=N), 1390 (m, C-N), 1230 (w, C-O) (Figure S16, Section S16, Supplementary Materials).
Table 2.
Catalytic Suzuki–Miyaura cross-coupling of compounds 3 with 7.
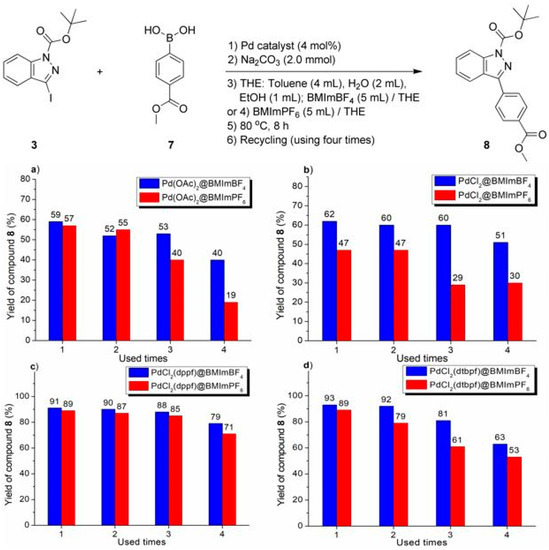
Figure 3.
Recycling experiments on Suzuki–Miyaura cross-coupling of compounds 3 with 7 catalyzed by Pd catalyst immobilized over imidazolium ionic liquid: (a) Pd(OAc)2 over ILs; (b) PdCl2 over ILs; (c) PdCl2(dppf) over ILs; (d) PdCl2(dtbpf) over ILs.
2.8. Synthesis of 4-(1H-Indazole-3-yl)benzoic Acid (Compound 9)
As shown in Scheme 1, compound 9 was obtained by saponification and subsequent deprotection of compound 8, according to a previous report, with some modifications [36]. In practice, compound 8 (400 mg, 1.136 mmol) was dissolved into a mixed solution of THF (5 mL) with methanol (1 mL) under magnetic stirring at 25 °C. Then, the LiOH solution (LiOH·H2O of 476 mg dissolved in distilled H2O of 3 mL) was introduced. The resulting reaction mixture was further stirred at 25 °C for 18 h. When the time was up, the reaction mixture was acidified by dropping HCl solution (3 mol L−1), until the pH value was adjusted to 2. The mixture thus obtained was stirred for 2 h, to remove the boc-group.
Next, the reaction mixture was extracted with CH2Cl2 (3 × 50 mL), and the combined organic layers were dried over anhydrous MgSO4 and then concentrated under reduced pressure. The residue was purified by flash chromatography (SiO2, 100–200 mesh; dichloromethane/methanol = 10/1, v/v, adding a few drops of triethylamine, 10 drops of triethylamine vs. 200 mL eluent) to afford compound 9 (247 mg, yield of 90%) as a white solid. Mp >280 °C. 1H NMR (400 MHz, DMSO-d6) δH, ppm: 13.49 (1H, s, COOH), 8.11 (5H, dd, J = 8.3, 16.0 Hz, ArH), 7.63 (1H, d, J = 8.4 Hz, ArH), 7.43 (1H, m, ArH), 7.24 (1H, m, ArH) (Figure S17, Section S17, Supplementary Materials). 13C NMR (101 MHz, DMSO-d6) δC, ppm: 167.78, 142.65, 142.23, 138.48, 130.55, 130.18, 127.08, 126.84, 122.04, 121.14, 120.68, 111.37 (Figure S18, Section S18, Supplementary Materials). ESI-HRMS (TOF-SIMS, positive, m/z): 239.0815 (Calcd. for [M+H]+ 239.2480) (Figure S19, Section S19, Supplementary Materials). Anal. Calcd. for C14H10N2O2: C, 70.58; H, 4.20; N, 11.76. Found: C, 71.33; H, 4.65; N, 12.25. FT-IR (KBr) σ, cm−1: 3396 (br m, N-H), 2925 (m, furyl ring), 1696 (vs, C=O), 1611 (m, C=N), 1383 (m, C-N), 1254 (m, C-O).
2.9. Synthesis of Tert-butyl-3-(3-(methoxycarbonyl)phenyl)-1H-indazole-1-carboxylate (Compound 11)
As shown in Scheme 1, compound 11 was obtained through Pd-catalyzed Suzuki–Miyaura coupling of compounds 3 with 10, which is comprehensively described in Section 2.11. Yellow oils (yields of 6–96%, Table 3 and Figure 4). 1H NMR (400 MHz, CDCl3) δH, ppm: 8.66 (1H, t, J = 1.8 Hz, ArH), 8.20 (2H, m, ArH), 8.13 (1H, d, J = 7.9 Hz, ArH), 7.98 (1H, d, J = 8.2 Hz, ArH), 7.57 (2H, m, ArH), 7.38 (1H, t, J = 7.6 Hz, ArH), 3.95 (3H, s, methyl), 1.74 (9H, s, tert-butyl) (Figure S20, Section S20, Supplementary Materials). 13C NMR (101 MHz, CDCl3) δC, ppm: 166.83, 149.40, 148.87, 141.12, 132.74, 132.50, 130.87, 130.39, 129.39, 129.09, 129.02, 124.19, 124.10, 121.33, 115.07, 85.18, 52.38, 28.28 (Figure S21, Section S21, Supplementary Materials). ESI-HRMS (positive, m/z): 353.1518 (Calcd. for [M+H]+ 353.3909) (Figure S22, Section S22, Supplementary Materials). Anal. Calcd. for C20H20N2O4: C, 68.18; H, 5.68; N, 7.95. Found: C, 68.75.01; H, 5.38; N, 6.61. FT-IR (KBr) σ, cm−1: 2983 (m, methyl on tert-butyl), 1729 (vs, C=O), 1649 (m, C=N), 1380 (m, C-N), 1246 (m, C-O) (Figure S23, Section S23, Supplementary Materials).
Table 3.
Catalytic Suzuki–Miyaura cross-coupling of compounds 3 with 10.
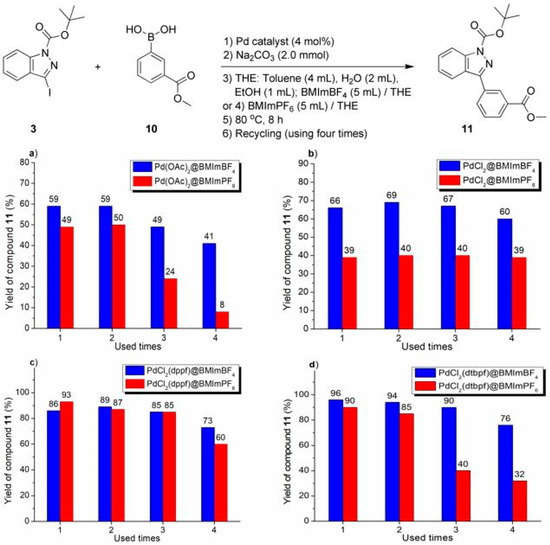
Figure 4.
Recycling experiments on Suzuki–Miyaura cross-coupling of compounds 3 with 10, catalyzed by Pd catalyst and immobilized over imidazolium ionic liquid: (a) Pd(OAc)2 over ILs; (b) PdCl2 over ILs; (c) PdCl2(dppf) over ILs; (d) PdCl2(dtbpf) over ILs.
2.10. Synthesis of 3-(1H-Indazole-3-yl)benzoic Acid (Compound 12)
As shown in Scheme 1, compound 12 was prepared by saponification and subsequent deprotection of compound 11, according to the same process employed for the synthesis of compound 9 (Section 2.8). Compound 12 was obtained as a light yellow solid, the yield was 89%. Mp 244.8–245.4 °C. 1H NMR (400 MHz, DMSO-d6) δH, ppm: 13.37 (1H, s, COOH), 8.55 (1H, t, J = 1.8 Hz, ArH), 8.21 (1H, dt, J = 1.5, 7.9 Hz, ArH), 8.03 (1H, d, J = 8.2 Hz, ArH), 7.95 (1H, dt, J = 1.4, 7.8 Hz, ArH), 7.61 (2H, m, ArH), 7.39 (1H, m, ArH), 7.20 (1H, dd, J = 6.9, 8.0 Hz, ArH) (Figure S24, Section S24, Supplementary Materials). 13C NMR (101 MHz, DMSO-d6) δC, ppm: 167.86, 142.77, 142.20, 134.62, 132.02, 131.36, 129.87, 128.93, 127.84, 126.81, 121.91, 120.84, 120.48, 111.35 (Figure S25, Section S25, Supplementary Materials). FT-IR (KBr) σ, cm−1: 3627 (br m, N-H), 2926 (s, furyl ring), 1698 (m, C=O), 1618 (m, C=N), 1380 (w, C-N), 1235 (m, C-O). ESI-HRMS (TOF-SIMS, positive, m/z): 239.0807 (Calcd. for [M+H]+ 239.2480) (Figure S26, Section S26, Supplementary Materials). Anal. Calcd. for C14H10N2O2: C, 70.58; H, 4.20; N, 11.76. Found: C, 70.97; H, 3.73; N, 12.03.
2.11. General Procedure for Catalytic Suzuki–Miyaura Coupling Reaction
2.11.1. Non-Ionic Liquid-Facilitated Reaction
As shown in annotations a-b of Table 1, Table 2 and Table 3, a mixture of compound 3 (1.0 mmol; Scheme 1; electrophile), compounds 4, 7, or 10 (1.2 mmol; Scheme 1; nucleophile), respectively, Pd catalyst (4 mol%, based on organic halide), anhydrous Na2CO3 (2.0 mmol), and THE solvent system (toluene, 4 mL; H2O, 2 mL; EtOH, 1 mL) were combined into a round-bottom flask (100 mL) with a condenser and balloon, and then vigorously stirred at 80 °C for 8 h. When the time was up, the mixture was extracted with diethyl ether (3 × 50 mL), and the organic layers were combined and washed with saturated NaHCO3 solution (50 mL), distilled H2O (50 mL), and brine (50 mL). After being dried over anhydrous Na2SO4 and filtration, the solution was concentrated under reduced pressure, and the residue was further purified using flash chromatography (SiO2, 100–200 mesh; petroleum ether/ethyl acetate, 4/1, v/v; adding a few drops of triethylamine in eluent, 10 drops of triethylamine vs. 200 mL eluent), to give cross-coupling product (compound 5, orange solid, Rf = 0.35; compound 8, white solid, Rf = 0.62; compound 11, light yellow solid, Rf = 0.50; all Rf values reported on TLC with the same eluent polarity as that used in flash chromatography).
2.11.2. Ionic Liquid-Facilitated Reaction
As shown in annotations a-b of Table 1, Table 2 and Table 3, compound 3 (1.0 mmol; Scheme 1; electrophile) and Pd catalyst (4 mol%, based on organic halide) were fully dispersed in imidazolium ILs (BMImBF4 or BMImPF6, 5 mL, respectively) in a round-bottom flask (100 mL) with a condenser and balloon under vigorous magnetic stirring at 25 °C. The mixture thus obtained was further stirred at 80 °C for 1 h, to achieve a uniform solution. Next, compounds 4, 7, or 10 (1.2 mmol; Scheme 1; nucleophile, respectively), anhydrous Na2CO3 (2.0 mmol), and THE solvent system (toluene, 4 mL; H2O, 2 mL; EtOH, 1 mL) were added, and then the mixture was further stirred at 80 °C for 8 h. The resulting mixture was carefully concentrated under reduced pressure to remove all solvents, and the residue (IL-containing phase) was extracted using diethyl ether (3 × 50 mL). Organic layers (diethyl ether phases) were decanted and collected, washed with saturated NaHCO3 solution (50 mL), distilled H2O (50 mL) and brine (50 mL), dried over anhydrous Na2SO4, filtrated, and concentrated under reduced pressure. After TLC monitoring, flash chromatography (SiO2, 100–200 mesh; petroleum ether/ethyl acetate, 4/1, v/v; adding a few drops of triethylamine, 10 drops of triethylamine vs. 200 mL eluent) afforded corresponding cross-coupled products (compounds 5, 8, 11, Scheme 1). Alternatively, the remaining oil (IL-containing phase) in flask was reloaded with all consumed components (except for Pd catalyst and ionic liquid), and recycling was carried out under the same reaction conditions.
2.12. Computational Methods
All relevant geometries were fully optimized using B3LYP density functional theory (DFT) in conjunction with the self-consistent reaction field (SCRF) method. The Def2-SVP basis set was used in the optimization. After geometry optimization, harmonic vibrational analyses were performed at the same level, to confirm that each minimum had no imaginary frequency or that each transition state (TS) had only one imaginary frequency. The minimum energy path (MEP) was also traced using the intrinsic reaction coordinate (IRC) method, to ensure that each TS structure was correctly linked with two minima. The alkaline THE solvent system (toluene, 4 mL; H2O, 2 mL; EtOH, 1 mL), Na2CO3 (2.0 mmol), ILs (BMImPF6 or BMImBF4, 5 mL) was used in the experiment. Accordingly, ethanol was selected as a compromise solvent in the computations. The implicit PCM solvent model was adopted to roughly evaluate the solvent effect on the reaction in ethanol (ε = 24.85). To refine energies, single-point energy computations were conducted at the PCM-B3LYP/Def2-TZVP level. Therefore, the total energy was the sum of the single-point energy and thermal corrections of 353.15 K. All computations were fulfilled with Gaussian 09 program [37]. All 3-D structures were generated with the CYLview program [38].
3. Results and Discussion
3.1. The C-3 Iodination and N-H Protection of 1H-indazole
According to previous studies, most functionalization of C-3 of 1H-indazole such as alkylation and arylation was carried out after the N-H group of 1H-indazole was protected or pre-substituted [34,39,40,41,42]. In this work, however, 1H-indazole was first iodized on the C-3 position without protecting the N-H group (Scheme 1) [34], but the subsequent Suzuki–Miyaura cross-coupling reaction required protection of the N-H group using (Boc)2O (Scheme 1). Additionally, as the reactivities of halogen substituents showed an order I >Br >>Cl (C–Cl = 96 kcal mol−1; C–Br = 81 kcal mol−1; C–I = 65 kcal mol−1 for Ph-X unit) [43], iodization was selected over bromination and chlorination, in order to accelerate the cross-coupling reaction.
3.2. The Catalytic Suzuki–Miyaura Cross-Coupling Reaction
3.2.1. Effect of Substrate
Overall, most catalytic Suzuki–Miyaura cross-coupling reactions proceeded smoothly within 8 h, and poor to excellent outputs were obtained for the coupling of compound 3 (electrophile) with compounds 4, 7, and 10 (all nucleophiles, Table 1, Table 2 and Table 3). Herein, furyl of compound 4 represents a structure having excessive π-electrons [44], but, comparatively, the aryls of compounds 7 and 10 had deficient π-electrons [45]. Furthermore, the π-electron density on the aryl of compound 10 was lower than that of 7, mainly due to 10 having an m-substituted methoxycarbonyl group, which was actually an o- and p-orienting substituent. Meanwhile, the nucleophilicity of aryl (heteroaryl) boronic acids (compounds 4, 7, 10) came from the negative charge at the carbon of C-B bond [46]. However, there was no obvious and definite order of reactivity among compounds 4, 7, and 10 (Table 1, Table 2 and Table 3), and it seemed that the catalyst along with the solvent dominated the cross-coupling yields more than the substrate structure (Table 1, Table 2 and Table 3), which probably means the present catalytic system has a wide scope for substrates.
3.2.2. Effect of Catalyst
When compound 3 was coupled with compound 4 during fresh catalysis, the activities of various Pa catalysts showed the order PdCl2(dtbpf) (averaged yield 86.66%, entries 13–15, Table 1) >PdCl2(dppf) (averaged yield 86.33%, entries 10–12, Table 1) >Pd(OAc)2 (averaged yield 42.33%, entries 4–6, Table 1) >PdCl2 (averaged yield 38.00%, entries 7–9, Table 1) >Pd(PPh3)4 (averaged yield 16.33%, entries 1–3, Table 1). Furthermore, the catalytic cross-coupling of compounds 3 with 7 and 10 showed much the same order (Table 2 and Table 3). Overall, the divalent Pd complexes (PdCl2(dtbpf) and PdCl2(dppf)) appeared better than the simple divalent Pd salts, such as Pd(OAc)2 and PdCl2, while the activity of the zero-valent Pd precursor (Pd(PPh3)4) was the lowest.
It was previously reported that the typical Suzuki–Miyaura catalytic cycle undergoes oxidative addition, transmetallation, and reductive elimination [21,47]. The catalytically active Pd(0) species are first generated in situ, coming from the Pd(0) or Pd(II) precursor (raw catalyst), and then oxidative addition of aryl halide ArX (electrophile) provides the Pd complex [ArPdXLn]. Next, the addition of [ArPdXLn] with the pre-added base (RO− or OH−) gives an [ArPdORLn] intermediate, which is quickly reacted with an organoboron compound Ar’B(OH)2, leading to the diaryl complex [ArPdAr’Ln]. This procedure is summarized as transmetallation. Lastly, reductive elimination of the diaryl complex produces the biaryl product Ar–Ar’, along with Pd(0) for recycling [21,47].
In association with the typical Suzuki–Miyaura cross-coupling mechanism, it can be seen that the Pd(PPh3)4, a zero-valent palladium precursor, could not afford sufficient catalytically-active Pd(0) species, probably because the PPh3 ligands were not able to stabilize the [ArPdXLn] intermediate complex very well in the present work [47]. In comparison, employment of OAc− and Cl− as ligands of Pd raw catalysts yielded better outputs (respective entries 4–9 vs. 1–3, Table 1, Table 2 and Table 3). However, the ferrocene-based ligands derived from PdCl2(dppf) and PdCl2(dtbpf) had the best effect on the stabilizing [ArPdXLn] intermediate, finally leading to much improved catalytic efficiencies (respective entries 10–15 vs. 1–9, Table 1, Table 2 and Table 3).
3.2.3. Effect of Ionic Liquid
With the catalytic results obtained to that point, it was significant to test the effects of the ionic liquids, including on the catalytic outputs, as well as catalyst recovery and recycling. First of all, all Pd catalysts were highly soluble in both BMImBF4 and BMImPF6, and the resulting mixture could be fully dispersed into the THE solvent system under vigorous magnetic stirring. These characters guaranteed the effects of “one-phase catalysis, two-phases separation” [48].
Next, for the cross-coupling of halogenated and protected indazole (compound 3) with 2-furanboronic acid (compound 4), the introduction of two imidazolium ILs increased the yields of coupled products in the reactions catalyzed by Pd(PPh3)4 (entries 2–3 vs. 1, Table 1), Pd(OAc)2 (entries 5–6 vs. 4, Table 1), and PdCl2 (entries 8–9 vs. 7, Table 1). The use of BMImBF4 in association with PdCl2(dppf) increased the yield of coupled product (entries 11 vs. 10, Table 1), but use of BMImPF6 showed negative effects on the catalytic yields (entries 12 vs. 10, Table 1). On the other hand, employing either BMImBF4 or BMImPF6 along with PdCl2(dtbpf) did not contribute to the catalytic outputs (entries 14–15 vs. 13, Table 1).
Therefore, it seemed that introduction of imidazolium ILs caused the coordination of N to Pd in the simple Pd salts (Pd(PPh3)4, Pd(OAc)2, PdCl2)-catalyzed reactions, probably leading to a more stable intermediate [ArPdXLn] (Pd complex) [47], further improving the catalytic yields. However, when the Pd catalysts contained sophisticated ligands, such as dppf and dtbpf, the original P-containing ferrocene-based ligands showed positive effects on stabilizing the catalytic intermediate, but the introduction of imidazolium ILs may affect this stability through coordination of N, sometimes decreasing the outputs.
When the substrate (nucleophile) was changed to 4-methoxycarbonylphenylboronic acid (compound 7, Scheme 1) and 3-methoxycarbonylphenylboronic acid (compound 10), use of two imidazolium ILs generally produced enhanced yields of cross-coupled products (Table 2 and Table 3), which could be ascribed to the unique structural and coordinating properties of the ILs.
In the meantime, there was another reasonable explanation for the roles of BMImBF4 and BMImPF6 in the present catalysis. It was previously reported that the combination of 1-n-butyl-3-methylimidazolium bromide (BMImBr) with Pd(OAc)2 in an alkaline environment produced 1-n-butyl-3-methylimidazol-2-ylidene (BMIy) complex of Pd (such as PdBr2(BMIy)2 and analogues), which showed a high activity in the Heck reaction [49]. This N-heterocyclic carbene (NHC) complex (PdBr2(BMIy)2) probably has influences on both the NHC and counterion (Br−), facilitating various catalytic reactions [49]. Furthermore, no palladium metal particles (Pd(0) black) appeared in this work, probably indicating the formation of N-heterocyclic carbene (NHC) complex of Pd. On the basis of the above analysis, the possible formation of NHC complex of Pd in this work is proposed in Scheme 2.
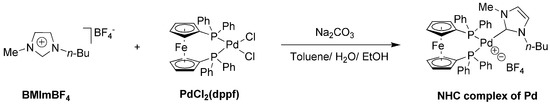
Scheme 2.
Possible formation of NHC complex of Pd in this work.
3.2.4. Effect of Catalyst Recycling
The imidazolium ILs-facilitated catalyst recycling experiments are shown in Figure 2, Figure 3 and Figure 4. In practice, several phenomena deserved attention. First of all, in the non-IL facilitated experiments, a lot of grey flakes appeared on the flask wall, which seemed to be amorphous Pd(0) black, but most of the IL-facilitated recycling reactions showed no Pd(0) black. Hence, the introduction of imidazolium ILs as recyclable media could stabilize the Pd intermediates, mainly due to coordination of N to Pd, which inhibited the formation of Pd(0) black and benefited the catalyst recovery (Figure 2, Figure 3 and Figure 4) [50], or to the formation of the NHC complex of Pd (Scheme 2).
The performance of BMImBF4 was usually much better than that of BMImPF6 for the conversion of all substrates (respective a-d, Figure 2, Figure 3 and Figure 4). Herein, BF4− was more hydrophilic than PF6− [29], which may show a better affinity to the base (Na2CO3) dissolved in H2O, probably stabilizing catalytic intermediates. Furthermore, taking into account the solubility of the solvent and its interaction with BMImPF6 or BMImBF4, some polar solvents such as diethyl ether, acetone, or DMF were able to extract cross-coupled products from ionic phases, but n-hexane was almost completely inert.
Moreover, the catalytic degradation of Pd-containing ionic liquids can be ascribed to the precipitation and coagulation of Pd complexes from BMImPF6 or BMImBF4. For instance, PdCl2(dtbpf) combined with BMImPF6 showed a 32% yield after being used four times (Figure 4d), where the ionic liquid layer became attenuated and the black solid coagulated and floated, probably leading to sharply declined outputs.
3.3. Theoretical Insights into the Catalytic Suzuki–Miyaura Cross-Coupling Mechanism
In order to further inspect the mechanism, the effective ligand dtbpf (entries 13–15, Table 3), compounds 3 and 10 (substrates 3 and 10, Figure 5), were selected for computation using density functional theory (optimized structures are shown in Section S27, Supplementary Materials). As shown in Figure 5, the computational results indicated that, after the formation of the Pd(0) precatalyst, the reaction successively underwent oxidative addition, transmetalation, and reductive elimination. In particular, herein, there were three OH groups in the coordinated boronic acid (Int-3, Figure 5): two of them came from the coordinated boronic acid (compound 10, Scheme 1), while the remaining one was derived from the alkaline reaction medium (Section 2.11), indicating that the P-Pd bond was hydrolyzed by OH− in the alkaline reaction medium (Int-3, Figure 5), which propelled a further reaction (Figure 5). The final reductive elimination towards compound 11 (substrate 11, Figure 5) was the rate-determining step (ΔG‡ = 10.1 kcal/mol). The overall reaction was strongly exothermic (ca. −70.5 kcal/mol). For comparison, an alternative ligand dppf (entries 10–12, Table 3) was also tested in the computations. It turned out that the barrier (17.3 kcal/mol) in the rate-determining step with dppf ligand was slightly higher than that with the dtbpf ligand.
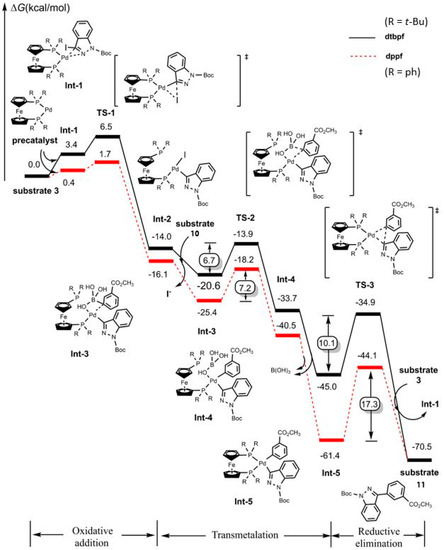
Figure 5.
Relative Gibbs free energy (kcal/mol) profiles in Suzuki–Miyaura cross-coupling of compounds 3 with 10 to yield compound 11 using Pa catalyst featuring dtbpf (balck solid line) or dppf (red dashed line) ligands.
As shown in Figure 6, the selected optimized structures showed that the bond angles of C-Pd-C in Int-5 were 78° and 82° for dtbpf and dppf liands in the reductive elimination, demonstrating that it suffered more constraint from the steric repulsion of two tert-butyl groups than that of two phenyl groups. In this sense, more constraint from the ligand led to a shorter distance and lower barrier in the reductive elimination. Note that, on the contrary, the strong repulsion would increase the barrier of oxidative addition (1.7 vs. 6.5 kcal/mol, TS1, Figure 5), although this step was somewhat rapid and could not strongly disturb the overall mechanism.
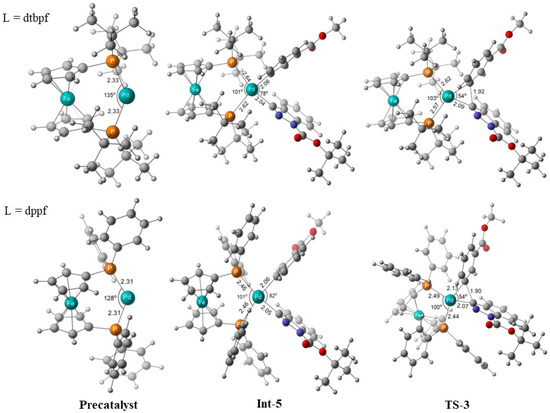
Figure 6.
Selected optimized geometries of Precatalyst, Int-5, and TS-3. Bond distances in Å and angles in degrees.
4. Conclusions
In conclusion, this work presented the Suzuki–Miyaura cross-coupling reactions of a halogenated and N-H protected indazole (electrophile) with organic boronic acids (nucleophiles), catalyzed by different divalent palladium complexes, with or without imidazolium ionic liquids as functional or recyclable media, eventually leading to three C3-functionalized 1H-indazole derivatives. The series of reaction parameters were fully discussed. In general, the averaged catalytic activity of the Pd catalysts exhibited the order PdCl2(dtbpf) >PdCl2(dppf) >Pd(OAc)2 >PdCl2 >Pd(PPh3)4. Moreover, using two imidazolium ionic liquids generally improved the yields of cross-coupled products, as compared to those obtained from non-ionic liquid-facilitated reactions. The BMImBF4 obviously performed better than BMImPF6 for fresh catalysis and recycling, which could be related to the hydrophilicity of anions such as BF4− and PF6−. Meanwhile, use of BMImBF4 and BMImPF6 as recycling media could avoid the formation of Pd(0) black to a large extent, clearly enhancing the catalyst recovery and recycling. In addition, on the basis of scientific calculations, it was seen that the dppf ligand coming from PdCl2(dtbpf) had four tert-butyl groups, which caused higher energy barriers during formation of the catalytic intermediates compared to those caused by dppf from PdCl2(dppf), also illustrating a reason why PdCl2(dppf) performed better than PdCl2(dtbpf). This work put forward some new insights into the interactions of Pd catalysts with ionic liquids and showed the potential for the large-scale production of valuable 1H-indazole derivatives.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/app13074095/s1, Section S1: 1H NMR spectrum of 3-iodo-1H-indazole (compound 2) (Figure S1); Section S2: 13C NMR spectrum of 3-iodo-1H-indazole (compound 2) (Figure S2); Section S3: 1H NMR spectrum of tert-butyl-3-iodo-1H-indazole-1-carboxylate (compound 3) (Figure S3); Section S4: 13C NMR spectrum of tert-butyl-3-iodo-1H-indazole-1-carboxylate (compound 3) (Figure S4); Section S5: ESI-HRMS of tert-butyl-3-iodo-1H-indazole-1-carboxylate (compound 3) (Figure S5); Section S6: FT-IR spectrum of tert-butyl-3-iodo-1H-indazole-1-carboxylate (compound 3) (Figure S6); Section S7: 1H NMR spectrum of tert-butyl-3-(2-furyl)-1H-indazole-1-carboxylate (compound 5) (Figure S7); Section S8: 13C NMR spectrum of tert-butyl-3-(2-furyl)-1H-indazole-1-carboxylate (compound 5) (Figure S8); Section S9: ESI-HRMS of tert-butyl-3-(2-furyl)-1H-indazole-1-carboxylate (compound 5) (Figure S9); Section S10: FT-IR spectrum of ESI-HRMS of tert-butyl-3-(2-furyl)-1H-indazole-1-carboxylate (compound 5) (Figure S10); Section S11: 1H NMR spectrum of 3-(2-furyl)-1H-indazole (compound 6) (Figure S11); Section S12: 13C NMR spectrum of 3-(2-furyl)-1H-indazole (compound 6) (Figure S12); Section S13: 1H NMR spectrum of tert-butyl-3-(4-(methoxycarbonyl)phenyl)-1H-indazole-1-carboxylate (compound 8) (Figure S13); Section S14: 13C NMR spectrum of tert-butyl-3-(4-(methoxycarbonyl)phenyl)-1H-indazole-1-carboxylate (compound 8) (Figure S14); Section S15: ESI-HRMS of tert-butyl-3-(4-(methoxycarbonyl)phenyl)-1H-indazole-1-carboxylate (compound 8) (Figure S15); Section S16: FT-IR spectrum of tert-butyl-3-(4-(methoxycarbonyl)phenyl)-1H-indazole-1-carboxylate (compound 8) (Figure S16); Section S17: 1H NMR spectrum of 4-(1H-indazole-3-yl)benzoic acid (compound 9) (Figure S17); Section S18: 13C NMR spectrum of 4-(1H-indazole-3-yl)benzoic acid (compound 9) (Figure S18); Section S19: ESI-HRMS (TOF-SIMS) of 4-(1H-indazole-3-yl)benzoic acid (compound 9) (Figure S19); Section S20: 1H NMR spectrum of tert-butyl-3-(3-(methoxycarbonyl)phenyl)-1H-indazole-1-carboxylate (compound 11) (Figure S20); Section S21: 13C NMR spectrum of tert-butyl-3-(3-(methoxycarbonyl)phenyl)-1H-indazole-1-carboxylate (compound 11) (Figure S21); Section S22: ESI-HRMS of tert-butyl-3-(3-(methoxycarbonyl)phenyl)-1H-indazole-1-carboxylate (compound 11) (Figure S22); Section S23: FT-IR spectrum of tert-butyl-3-(3-(methoxycarbonyl)phenyl)-1H-indazole-1-carboxylate (compound 11) (Figure S23); Section S24: 1H NMR spectrum of 3-(1H-indazole-3-yl)benzoic acid (compound 12) (Figure S24); Section S25: 13C NMR spectrum of 3-(1H-indazole-3-yl)benzoic acid (compound 12) (Figure S25); Section S26: ESI-HRMS (TOF-SIMS) of 3-(1H-indazole-3-yl)benzoic acid (compound 12) (Figure S26); Section S27: Optimized structures from calculations.
Author Contributions
Experimental and sample analysis, J.Y., A.Z. and L.J.; scientific calculation, Y.W.; material characterization, Q.P.; sample analysis and funding acquisition, X.W.; experiment design and methodology, X.L.; catalytic reaction, W.W.; experiment design and methodology, M.G.; conceptualization and original draft preparation, Y.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Natural Science Basic Research Program of Shaanxi Province (No. 2020JM-019).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Büchel, G.E.; Kossatz, S.; Sadique, A.; Rapta, P.; Zalibera, M.; Bucinsky, L.; Komorovsky, S.; Telser, J.; Eppinger, J.; Reiner, T.; et al. cis-Tetrachlorido-bis(indazole)osmium(IV) and its osmium(III) analogues: Paving the way towards the cis-isomer of the ruthenium anticancer drugs KP1019 and/or NKP1339. Dalton Trans. 2017, 46, 11925–11941. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yu, J.-T.; Pan, C. Recent advances in C–H functionalization of 2H-indazoles. Org. Biomol. Chem. 2022, 20, 7746–7764. [Google Scholar] [CrossRef] [PubMed]
- Tandon, N.; Luxami, V.; Kant, D.; Tandon, R.; Paul, K. Current progress, challenges and future prospects of indazoles as protein kinase inhibitors for the treatment of cancer. RSC Adv. 2021, 11, 25228–25257. [Google Scholar] [CrossRef] [PubMed]
- Yadav, L.; Chaudhary, S. Bu4NI-catalyzed, oxidative C(sp2)–C(sp3) cross dehydrogenative coupling for the regioselective direct C-3 benzylation of 2H-indazoles. Org. Biomol. Chem. 2020, 18, 5927–5936. [Google Scholar] [CrossRef] [PubMed]
- Baraldi, P.G.; Balboni, G.; Pavani, M.G.; Spalluto, G.; Tabrizi, M.A.; De Clercq, E.; Balzarini, J.; Bando, T.; Sugiyama, H.; Romagnoli, R. Design, synthesis, DNA binding, and biological evaluation of water-soluble hybrid molecules containing two pyrazole analogues of the alkylating cyclopropylpyrroloindole (CPI) subunit of the antitumor agent CC-1065 and polypyrrole minor groove binders. J. Med. Chem. 2001, 44, 2536–2543. [Google Scholar] [CrossRef]
- Qian, S.; Cao, J.; Yan, Y.; Sun, M.; Zhu, H.; Hu, Y.; He, Q.; Yang, B. SMT-A07 a 3-(indol-2-yl) indazole derivative, induces apoptosis of leukemia cells in vitro. Mol. Cell. Biochem. 2010, 345, 13–21. [Google Scholar] [CrossRef]
- Ghosh, D.; Ghosh, S.; Ghosh, A.; Pyne, P.; Majumder, S.; Hajra, A. Visible light-induced functionalization of indazole and pyrazole: A recent update. Chem. Commun. 2022, 58, 4435–4455. [Google Scholar] [CrossRef]
- Fustero, S.; Sánchez-Roselló, M.; Barrio, P.; Simón-Fuentes, A. From 2000 to mid-2010: A fruitful decade for the synthesis of pyrazoles. Chem. Rev. 2011, 111, 6984–7034. [Google Scholar] [CrossRef]
- Ma, C.; Feng, Z.; Li, J.; Zhang, D.; Li, W.; Jiang, Y.; Yu, B. Photocatalytic transition-metal-free direct 3-alkylation of 2-aryl-2H-indazoles in dimethyl carbonate. Org. Chem. Front. 2021, 8, 3286–3291. [Google Scholar] [CrossRef]
- Vidyacharan, S.; Ramanjaneyulu, B.T.; Jang, S.; Kim, D.-P. Continuous-flow visible light organophotocatalysis for direct arylation of 2H-indazoles: Fast access to drug molecules. ChemSusChem 2019, 12, 2581–2586. [Google Scholar] [CrossRef]
- Sun, M.; Li, L.; Wang, L.; Huo, J.; Sun, M.; Li, P. Controllable chemoselectivity in the reaction of 2H-indazoles with alcohols under visible-light irradiation: Synthesis of C3-alkoxylated 2H-indazoles and ortho-alkoxycarbonylated azobenzenes. Org. Chem. Front. 2021, 8, 4230–4236. [Google Scholar] [CrossRef]
- Park, B.Y.; Pirnot, M.T.; Buchwald, S.L. Visible light-mediated (hetero)aryl amination using Ni(II) salts and photoredox catalysis in flow: A synthesis of tetracaine. J. Org. Chem. 2020, 85, 3234–3244. [Google Scholar] [CrossRef] [PubMed]
- Petrone, D.A.; Ye, J.; Lautens, M. Modern transition-metal-catalyzed carbon-halogen bond formation. Chem. Rev. 2016, 116, 8003–8104. [Google Scholar] [CrossRef]
- Singsardar, M.; Dey, A.; Sarkar, R.; Hajra, A. Visible-light-induced organophotoredox-catalyzed phosphonylation of 2H-indazoles with diphenylphosphine oxide. J. Org. Chem. 2018, 83, 12694–12701. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Xu, Z.; Cai, Y.; Wang, L. Visible-light-driven multicomponent cyclization by trapping a 1,3-vinylimine ion intermediate: A direct approach to pyrimido[1,2-b]indazole derivatives. Org. Lett. 2021, 23, 8343–8347. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.Y.W.; Kir, S.; Tooze, S.A. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J. Biol. Chem. 2007, 282, 25464–25474. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, M.B.; Novotny, C.J.; Shokat, K.M. Structure of the human autophagy initiating kinase ULK1 in complex with potent inhibitors. ACS Chem. Biol. 2015, 10, 257–261. [Google Scholar] [CrossRef]
- Wood, S.D.; Grant, W.; Adrados, I.; Choi, J.Y.; Alburger, J.M.; Duckett, D.R.; Roush, W.R. In silico HTS and structure based optimization of indazole-derived ULK1 inhibitors. ACS Med. Chem. Lett. 2017, 8, 1258–1263. [Google Scholar] [CrossRef]
- Clutterbuck, L.A.; Posada, C.G.; Visintin, C.; Riddal, D.R.; Lancaster, B.; Gane, P.J.; Garthwaite, J.; Selwood, D.L. Oxadiazolylindazole sodium channel modulators are neuroprotective toward hippocampal neurones. J. Med. Chem. 2009, 52, 2694–2707. [Google Scholar] [CrossRef]
- Collot, V.; Varlet, D.; Rault, S. Heck cross-coupling reaction of 3-iodoindazoles with methyl acrylate: A mild and flexible strategy to design 2-azatryptamines. Tetrahedron Lett. 2000, 41, 4363–4366. [Google Scholar] [CrossRef]
- Han, F.-S. Transition-metal-catalyzed Suzuki–Miyaura cross-coupling reactions: A remarkable advance from palladium to nickel catalysts. Chem. Soc. Rev. 2013, 42, 5270–5298. [Google Scholar] [CrossRef] [PubMed]
- Beletskaya, I.P.; Alonso, F.; Tyurin, V. The Suzuki-Miyaura reaction after the Nobel prize. Coord. Chem. Rev. 2019, 385, 137–173. [Google Scholar] [CrossRef]
- Miyaura, N. Cross-coupling reaction of organoboron compounds via base-assisted transmetalation to palladium(II) complexes. J. Organomet. Chem. 2002, 653, 54–57. [Google Scholar] [CrossRef]
- Boit, T.B.; Weires, N.A.; Kim, J.; Garg, N.K. Nickel-catalyzed Suzuki–Miyaura coupling of aliphatic amides. ACS Catal. 2018, 8, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Weinstein, A.B.; White, P.B.; Stahl, S.S. Ligand-promoted palladium-catalyzed aerobic oxidation reactions. Chem. Rev. 2018, 118, 2636–2679. [Google Scholar] [CrossRef]
- Chernyshev, V.M.; Khazipov, O.V.; Eremin, D.B.; Denisova, E.A.; Ananikov, V.P. Formation and stabilization of nanosized Pd particles in catalytic systems: Ionic nitrogen compounds as catalytic promoters and stabilizers of nanoparticles. Coord. Chem. Rev. 2021, 437, 213860. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, S.; Deng, Y. Recent advances in ionic liquid catalysis. Green Chem. 2011, 13, 2619–2637. [Google Scholar] [CrossRef]
- Mathews, C.J.; Smith, P.J.; Welton, T. Palladium catalysed Suzuki cross-coupling reactions in ambient temperature ionic liquids. Chem. Commun. 2000, 1249–1250. [Google Scholar] [CrossRef]
- Lombardo, M.; Chiarucci, M.; Trombini, C. A recyclable triethylammonium ion-tagged diphenylphosphine palladium complex for the Suzuki–Miyaura reaction in ionic liquids. Green Chem. 2009, 11, 574–579. [Google Scholar] [CrossRef]
- Mehnert, C.P.; Cook, R.A.; Dispenziere, N.C.; Afeworki, A.M. Supported ionic liquid catalysis-a new concept for homogeneous hydroformylation catalysis. J. Am. Chem. Soc. 2002, 124, 12932–12933. [Google Scholar] [CrossRef]
- Muthayala, M.K.; Chhikara, B.S.; Parang, K.; Kumar, A. Ionic liquid-supported synthesis of sulfonamides and carboxamides. ACS Comb. Sci. 2012, 14, 60–65. [Google Scholar] [CrossRef]
- Bocchi, V.; Palla, G. High yield selective bromination and iodination of indoles in N,N-dimethylformamide. Synthesis 1982, 1096–1097. [Google Scholar] [CrossRef]
- Collot, V.; Dallemagne, P.; Bovy, P.R.; Rault, S. Suzuki-type cross-coupling reaction of 3-iodoindazoles with aryl boronic acids: A general and flexible route to 3-arylindazoles. Tetrahedron 1999, 50, 6917–6922. [Google Scholar] [CrossRef]
- Wentrup, C.; Benedikt, J. Nitrile imine and carbene rearrangements. From furfural to benzofulvene-8-carboxaldehyde, 8-benzofulvenylcarbene, and 1-vinylideneindene. J. Org. Chem. 1980, 45, 1407–1409. [Google Scholar] [CrossRef]
- Vazquez, J.; De, S.K.; Chen, L.-H.; Riel-Mehan, M.; Emdadi, A.; Cellitti, J.; Stebbins, J.L.; Rega, M.F.; Pellecchia, M. Development of paramagnetic probes for molecular recognition studies in protein kinases. J. Med. Chem. 2008, 51, 3460–3465. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision, D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Legault, C.Y. CYLview, 1.0b; Université de Sherbrooke. 2009. Available online: http://www.cylview.org (accessed on 15 July 2022).
- Ye, M.; Edmunds, A.J.F.; Morris, J.A.; Sale, D.; Zhang, Y.; Yu, J.-Q. A robust protocol for Pd(II)-catalyzed C-3 arylation of (1H) indazoles and pyrazoles: Total synthesis of nigellidine hydrobromide. Chem. Sci. 2013, 4, 2374–2379. [Google Scholar] [CrossRef] [PubMed]
- Ben-Yahia, A.; Naas, M.; Kazzouli, S.E.; Essassi, E.M.; Guillaumet, G. Direct C-3-arylations of 1H-indazoles. Eur. J. Org. Chem. 2012, 2012, 7075–7081. [Google Scholar] [CrossRef]
- Gambouz, K.; Abbouchi, A.E.; Nassiri, S.; Suzenet, F.; Bousmina, M.; Akssira, M.; Guillaumet, G.; Kazzouli, S.E. “On water” palladium catalyzed direct arylation of 1H-indazole and 1H-7-azaindazole. Molecules 2020, 25, 2820. [Google Scholar] [CrossRef]
- Hattori, K.; Yamaguchi, K.; Yamaguchi, J.; Itami, K. Pd- and Cu-catalyzed C-H arylation of indazoles. Tetrahedron 2012, 68, 7605–7612. [Google Scholar] [CrossRef]
- Grushin, V.V.; Alper, H. Transformations of chloroarenes, catalyzed by transition-metal complexes. Chem. Rev. 1994, 94, 1047–1062. [Google Scholar] [CrossRef]
- Ackermann, M.; Pascariu, A.; Höcher, T.; Siehl, H.-U.; Berger, S. Electronic properties of furyl substituents at phosphorus and their influence on 31P NMR chemical shifts. J. Am. Chem. Soc. 2006, 128, 8434–8440. [Google Scholar] [CrossRef]
- Ribas, J.; Cubero, E.; Luque, F.J.; Orozco, M. Theoretical study of alkyl-π and aryl-π interactions. Reconciling theory and experiment. J. Org. Chem. 2002, 67, 7057–7065. [Google Scholar] [CrossRef]
- Wang, J.; Zheng, W.; Ding, L.; Wang, Y. Computational study on C–B homolytic bond dissociation enthalpies of organoboron compounds. New J. Chem. 2017, 41, 1346–1362. [Google Scholar] [CrossRef]
- Jana, R.; Pathak, T.P.; Sigman, M.S. Advances in transition metal (Pd,Ni,Fe)-catalyzed cross-coupling reactions using alkyl-organometallics as reaction partners. Chem. Rev. 2011, 111, 1417–1492. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Hu, J.; Fan, J.; Chang, J. One-pot conversion of sugars and lignin in ionic liquid and recycling of ionic liquid. Ind. Eng. Chem. Res. 2012, 51, 3452–3457. [Google Scholar] [CrossRef]
- Xu, L.; Chen, W.; Xiao, J. Heck reaction in ionic liquids and the in situ identification of N-heterocyclic carbene complexes of palladium. Organometallics 2000, 19, 1123–1127. [Google Scholar] [CrossRef]
- Slagt, V.F.; de Vires, A.H.M.; de Vires, J.G.; Kellogg, R.M. Practical aspects of carbon-carbon cross-coupling reactions using heteroarenes. Org. Proc. Res. Dev. 2010, 14, 30–47. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).