

First Theoretical Realization of a Stable Two-Dimensional Boron Fullerene Network


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Computational Methods
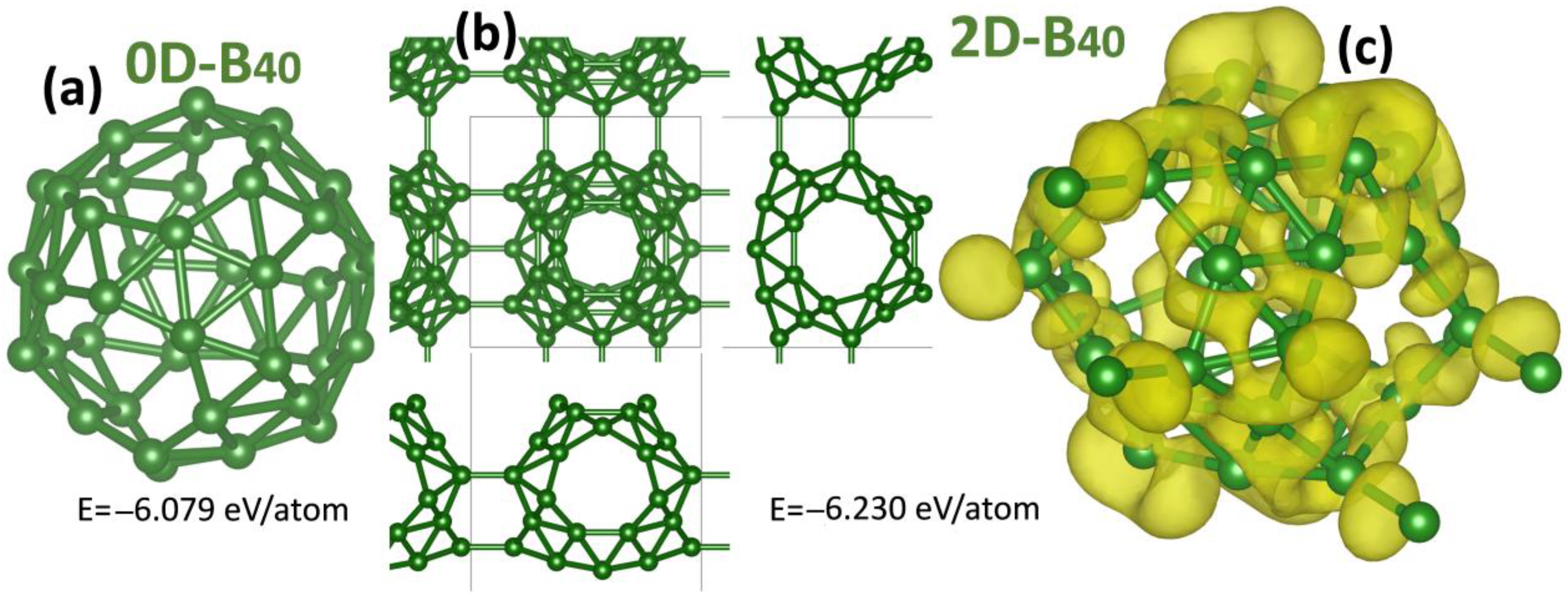
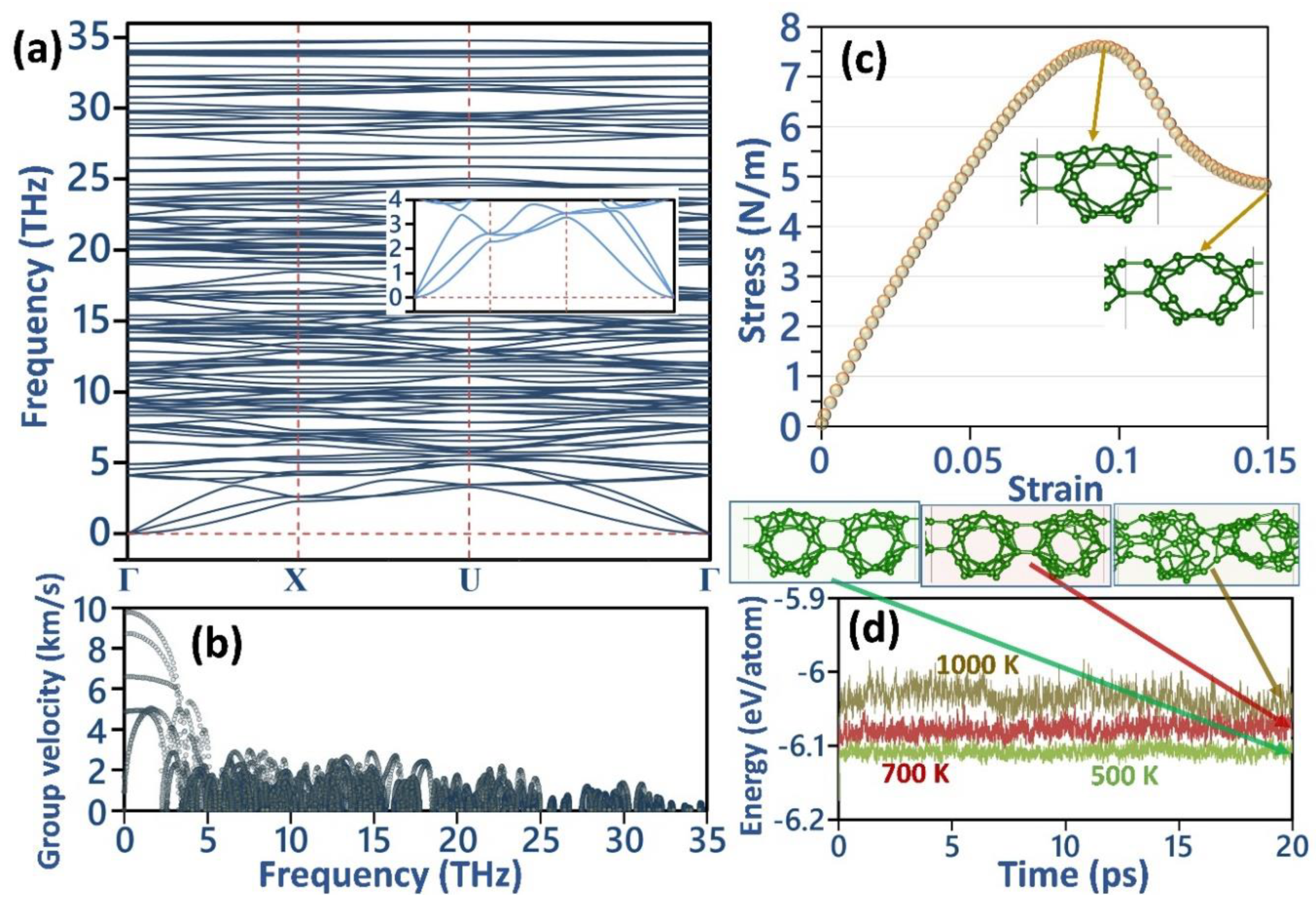
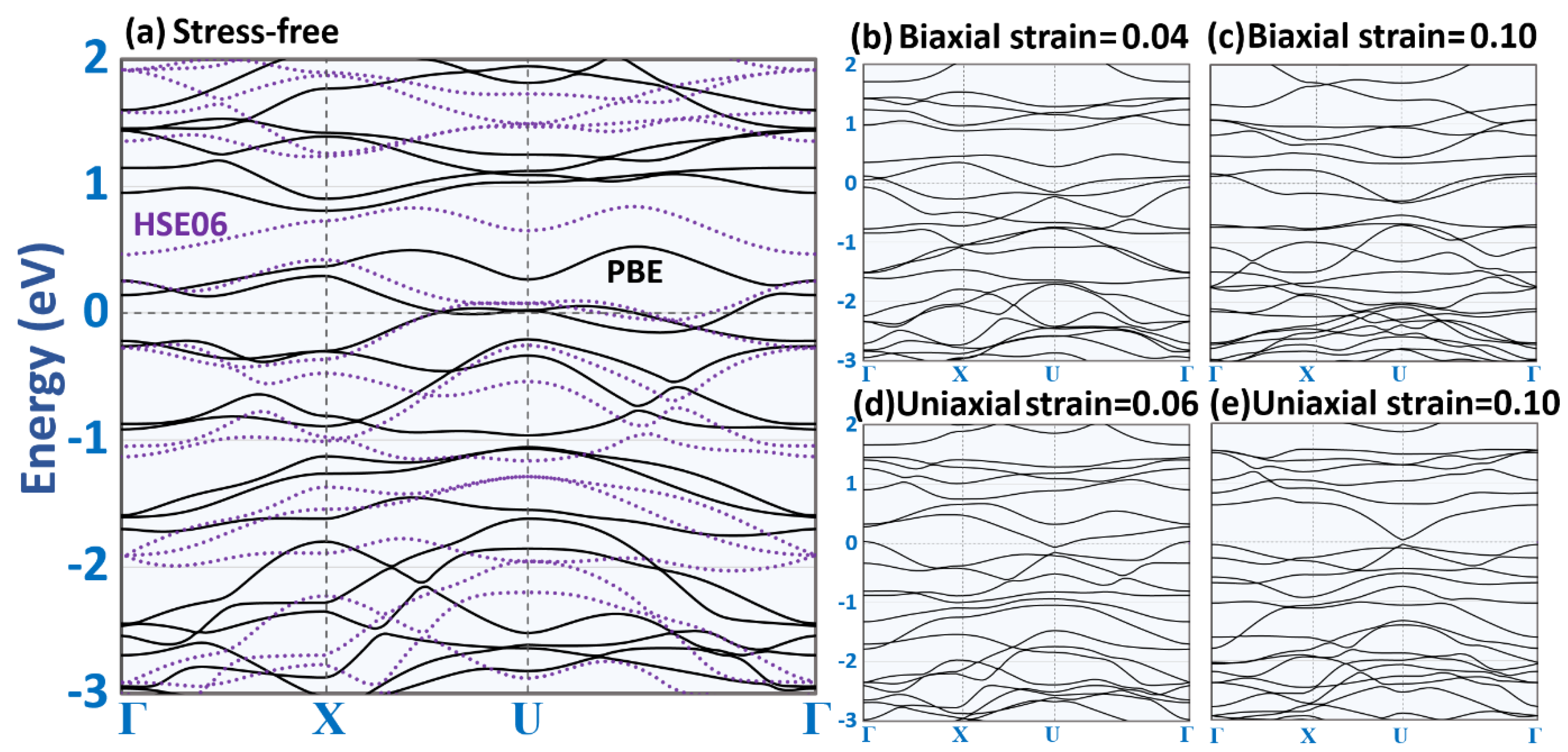
3. Results and Discussion
4. Concluding Remarks
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schwerdtfeger, P.; Wirz, L.N.; Avery, J. The topology of fullerenes. WIREs Comput. Mol. Sci. 2015, 5, 96–145. [Google Scholar] [CrossRef] [PubMed]
- Ge, M.; Sattler, K. Observation of fullerene cones. Chem. Phys. Lett. 1994, 220, 192–196. [Google Scholar] [CrossRef]
- Schwerdtfeger, P.; Wirz, L.; Avery, J. Program Fullerene: A software package for constructing and analyzing structures of regular fullerenes. J. Comput. Chem. 2013, 34, 1508–1526. [Google Scholar] [CrossRef] [PubMed]
- Shakirova, A.A.; Tomilin, F.N.; Pomogaev, V.A.; Vnukova, N.G.; Churilov, G.N.; Kudryasheva, N.S.; Tchaikovskaya, O.N.; Ovchinnikov, S.G.; Avramov, P. Synthesis, Mass Spectroscopy Detection, and Density Functional Theory Investigations of the Gd Endohedral Complexes of C82 Fullerenols. Computation 2021, 9, 58. [Google Scholar] [CrossRef]
- Melchakova, I.A.; Tenev, T.G.; Vitanov, N.V.; Tchaikovskaya, O.N.; Chernozatonskii, L.A.; Yakobson, B.I.; Avramov, P.V. Extreme structure and spontaneous lift of spin degeneracy in doped perforated bilayer graphenes. Carbon 2022, 192, 61–70. [Google Scholar] [CrossRef]
- Mortazavi, B.; Shojaei, F.; Zhuang, X. A novel two-dimensional C36 fullerene network; an isotropic, auxetic semiconductor with low thermal conductivity and remarkable stiffness. Mater. Today Nano. 2023, 21, 100280. [Google Scholar] [CrossRef]
- Hou, L.; Cui, X.; Guan, B.; Wang, S.; Li, R.; Liu, Y.; Zhu, D.; Zheng, J. Synthesis of a monolayer fullerene network. Nature 2022, 606, 507–510. [Google Scholar] [CrossRef]
- Pan, F.; Ni, K.; Xu, T.; Chen, H.; Wang, Y.; Gong, K.; Liu, C.; Li, X.; Lin, M.-L.; Li, S.; et al. Long-range ordered porous carbons produced from C60. Nature 2023, 1–7. [Google Scholar] [CrossRef]
- Meirzadeh, E.; Evans, A.M.; Rezaee, M.; Milich, M.; Dionne, C.J.; Darlington, T.P.; Bao, S.T.; Bartholomew, A.K.; Handa, T.; Rizzo, D.J.; et al. A few-layer covalent network of fullerenes. Nature 2023, 613, 71–76. [Google Scholar] [CrossRef]
- Mortazavi, B.; Zhuang, X. Low and Anisotropic Tensile Strength and Thermal Conductivity in the Single-Layer Fullerene Network Predicted by Machine-Learning Interatomic Potentials. Coatings 2022, 12, 1171. [Google Scholar] [CrossRef]
- Ying, P.; Dong, H.; Liang, T.; Fan, Z.; Zhong, Z.; Zhang, J. Atomistic insights into the mechanical anisotropy and fragility of monolayer fullerene networks using quantum mechanical calculations and machine-learning molecular dynamics simulations. Extrem. Mech. Lett. 2023, 58, 101929. [Google Scholar] [CrossRef]
- Shen, G.; Li, L.; Tang, S.; Jin, J.; Chen, X.; Peng, Q. Stability and Elasticity of Quasi-Hexagonal Fullerene Monolayer from First-Principles Study. Crystals 2023, 13, 224. [Google Scholar] [CrossRef]
- Zhai, H.-J.; Zhao, Y.-F.; Li, W.-L.; Chen, Q.; Bai, H.; Hu, H.-S.; Piazza, Z.; Tian, W.-J.; Lu, H.-G.; Wu, Y.-B.; et al. Observation of an all-boron fullerene. Nat. Chem. 2014, 6, 727–731. [Google Scholar] [CrossRef]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef]
- Neto, A.H.C.; Peres, N.M.R.; Novoselov, K.S.; Geim, A.K.; Guinea, F. The electronic properties of graphene. Rev. Mod. Phys. 2009, 81, 109–162. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef]
- Mannix, A.J.; Zhou, X.-F.; Kiraly, B.; Wood, J.D.; Alducin, D.; Myers, B.D.; Liu, X.; Fisher, B.L.; Santiago, U.; Guest, J.R.; et al. Synthesis of borophenes: Anisotropic, two-dimensional boron polymorphs. Science 2015, 350, 1513–1516. [Google Scholar] [CrossRef]
- Feng, B.; Zhang, J.; Zhong, Q.; Li, W.; Li, S.; Li, H.; Cheng, P.; Meng, S.; Chen, L.; Wu, K. Experimental Realization of Two-Dimensional Boron Sheets. Nat. Chem. 2016, 8, 563–568. [Google Scholar] [CrossRef]
- Zhou, X.F.; Dong, X.; Oganov, A.R.; Zhu, Q.; Tian, Y.; Wang, H.T. Semimetallic two-dimensional boron allotrope with massless Dirac fermions. Phys. Rev. Lett. 2014, 112, 085502. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, Y.; Gao, G.; Yakobson, B.I. Two-Dimensional Boron Monolayers Mediated by Metal Substrates. Angew. Chemie 2015, 127, 13214–13218. [Google Scholar] [CrossRef]
- Yang, L.; Li, Y.; Hao, D.; Li, L.; Peng, H.; Jin, P. Aggregation behavior and non-covalent functionalization of borofullerenes B28, B38, and B40: A density functional theory investigation. Int. J. Quantum Chem. 2019, 119, e25921. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.; Pack, J. Special points for Brillouin zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. [Google Scholar] [CrossRef]
- Shapeev, A.V. Moment tensor potentials: A class of systematically improvable interatomic potentials. Multiscale Model. Simul. 2016, 14, 1153–1173. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef]
- Mortazavi, B.; Novikov, I.S.; Podryabinkin, E.V.; Roche, S.; Rabczuk, T.; Shapeev, A.V.; Zhuang, X. Exploring phononic properties of two-dimensional materials using machine learning interatomic potentials. Appl. Mater. Today 2020, 20, 100685. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of Chemical-Bonds Based on Topological Analysis of Electron Localization Functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Gardeh, M.G.; Kistanov, A.A.; Nguyen, H.; Manzano, H.; Cao, W.; Kinnunen, P. Exploring Mechanisms of Hydration and Carbonation of MgO and Mg(OH)2 in Reactive Magnesium Oxide-Based Cements. J. Phys. Chem. C 2022, 126, 6196–6206. [Google Scholar] [CrossRef]
- Kistanov, A.A.; Rani, E.; Singh, H.; Fabritius, T.; Huttula, M.; Cao, W. Discerning phase-matrices for individual nitride inclusions within ultra-high-strength steel: Experiment driven DFT investigation. Phys. Chem. Chem. Phys. 2022, 24, 1456–1461. [Google Scholar] [CrossRef]
- Kistanov, A.A.; Shcherbinin, S.A.; Botella, R.; Davletshin, A.; Cao, W. Family of Two-Dimensional Transition Metal Dichlorides: Fundamental Properties, Structural Defects, and Environmental Stability. J. Phys. Chem. Lett. 2022, 13, 2165–2172. [Google Scholar] [CrossRef]
- Arabha, S.; Rajabpour, A. Thermo-mechanical properties of nitrogenated holey graphene (C2N): A comparison of machine-learning-based and classical interatomic potentials. Int. J. Heat Mass Transf. 2021, 178, 121589. [Google Scholar] [CrossRef]
- Arabha, S.; Aghbolagh, Z.S.; Ghorbani, K.; Hatam-Lee, M.; Rajabpour, A. Recent advances in lattice thermal conductivity calculation using machine-learning interatomic potentials. J. Appl. Phys. 2021, 130, 210903. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mortazavi, B. First Theoretical Realization of a Stable Two-Dimensional Boron Fullerene Network. Appl. Sci. 2023, 13, 1672. https://doi.org/10.3390/app13031672
Mortazavi B. First Theoretical Realization of a Stable Two-Dimensional Boron Fullerene Network. Applied Sciences. 2023; 13(3):1672. https://doi.org/10.3390/app13031672
Chicago/Turabian StyleMortazavi, Bohayra. 2023. "First Theoretical Realization of a Stable Two-Dimensional Boron Fullerene Network" Applied Sciences 13, no. 3: 1672. https://doi.org/10.3390/app13031672
APA StyleMortazavi, B. (2023). First Theoretical Realization of a Stable Two-Dimensional Boron Fullerene Network. Applied Sciences, 13(3), 1672. https://doi.org/10.3390/app13031672