
Genomic Approaches for Monogenic Kidney Diseases: A Comparative Review of Diagnostic Methods and Precision Medicine Implications

Abstract
:1. Introduction
2. Genetic Testing: The Current Framework
2.1. Sanger Technology
2.2. Targeted Panel Technology
2.3. Whole-Exome Sequencing
Studies Including Direct Comparisons of 2 Technologies
2.4. Whole-Genome Sequencing
2.5. Ethical Implications of Genetic Testing
3. Discussion
4. Conclusions and Future Prospections
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.A.; O’Callaghan, C.A.; Lasserson, D.S.; Hobbs, F.D.R. Global Prevalence of Chronic Kidney Disease—A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef] [PubMed]
- Cockwell, P.; Fisher, L.A. The global burden of chronic kidney disease. Lancet 2020, 395, 662–664. [Google Scholar] [CrossRef] [PubMed]
- Kovesdy, C.P. Epidemiology of chronic kidney disease: An update 2022. Kidney Int. Suppl. 2022, 12, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, T.; Ninomiya, T.; Jha, V.; Neal, B.; Patrice, H.M.; Okpechi, I.; Zhao, M.; Lv, J.; Garg, A.X.; Knight, J.; et al. Worldwide access to treatment for end-stage kidney disease: A systematic review. Lancet 2015, 385, 1975–1982. [Google Scholar] [CrossRef] [PubMed]
- Bello, A.K.; Levin, A.; Lunney, M.; Osman, M.A.; Ye, F.; Ashuntantang, G.; Bellorin-Font, E.; Benghanem Gharbi, M.; Ghnaimat, M.; Harden, P.; et al. Global Kidney Health Atlas: A report by the International Society of Nephrology on the Global Burden of End-stage Kidney Disease and Capacity for Kidney Replacement Therapy and Conservative Care across. World Countries and Regions; International Society of Nephrology: Brussels, Belgium, 2019. [Google Scholar]
- Nordio, M.; Limido, A.; Maggiore, U.; Nichelatti, M.; Postorino, M.; Quintaliani, G. Survival in patients treated by long-term dialysis compared with the general population. Am. J. Kidney Dis. 2012, 59, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Sundström, J.; Bodegard, J.; Bollmann, A.; Vervloet, M.G.; Mark, P.B.; Karasik, A.; Taveira-Gomes, T.; Botana, M.; Birkeland, K.I.; Thuresson, M.; et al. Prevalence, outcomes, and cost of chronic kidney disease in a contemporary population of 2·4 million patients from 11 countries: The CaReMe CKD study. Lancet Reg. Health Eur. 2022, 20, 100438. [Google Scholar] [CrossRef]
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.-M.; Yang, C.-W. Chronic kidney disease: Global dimension and perspectives. Lancet 2013, 382, 260–272. [Google Scholar] [CrossRef]
- Al-Awaida, W.J.; Hameed, W.S.; Al Hassany, H.J.; Al-Dabet, M.M.; Al-Bawareed, O.; Hadi, N.R. Evaluation of the Genetic Association and Expressions of Notch-2 /Jagged-1 in Patients with Type 2 Diabetes Mellitus. Med. Arch. 2021, 75, 101–108. [Google Scholar] [CrossRef]
- Neild, G.H. Primary renal disease in young adults with renal failure. Nephrol. Dial. Transplant. 2010, 25, 1025–1032. [Google Scholar] [CrossRef]
- Connaughton, D.M.; Hildebrandt, F. Personalized medicine in chronic kidney disease by detection of monogenic mutations. Nephrol. Dial. Transplant. 2020, 35, 390–397. [Google Scholar] [CrossRef]
- Torra, R.; Furlano, M.; Ortiz, A.; Ars, E. Genetic kidney diseases as an underrecognized cause of chronic kidney disease: The key role of international registry reports. Clin. Kidney J. 2021, 14, 1879–1885. [Google Scholar] [CrossRef]
- Devuyst, O.; Knoers, N.V.; Remuzzi, G.; Schaefer, F. Association BotWGfIKDotERAaEDaT. Rare inherited kidney diseases: Challenges, opportunities, and perspectives. Lancet 2014, 383, 1844–1859. [Google Scholar] [CrossRef] [PubMed]
- Rasouly, H.M.; Groopman, E.E.; Heyman-Kantor, R.; Fasel, D.A.; Mitrotti, A.; Westland, R.; Bier, L.; Weng, C.; Ren, Z.; Copeland, B.; et al. The Burden of Candidate Pathogenic Variants for Kidney and Genitourinary Disorders Emerging From Exome Sequencing. Ann. Intern. Med. 2019, 170, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Vivante, A.; Hildebrandt, F. Exploring the genetic basis of early-onset chronic kidney disease. Nat. Rev. Nephrol. 2016, 12, 133–146. [Google Scholar] [CrossRef]
- Knoers, N.; Antignac, C.; Bergmann, C.; Dahan, K.; Giglio, S.; Heidet, L.; Lipska-Ziętkiewicz, B.S.; Noris, M.; Remuzzi, G.; Vargas-Poussou, R.; et al. Genetic testing in the diagnosis of chronic kidney disease: Recommendations for clinical practice. Nephrol. Dial. Transplant. 2022, 37, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Liu, J.; Chen, J.; Zhou, S.; Wang, Y.; Yu, L.; Sun, L.; Wang, L.; Wu, B.; Liu, F.; et al. Multidisciplinary approach to screening and management of children with Fabry disease: Practice at a Tertiary Children’s Hospital in China. Orphanet J. Rare Dis. 2021, 16, 509. [Google Scholar] [CrossRef]
- Favalli, V.; Disabella, E.; Molinaro, M.; Tagliani, M.; Scarabotto, A.; Serio, A.; Grasso, M.; Narula, N.; Giorgianni, C.; Caspani, C.; et al. Genetic Screening of Anderson-Fabry Disease in Probands Referred From Multispecialty Clinics. J. Am. Coll. Cardiol. 2016, 68, 1037–1050. [Google Scholar] [CrossRef]
- Trautmann, A.; Lipska-Ziętkiewicz, B.S.; Schaefer, F. Exploring the Clinical and Genetic Spectrum of Steroid Resistant Nephrotic Syndrome: The PodoNet Registry. Front. Pediatr. 2018, 6, 200. [Google Scholar] [CrossRef]
- Bullich, G.; Domingo-Gallego, A.; Vargas, I.; Ruiz, P.; Lorente-Grandoso, L.; Furlano, M.; Fraga, G.; Madrid, A.; Ariceta, G.; Borregán, M.; et al. A kidney-disease gene panel allows a comprehensive genetic diagnosis of cystic and glomerular inherited kidney diseases. Kidney Int. 2018, 94, 363–371. [Google Scholar] [CrossRef]
- Gefen, A.M.; Sethna, C.B.; Cil, O.; Perwad, F.; Schoettler, M.; Michael, M.; Angelo, J.R.; Safdar, A.; Amlie-Wolf, L.; Hunley, T.E.; et al. Genetic testing in children with nephrolithiasis and nephrocalcinosis. Pediatr. Nephrol. 2023, 38, 2615–2622. [Google Scholar] [CrossRef]
- Obeidova, L.; Seeman, T.; Fencl, F.; Blahova, K.; Hojny, J.; Elisakova, V.; Reiterova, J.; Stekrova, J. Results of targeted next-generation sequencing in children with cystic kidney diseases often change the clinical diagnosis. PLoS ONE 2020, 15, e0235071. [Google Scholar] [CrossRef]
- Ashton, E.J.; Legrand, A.; Benoit, V.; Roncelin, I.; Venisse, A.; Zennaro, M.C.; Jeunemaitre, X.; Iancu, D.; Van’t Hoff, W.G.; Walsh, S.B.; et al. Simultaneous sequencing of 37 genes identified causative mutations in the majority of children with renal tubulopathies. Kidney Int. 2018, 93, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, K.V.; Chang, A.R.; Block, G.A.; Robinson, K.; Bristow, S.L.; Morales, A.; Mitchell, A.; McCalley, S.; McKay, J.; Pollak, M.R.; et al. The KIDNEYCODE Program: Diagnostic Yield and Clinical Features of Individuals with CKD. Kidney360 2022, 3, 900–909. [Google Scholar] [CrossRef]
- Rush, E.T.; Johnson, B.; Aradhya, S.; Beltran, D.; Bristow, S.L.; Eisenbeis, S.; Guerra, N.E.; Krolczyk, S.; Miller, N.; Morales, A.; et al. Molecular Diagnoses of X-Linked and Other Genetic Hypophosphatemias: Results From a Sponsored Genetic Testing Program. J. Bone Miner. Res. 2022, 37, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Schönauer, R.; Baatz, S.; Nemitz-Kliemchen, M.; Frank, V.; Petzold, F.; Sewerin, S.; Popp, B.; Münch, J.; Neuber, S.; Bergmann, C.; et al. Matching clinical and genetic diagnoses in autosomal dominant polycystic kidney disease reveals novel phenocopies and potential candidate genes. Genet. Med. 2020, 22, 1374–1383. [Google Scholar] [CrossRef] [PubMed]
- Bao, M.; Cai, J.; Yang, X.; Ma, W. Genetic screening for Bartter syndrome and Gitelman syndrome pathogenic genes among individuals with hypertension and hypokalemia. Clin. Exp. Hypertens. 2019, 41, 381–388. [Google Scholar] [CrossRef]
- Schönauer, R.; Scherer, L.; Nemitz-Kliemchen, M.; Hagemann, T.; Hantmann, E.; Seidel, A.; Müller, L.; Kehr, S.; Voigt, C.; Stolzenburg, J.U.; et al. Systematic assessment of monogenic etiology in adult-onset kidney stone formers undergoing urological intervention-evidence for genetic pretest probability. Am. J. Med. Genet. C Semin. Med. Genet. 2022, 190, 279–288. [Google Scholar] [CrossRef]
- Gribouval, O.; Boyer, O.; Hummel, A.; Dantal, J.; Martinez, F.; Sberro-Soussan, R.; Etienne, I.; Chauveau, D.; Delahousse, M.; Lionet, A.; et al. Identification of genetic causes for sporadic steroid-resistant nephrotic syndrome in adults. Kidney Int. 2018, 94, 1013–1022. [Google Scholar] [CrossRef]
- Gast, C.; Pengelly, R.J.; Lyon, M.; Bunyan, D.J.; Seaby, E.G.; Graham, N.; Venkat-Raman, G.; Ennis, S. Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2016, 31, 961–970. [Google Scholar] [CrossRef]
- Jayasinghe, K.; Wu, Y.; Stark, Z.; Kerr, P.G.; Mallett, A.J.; Gaff, C.; Martyn, M.; Goranitis, I.; Quinlan, C. Cost-Effectiveness of Targeted Exome Analysis as a Diagnostic Test in Glomerular Diseases. Kidney Int. Rep. 2021, 6, 2850–2861. [Google Scholar] [CrossRef]
- Shanks, J.; Butler, G.; Cheng, D.; Jayasinghe, K.; Quinlan, C. Clinical and diagnostic utility of genomic sequencing for children referred to a Kidney Genomics Clinic with microscopic haematuria. Pediatr. Nephrol. 2023, 38, 2623–2630. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Udwan, K.; John, R.; Rana, A.; Haghighi, A.; Xu, L.; Hack, S.; Reich, H.N.; Hladunewich, M.A.; Cattran, D.C.; et al. Integration of Genetic Testing and Pathology for the Diagnosis of Adults with FSGS. Clin. J. Am. Soc. Nephrol. 2019, 14, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Braunisch, M.C.; Riedhammer, K.M.; Herr, P.M.; Draut, S.; Günthner, R.; Wagner, M.; Weidenbusch, M.; Lungu, A.; Alhaddad, B.; Renders, L.; et al. Identification of disease-causing variants by comprehensive genetic testing with exome sequencing in adults with suspicion of hereditary FSGS. Eur. J. Hum. Genet. 2021, 29, 262–270. [Google Scholar] [CrossRef]
- Wopperer, F.J.; Knaup, K.X.; Stanzick, K.J.; Schneider, K.; Jobst-Schwan, T.; Ekici, A.B.; Uebe, S.; Wenzel, A.; Schliep, S.; Schürfeld, C.; et al. Diverse molecular causes of unsolved autosomal dominant tubulointerstitial kidney diseases. Kidney Int. 2022, 102, 405–420. [Google Scholar] [CrossRef]
- Mallawaarachchi, A.C.; Lundie, B.; Hort, Y.; Schonrock, N.; Senum, S.R.; Gayevskiy, V.; Minoche, A.E.; Hollway, G.; Ohnesorg, T.; Hinchcliffe, M.; et al. Genomic diagnostics in polycystic kidney disease: An assessment of real-world use of whole-genome sequencing. Eur. J. Hum. Genet. 2021, 29, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Rheault, M.N.; McLaughlin, H.M.; Mitchell, A.; Blake, L.E.; Devarajan, P.; Warady, B.A.; Gibson, K.L.; Lieberman, K.V. COL4A gene variants are common in children with hematuria and a family history of kidney disease. Pediatr. Nephrol. 2023, 38, 3625–3633. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lin, F.; Zhai, Y.; Wang, C.; Wu, B.; Ma, D.; Rao, J.; Liu, J.; Liu, J.; Yu, M.; et al. Diagnostic and clinical utility of genetic testing in children with kidney failure. Pediatr. Nephrol. 2021, 36, 3653–3662. [Google Scholar] [CrossRef]
- Domingo-Gallego, A.; Pybus, M.; Bullich, G.; Furlano, M.; Ejarque-Vila, L.; Lorente-Grandoso, L.; Ruiz, P.; Fraga, G.; González, M.L.; Piñero-Fernández, J.A.; et al. Clinical utility of genetic testing in early-onset kidney disease: Seven genes are the main players. Nephrol. Dial. Transplant. 2022, 37, 687–696. [Google Scholar] [CrossRef]
- Bleyer, A.J.; Westemeyer, M.; Xie, J.; Bloom, M.S.; Brossart, K.; Eckel, J.J.; Jones, F.; Molnar, M.Z.; Kotzker, W.; Anand, P.; et al. Genetic Etiologies for Chronic Kidney Disease Revealed through Next-Generation Renal Gene Panel. Am. J. Nephrol. 2022, 53, 297–306. [Google Scholar] [CrossRef]
- Oh, J.; Shin, J.I.; Lee, K.; Lee, C.; Ko, Y.; Lee, J.S. Clinical application of a phenotype-based NGS panel for differential diagnosis of inherited kidney disease and beyond. Clin. Genet. 2021, 99, 236–249. [Google Scholar] [CrossRef]
- Mansilla, M.A.; Sompallae, R.R.; Nishimura, C.J.; Kwitek, A.E.; Kimble, M.J.; Freese, M.E.; Campbell, C.A.; Smith, R.J.; Thomas, C.P. Targeted broad-based genetic testing by next-generation sequencing informs diagnosis and facilitates management in patients with kidney diseases. Nephrol. Dial. Transplant. 2021, 36, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Schrezenmeier, E.; Kremerskothen, E.; Halleck, F.; Staeck, O.; Liefeldt, L.; Choi, M.; Schüler, M.; Weber, U.; Bachmann, N.; Grohmann, M.; et al. The underestimated burden of monogenic kidney disease in adults waitlisted for kidney transplantation. Genet. Med. 2021, 23, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Ottlewski, I.; Münch, J.; Wagner, T.; Schönauer, R.; Bachmann, A.; Weimann, A.; Hentschel, J.; Lindner, T.H.; Seehofer, D.; Bergmann, C.; et al. Value of renal gene panel diagnostics in adults waiting for kidney transplantation due to undetermined end-stage renal disease. Kidney Int. 2019, 96, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, Y.; Huang, J.; Zeng, Y.; Qian, Y.; Chen, J.; Chen, G.; Xia, G.; Wang, C.; Feng, A.; et al. New insights from trio whole-exome sequencing in the children with kidney disease: A single-center retrospective cohort study. Mol. Genet. Genom. Med. 2023, 11, e2163. [Google Scholar] [CrossRef] [PubMed]
- El Naofal, M.; Ramaswamy, S.; Alsarhan, A.; Nugud, A.; Sarfraz, F.; Janbaz, H.; Taylor, A.; Jain, R.; Halabi, N.; Yaslam, S.; et al. The genomic landscape of rare disorders in the Middle East. Genome Med. 2023, 15, 5. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Yu, F.; Dong, R.; Zhang, K.; Lv, Y.; Ma, J.; Wang, D.; Zhang, H.; Gai, Z.; Liu, Y.; et al. Diagnostic application of exome sequencing in Chinese children with suspected inherited kidney diseases. Front. Genet. 2022, 13, 933636. [Google Scholar] [CrossRef] [PubMed]
- Mann, N.; Braun, D.A.; Amann, K.; Tan, W.; Shril, S.; Connaughton, D.M.; Nakayama, M.; Schneider, R.; Kitzler, T.M.; van der Ven, A.T.; et al. Whole-Exome Sequencing Enables a Precision Medicine Approach for Kidney Transplant Recipients. J. Am. Soc. Nephrol. 2019, 30, 201–215. [Google Scholar] [CrossRef]
- Snoek, R.; van Jaarsveld, R.H.; Nguyen, T.Q.; Peters, E.D.J.; Elferink, M.G.; Ernst, R.F.; Rookmaaker, M.B.; Lilien, M.R.; Spierings, E.; Goldschmeding, R.; et al. Genetics-first approach improves diagnostics of ESKD patients <50 years old. Nephrol. Dial. Transplant. 2022, 37, 349–357. [Google Scholar]
- Al-Hamed, M.H.; Hussein, M.H.; Shah, Y.; Al-Mojalli, H.; Alsabban, E.; Alshareef, T.; Altayyar, A.; Elshouny, S.; Ali, W.; Abduljabbar, M.; et al. Exome sequencing unravels genetic variants associated with chronic kidney disease in Saudi Arabian patients. Hum. Mutat. 2022, 43, e24–e37. [Google Scholar] [CrossRef]
- Jayasinghe, K.; Stark, Z.; Kerr, P.G.; Gaff, C.; Martyn, M.; Whitlam, J.; Creighton, B.; Donaldson, E.; Hunter, M.; Jarmolowicz, A.; et al. Clinical impact of genomic testing in patients with suspected monogenic kidney disease. Genet. Med. 2021, 23, 183–191. [Google Scholar] [CrossRef]
- Riedhammer, K.M.; Braunisch, M.C.; Günthner, R.; Wagner, M.; Hemmer, C.; Strom, T.M.; Schmaderer, C.; Renders, L.; Tasic, V.; Gucev, Z.; et al. Exome Sequencing and Identification of Phenocopies in Patients With Clinically Presumed Hereditary Nephropathies. Am. J. Kidney Dis. 2020, 76, 460–470. [Google Scholar] [CrossRef]
- Lata, S.; Marasa, M.; Li, Y.; Fasel, D.A.; Groopman, E.; Jobanputra, V.; Rasouly, H.; Mitrotti, A.; Westland, R.; Verbitsky, M.; et al. Whole-Exome Sequencing in Adults With Chronic Kidney Disease: A Pilot Study. Ann. Intern. Med. 2018, 168, 100–109. [Google Scholar] [CrossRef]
- Groopman, E.E.; Marasa, M.; Cameron-Christie, S.; Petrovski, S.; Aggarwal, V.S.; Milo-Rasouly, H.; Li, Y.; Zhang, J.; Nestor, J.; Krithivasan, P.; et al. Diagnostic Utility of Exome Sequencing for Kidney Disease. N. Engl. J. Med. 2019, 380, 142–151. [Google Scholar] [CrossRef]
- Connaughton, D.M.; Kennedy, C.; Shril, S.; Mann, N.; Murray, S.L.; Williams, P.A.; Conlon, E.; Nakayama, M.; van der Ven, A.T.; Ityel, H.; et al. Monogenic causes of chronic kidney disease in adults. Kidney Int. 2019, 95, 914–928. [Google Scholar] [CrossRef]
- Mallett, A.J.; McCarthy, H.J.; Ho, G.; Holman, K.; Farnsworth, E.; Patel, C.; Fletcher, J.T.; Mallawaarachchi, A.; Quinlan, C.; Bennetts, B.; et al. Massively parallel sequencing and targeted exomes in familial kidney disease can diagnose underlying genetic disorders. Kidney Int. 2017, 92, 1493–1506. [Google Scholar] [CrossRef] [PubMed]
- Bekheirnia, N.; Glinton, K.E.; Rossetti, L.; Manor, J.; Chen, W.; Lamb, D.J.; Braun, M.C.; Bekheirnia, M.R. Clinical Utility of Genetic Testing in the Precision Diagnosis and Management of Pediatric Patients with Kidney and Urinary Tract Diseases. Kidney360 2021, 2, 90–104. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.Y.; Borden, C.; Roberts, M.B.; Mazzola, S.; Tan, Q.K.; Fatica, R.; Simon, J.; Calle, J.; Taliercio, J.; Dell, K.; et al. Renal Genetics Clinic: 3-Year Experience in the Cleveland Clinic. Kidney Med. 2023, 5, 100585. [Google Scholar] [CrossRef] [PubMed]
- Pode-Shakked, B.; Ben-Moshe, Y.; Barel, O.; Regev, L.C.; Kagan, M.; Eliyahu, A.; Marek-Yagel, D.; Atias-Varon, D.; Lahav, E.; Issler, N.; et al. A multidisciplinary nephrogenetic referral clinic for children and adults-diagnostic achievements and insights. Pediatr. Nephrol. 2022, 37, 1623–1646. [Google Scholar] [CrossRef]
- Pinto, E.V.F.; Prochnow, C.; Kemppainen, J.L.; Lisi, E.C.; Steyermark, J.M.; Kruisselbrink, T.M.; Pichurin, P.N.; Dhamija, R.; Hager, M.M.; Albadri, S.; et al. Genomics Integration Into Nephrology Practice. Kidney Med. 2021, 3, 785–798. [Google Scholar] [CrossRef]
- Bogyo, K.; Vena, N.; May, H.; Rasouly, H.M.; Marasa, M.; Sanna-Cherchi, S.; Kiryluk, K.; Nestor, J.; Gharavi, A. Incorporating genetics services into adult kidney disease care. Am. J. Med. Genet. C Semin. Med. Genet. 2022, 190, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Murray, S.L.; Dorman, A.; Benson, K.A.; Connaughton, D.M.; Stapleton, C.P.; Fennelly, N.K.; Kennedy, C.; McDonnell, C.A.; Kidd, K.; Cormican, S.M.; et al. Utility of Genomic Testing after Renal Biopsy. Am. J. Nephrol. 2020, 51, 43–53. [Google Scholar] [CrossRef]
- Elhassan, E.A.E.; Murray, S.L.; Connaughton, D.M.; Kennedy, C.; Cormican, S.; Cowhig, C.; Stapleton, C.; Little, M.A.; Kidd, K.; Bleyer, A.J.; et al. The utility of a genetic kidney disease clinic employing a broad range of genomic testing platforms: Experience of the Irish Kidney Gene Project. J. Nephrol. 2022, 35, 1655–1665. [Google Scholar] [CrossRef]
- Pasqualim, G.; Dos Santos, B.A.; Giugliani, R.; Matte, U. Simple and efficient screening of patients with Fabry disease with high resolution melting. Clin. Biochem. 2018, 53, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Groopman, E.E.; Rasouly, H.M.; Gharavi, A.G. Genomic medicine for kidney disease. Nat. Rev. Nephrol. 2018, 14, 83–104. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Ankala, A.; Wilcox, W.R.; Hegde, M.R. Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing: Single-gene, gene panel, or exome/genome sequencing. Genet. Med. 2015, 17, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing depth and coverage: Key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Barbitoff, Y.A.; Polev, D.E.; Glotov, A.S.; Serebryakova, E.A.; Shcherbakova, I.V.; Kiselev, A.M.; Kostareva, A.A.; Glotov, O.S.; Predeus, A.V. Systematic dissection of biases in whole-exome and whole-genome sequencing reveals major determinants of coding sequence coverage. Sci. Rep. 2020, 10, 2057. [Google Scholar] [CrossRef] [PubMed]
- Ewans, L.J.; Minoche, A.E.; Schofield, D.; Shrestha, R.; Puttick, C.; Zhu, Y.; Drew, A.; Gayevskiy, V.; Elakis, G.; Walsh, C.; et al. Whole exome and genome sequencing in mendelian disorders: A diagnostic and health economic analysis. Eur. J. Hum. Genet. 2022, 30, 1121–1131. [Google Scholar] [CrossRef]
- Schwarze, K.; Buchanan, J.; Taylor, J.C.; Wordsworth, S. Are whole-exome and whole-genome sequencing approaches cost-effective? A systematic review of the literature. Genet. Med. 2018, 20, 1122–1130. [Google Scholar] [CrossRef]
- Rao, J.; Liu, X.; Mao, J.; Tang, X.; Shen, Q.; Li, G.; Sun, L.; Bi, Y.; Wang, X.; Qian, Y.; et al. Genetic spectrum of renal disease for 1001 Chinese children based on a multicenter registration system. Clin. Genet. 2019, 96, 402–410. [Google Scholar] [CrossRef]
- Wilson, P.C.; Love-Gregory, L.; Corliss, M.; McNulty, S.; Heusel, J.W.; Gaut, J.P. Beyond Panel-Based Testing: Exome Analysis Increases Sensitivity for Diagnosis of Genetic Kidney Disease. Kidney360 2020, 1, 772–780. [Google Scholar] [CrossRef]
- Becherucci, F.; Landini, S.; Palazzo, V.; Cirillo, L.; Raglianti, V.; Lugli, G.; Tiberi, L.; Dirupo, E.; Bellelli, S.; Mazzierli, T.; et al. A Clinical Workflow for Cost-Saving High-Rate Diagnosis of Genetic Kidney Diseases. J. Am. Soc. Nephrol. 2023, 34, 706–720. [Google Scholar] [CrossRef]
- Chapman, A.B.; Devuyst, O.; Eckardt, K.U.; Gansevoort, R.T.; Harris, T.; Horie, S.; Kasiske, B.L.; Odland, D.; Pei, Y.; Perrone, R.D.; et al. Autosomal-dominant polycystic kidney disease (ADPKD): Executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2015, 88, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Mantere, T.; Kersten, S.; Hoischen, A. Long-Read Sequencing Emerging in Medical Genetics. Front. Genet. 2019, 10, 426. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, P.; Frommolt, P. Challenges in the Setup of Large-scale Next-Generation Sequencing Analysis Workflows. Comput. Struct. Biotechnol. J. 2017, 15, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Beyond One Million Genomes. Available online: https://b1mg-project.eu/ (accessed on 12 September 2023).
- Bertoli-Avella, A.M.; Beetz, C.; Ameziane, N.; Rocha, M.E.; Guatibonza, P.; Pereira, C.; Calvo, M.; Herrera-Ordonez, N.; Segura-Castel, M.; Diego-Alvarez, D.; et al. Successful application of genome sequencing in a diagnostic setting: 1007 index cases from a clinically heterogeneous cohort. Eur. J. Hum. Genet. 2021, 29, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Battke, F.; Schulte, B.; Schulze, M.; Biskup, S. The question of WGS’s clinical utility remains unanswered. Eur. J. Hum. Genet. 2021, 29, 722–723. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.F.; FitzPatrick, D.R.; Firth, H.V. Paediatric genomics: Diagnosing rare disease in children. Nat. Rev. Genet. 2018, 19, 325. [Google Scholar] [CrossRef]
- Okada, E.; Morisada, N.; Horinouchi, T.; Fujii, H.; Tsuji, T.; Miura, M.; Katori, H.; Kitagawa, M.; Morozumi, K.; Toriyama, T.; et al. Detecting MUC1 Variants in Patients Clinicopathologically Diagnosed With Having Autosomal Dominant Tubulointerstitial Kidney Disease. Kidney Int. Rep. 2022, 7, 857–866. [Google Scholar] [CrossRef]
- Kalia, S.S.; Adelman, K.; Bale, S.J.; Chung, W.K.; Eng, C.; Evans, J.P.; Herman, G.E.; Hufnagel, S.B.; Hufnagel, S.B.; Klein, T.E.; et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of Medical Genetics and Genomics. Genet. Med. 2017, 19, 249–255. [Google Scholar] [CrossRef]
- Miller, D.T.; Lee, K.; Chung, W.K.; Gordon, A.S.; Herman, G.E.; Klein, T.E.; Stewart, D.R.; Amendola, L.M.; Adelman, K.; Bale, S.J.; et al. ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Cassa, C.A.; Savage, S.K.; Taylor, P.L.; Green, R.C.; McGuire, A.L.; Mandl, K.D. Disclosing pathogenic genetic variants to research participants: Quantifying an emerging ethical responsibility. Genome Res. 2012, 22, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Recommendation CM/Rec(2016)8 of the Committee of Ministers to the Member States on the Processing of Personal Health-Related Data for Insurance Purposes, Including Data Resulting from Genetic Tests. 1269th Meeting of the Ministers’ Deputies. 2016. Available online: https://www.quotidianosanita.it/allegati/allegato2027308.pdf (accessed on 12 September 2023).
- Clissold, R.L.; Shaw-Smith, C.; Turnpenny, P.; Bunce, B.; Bockenhauer, D.; Kerecuk, L.; Waller, S.; Bowman, P.; Bowman, P.; Bowman, P.; et al. Chromosome 17q12 microdeletions but not intragenic HNF1B mutations link developmental kidney disease and psychiatric disorder. Kidney Int. 2016, 90, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Bertier, G.; Hétu, M.; Joly, Y. Unsolved challenges of clinical whole-exome sequencing: A systematic literature review of end-users’ views. BMC Med. Genom. 2016, 9, 52. [Google Scholar] [CrossRef]
- Hook, P.W.; Timp, W. Beyond assembly: The increasing flexibility of single-molecule sequencing technology. Nat. Rev. Genet. 2023, 24, 627–641. [Google Scholar] [CrossRef]
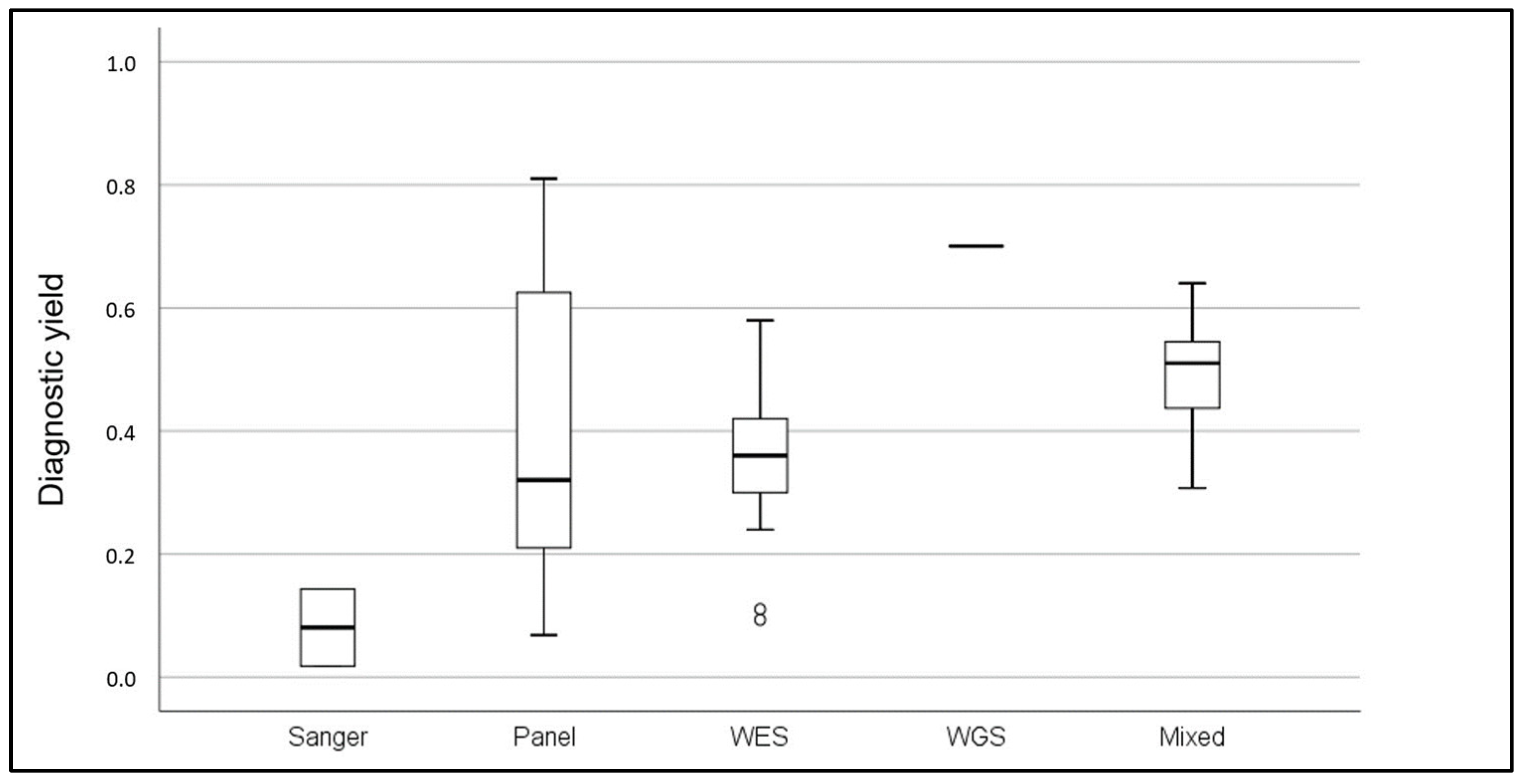

{kind=link}
| CKD Clinical | Technologies | Pediatric | Pediatric/Adult | Adult | |||
|---|---|---|---|---|---|---|---|
| Presentation | N° Cases | Yield (%) | N° Cases | Yield (%) | N° Cases | Yield (%) | |
| Specific | Sanger [17,18] | 35 | 14 | - | - | 2034 | 2 |
| Targeted panel [19,20,21,22,23,24,25,26,27,28,29,30] | 31–1554 | 24–78 | 34–859 | 18–81 | 81–236 | 7–22 | |
| WES [31,32,33,34] | 24–60 | 42–58 | - | - | 24–193 | 11–36.5 | |
| Mixed [35] | - | - | 45 | 64 | - | - | |
| WGS [36] | 144 | 70 | |||||
| Non-specific | Sanger | - | - | - | - | - | - |
| Targeted panel [37,38,39,40,41,42,43,44] | 188–832 | 28–40 | 50–1007 | 21–65 | 135–416 | 12–56 | |
| WES [45,46,47,48,49,50,51,52,53,54,55,56] | 104–1000 | 32.5–52 | 80–174 | 30–51 | 92–3315 | 9.3–34 | |
| Mixed [57,58,59,60,61,62,63] | 158 | 51 | 74–309 | 31–57 | 231 | 42 | |
| Technology | N° Cases | Diagnostic Yield (%) | Incremental (%) | References |
|---|---|---|---|---|
| Panel (2703 genes) | 482 | 42.6 | No statistical differences (p > 0.05) | [71] |
| Singleton-WES | 196 | 36.2 | ||
| Trio-WES | 317 | 44.8 | ||
| Panels (virtual analysis) | 3315 | 4.1 | 5 (p < 0.01) | [54] |
| WES | 3315 | 9.3 | ||
| Panels (disease specific) | 324 | 20.0 | 10 (p < 0.01) | [72] |
| WES | 324 | 30.0 |
| Advantages | Disadvantages | |
|---|---|---|
| Whole-Exome Sequencing | Detection of the coding-region variants. | Low coverage, mainly in GC rich and homology sequences. |
| Applicability in unclear CKD case. | Need for gap filling with Sanger. | |
| Cost effective options compared to WGS. | Burden of incidental findings. | |
| Possibility to create virtual panel of analysis to reduce the burden of VUS variants detected. | ||
| Whole-Genome Sequencing | Detection of the coding-region and non-coding variants. | Higher costs compared to WES. |
| Detection of structural variants. | Need for dedicated infrastructure and trained specialists. | |
| Applicability in unclear CKD case. | Relevant burden for VUS variants and incidental findings. | |
| Ability to detect CNVs and variants in high-homology regions (PKD1): one strategy for all the detections. | The superiority in diagnostic yield is debated (from 2% to 30%). |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giovanella, S.; Ligabue, G.; Chester, J.; Magistroni, R. Genomic Approaches for Monogenic Kidney Diseases: A Comparative Review of Diagnostic Methods and Precision Medicine Implications. Appl. Sci. 2023, 13, 12733. https://doi.org/10.3390/app132312733
Giovanella S, Ligabue G, Chester J, Magistroni R. Genomic Approaches for Monogenic Kidney Diseases: A Comparative Review of Diagnostic Methods and Precision Medicine Implications. Applied Sciences. 2023; 13(23):12733. https://doi.org/10.3390/app132312733
Chicago/Turabian StyleGiovanella, Silvia, Giulia Ligabue, Johanna Chester, and Riccardo Magistroni. 2023. "Genomic Approaches for Monogenic Kidney Diseases: A Comparative Review of Diagnostic Methods and Precision Medicine Implications" Applied Sciences 13, no. 23: 12733. https://doi.org/10.3390/app132312733
APA StyleGiovanella, S., Ligabue, G., Chester, J., & Magistroni, R. (2023). Genomic Approaches for Monogenic Kidney Diseases: A Comparative Review of Diagnostic Methods and Precision Medicine Implications. Applied Sciences, 13(23), 12733. https://doi.org/10.3390/app132312733