Abstract
Congenital muscular dystrophies (CMDs) are a heterogeneous group of genetic neuromuscular disorders. They usually occur at birth or in early childhood, with delayed acquisition of motor milestones, and diffuse muscle weakness. A dystrophic pattern is evident on the muscle biopsy. They are highly variable both in terms of severity and clinical evolution and in terms of pathogenetic biochemical mechanisms. The aim of this review is to collect and summarize the current knowledge of motor function in pediatric patients with congenital muscular dystrophies and the instruments used to assess it. This scoping review was conducted using the methodology of PRISMA (extension for Scoping Reviews, PRISMA-ScR). Two databases were queried from January 2002 to November 2022. Articles were identified based on title and abstract. Full-text papers published in peer-reviewed English-language journals were selected. It emerged that motor functional aspects are still underinvestigated in CMD patients, probably due to the rarity of these conditions and the phenotypic variability. The scales used to assess motor function are heterogeneous, as are the age groups considered. Finally, the predominant type of research design is cross-sectional; few studies analyze the progression of motor function over time. All these factors make it difficult to correlate the results of different publications and stress the need for more accurate and shared protocols for assessing motor function in these patients.
1. Introduction
Congenital muscular dystrophies are a heterogeneous group of rare neuromuscular disorders. The overall prevalence of CMD is estimated at between 0.6–0.9 cases per 100,000 births. By definition, onset is usually at birth or in early childhood [1]. Suspicion of CMD in later life may be due to the presence of mild or previously overlooked symptoms [2].
Common symptoms of the various forms of CMD are early muscle weakness, hypotonia, and joint contractures. These features may be more or less prominent. The typical floppy infant presentation may be observed in severe cases; in milder cases, antigravity movements of the limbs may be preserved, with greater impairment at the axial level. Joint contractures may sometimes be associated with ligamentous laxity and may be more or less diffuse.
In the advanced stages of the disease, cardiac and respiratory failure may occur. Brain involvement may also be present, usually manifesting as an intellectual disability [2].
Differential diagnosis in children with suspected congenital muscular dystrophy or congenital myopathy is determined by clinical features, age of onset, and inheritance pattern [3].
Congenital muscular dystrophies are characterized by dystrophic components on muscle biopsy [4]. However, heterogeneity in findings at biopsy has blurred the distinctions between disorders once thought to be distinct.
Today, with improved sequencing technology, genetic testing is the gold standard, as it speeds up the diagnostic process and reduces costs [3]. Thirty-seven genes are currently associated with congenital muscular dystrophy phenotypes. The most common types are collagen VI-related disorders (12–19%), dystroglycanopathies (12–25%), laminin-alpha2-related dystrophies (10–37%), and selenoprotein N (11.65%). Fukuyama CMD is the most prevalent form of CMD in Japan [5].
Although advances in research have improved the understanding of the disease mechanisms, there is still no commonly accepted approach to the treatment and care of CMD patients. The causes could be attributed to the rarity of the diseases and the great variability of clinical phenotypes.
In this scoping review, the current knowledge of the natural history and trends of motor function in pediatric patients with congenital muscular dystrophy is collected and summarized. In addition, the assessment tools used to describe motor function according to the form or stage of the disease are also described.
Collecting and sharing data on disease trajectories using longitudinal studies could add significant information to what is currently available. Therefore, we aim to make the scientific community aware of the lack of data and the future efforts needed to increase evidence, also considering possible innovative drug treatments.
2. Materials and Methods
2.1. Protocol and Registration
The PRISMA methodology for scoping reviews was employed to conduct the review. The results are presented following the Preferred Reporting Items for Systematic reviews and Meta-Analyses extension for Scoping Reviews (PRISMA-ScR) Checklist [6].
No a priori protocol was registered. Additional information about the process can be obtained from the corresponding author on request.
2.2. Inclusion Criteria
The inclusion criteria were categorized according to the scheme Population, Concept, and Context (PCC) recommended by the Joanna Briggs Institute for scoping reviews [7].
2.3. Population
We searched for articles including descriptions of motor function in patients with congenital muscular dystrophies (CMD), including alpha-dystroglycanopathies, Emery–Dreifuss muscular dystrophies, collagen VI-related dystrophy, and Merosin-deficient congenital muscular dystrophies. For this review, we did not consider non-congenital dystrophies and other neuromuscular disorders. We included articles that focused on pediatric populations.
2.4. Concept
We selected articles that analyzed and described motor function and its progression over time.
2.5. Context
No cultural, geographical, race, or gender-specific limits were considered for our review.
2.6. Information Sources
We considered articles available from 2002 to November 2022. Selected keywords were combined to create search strategies, adjusted for each screened database.
Articles were searched in the following databases: PubMed/MEDLINE, Embase.
2.7. Search Methods
We used a combination of Medical Subject Headings (MeSH) and free text (where terms were not present in the MeSH Database) to search the entries.
In consultation with a librarian from the Federated Library of Medicine (BFM) at the University of Turin, we developed a working framework for a search strategy (see Appendix A).
The Appendix A show the search process (search strings) used to retrieve the final articles from PubMed/MEDLINE and Embase. References from relevant articles were searched for the inclusion of additional papers not previously identified through the systematic search.
2.8. Level of Evidence and Qualitative Analysis of Eligible Articles
The quality of the eligible articles was assessed according to the levels of evidence established by the JBI. The qualitative assessment was performed by two authors (I.C. and F.R.) independently. All selected papers are case series so the level of evidence is 4b.
3. Results
A total of 16 articles focusing on motor outcome measures in pediatric patients with congenital muscular dystrophies were included in the review after selection.
There were specific articles on collagen VI-related dystrophies (COL6-RD) (n = 2), Fukuyama congenital muscular dystrophies (FCMD) (n = 2), laminopathies or LMNA-related congenital muscular dystrophies (LMNA-RD) (n = 1), LAMA2-related (Merosin-deficient) congenital muscular dystrophies (LAMA2-RD) (n = 2), SEPN1-related myopathies (SEPN1-RM) (n = 2), and a-dystroglycanopathies–FKRP mutations (FKRP) (n = 1), as well as other articles in which different pathologies were analyzed (n = 6).
3.1. Selection of Sources of Evidence
The articles were examined by two authors (I.C. and F.R.), and eligibility for inclusion was determined independently; in the case of discordant opinions between the reviewers, the eligibility of the article was discussed until consensus was reached.
Articles were initially screened based on titles and abstracts according to the population, concept, and context elements previously described. Duplicates were identified and removed. Only full-text papers published in peer-reviewed journals and English were selected. A total of 2690 articles were found: 912 in PubMed/MEDLINE, 1788 in Embase (see Figure 1). The flowchart of the selection of evidence is shown.
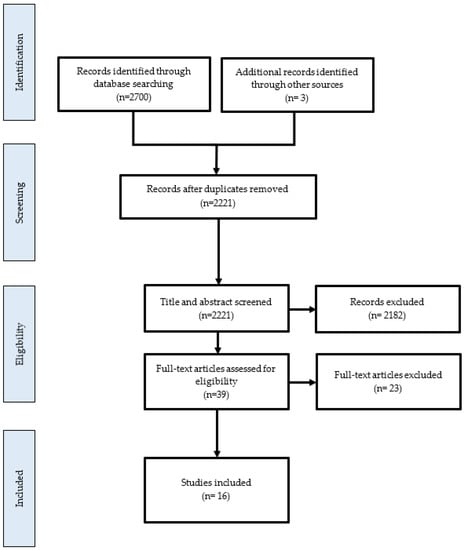
Figure 1.
Flowchart of selection of sources of evidence.
3.2. Synthesis of Results
3.2.1. Studies in Patients with COL6-RD Diseases
The data were grouped according to the type of congenital muscular dystrophy. Articles including different forms were grouped in a separate table. For each article, the author, year of publication, type of congenital muscular dystrophy, instruments used for the evaluation, and results are reported.
We found two articles regarding motor outcomes in patients with COLVI-RD. The details of each article are reported in Table 1.

Table 1.
Motor outcome measures in patients with COL6-RD (UCMD: Ullrich congenital muscular dystrophy, BM: Bethlem myopathy; COL6-RD: collagen VI-related dystrophies; MFM32: Motor Function Measure; 6 MWT: 6 min walk test; NSAA: North Star Ambulatory Assessment; LoA: Loss of Ambulation).
3.2.2. Studies in Patients with LMNA-RD
We found one article regarding motor outcomes in patients with LMNA-RD. The details of the article are reported in Table 2.
Table 2.
Motor outcome measures in patients with LMNA-RD (EDMD: Emery–Dreifuss muscular dystrophy, LoA: Loss of Ambulation).
3.2.3. Studies in Patients with LAMA2—CMD
We found two articles regarding motor outcomes in patients with LAMA2-CMD. The details of the articles are reported in Table 3.
Table 3.
Motor outcome measures in patients with LAMA2-RD (KAFO: Knee–Ankle–Foot Orthosis).
3.2.4. Studies in Patients with FCMD
We found two articles regarding motor outcomes in patients with FCMD. The details of the articles are reported in Table 4.
Table 4.
Motor outcome measures in patients with FCMD (FCMD: Fukuyama; GMFM: Gross Motor Function Measure; HMFS: Hammersmith Motor Function).
3.2.5. Studies in Patients with SEPN1-RM
We found two articles regarding motor outcomes in patients with SEPN1-RM. The details of the articles are reported in Table 5.
Table 5.
Motor outcome measures in patients with SEPN1-RM (SPN1-RM: SEPN1-related myopathies HFMS: Hammersmith Motor Function).
3.2.6. Studies in Patients with FKRP Mutations
We found one article regarding motor outcomes in patients with FKRP mutations. The details of the article are reported in Table 6.
Table 6.
Motor outcome measures in patients with FKRP mutations (6MWT: 6 min walk test; MHFMS: Modified Hammersmith Motor Function; PEDI: Pediatric Evaluation of Disability Inventory; PUL: Performance of Upper Limb; TFTs: timed function tests).
3.2.7. Studies in Patients with Various Forms of CMD
We found six articles regarding motor outcomes in patients with various forms of CMD. The details of the articles are reported in Table 7.
Table 7.
Motor outcome measures in patients with congenital muscular dystrophies (MFM32: Motor Function Measure; Jebsen: Jebsen–Taylor Hand Function Test; QUEST: Quality of Upper Extremity Skills Test; HHD: Hand-Held Dynamometry; 6MWT: 6 min walk test; HFMS: Hammersmith Motor Function; NSAA: North Star Ambulatory Assessment; TFTs: timed function tests).
4. Discussion
The scoping review method enabled an exhaustive literature search, a definition of the current level of knowledge, and the identification of gaps with respect to the current knowledge of motor function in pediatric patients with congenital muscular dystrophies.
This study confirms that data published in the last 20 years on motor outcomes in patients with CMD are limited. Only 16 articles on this topic were found, of which 2 focused on COLVI-RD [8,9], 1 on LMNA-RD [10], 2 on LAMA2-RD [11,12], 2 on FCMD [14,15], 2 on SEPN1-MD [15,16], 1 on FKRP mutations [17], and 6 on different forms of CMD [18,19,20,21,22,23].
As already reported by Zambon et al., the rarity of these diseases and the great phenotypic heterogeneity make large and comparable studies complex [5]. In this regard, attention is drawn to the pathologies analyzed in the selected articles. Despite the presence of numerous forms of CMD, most of the studies focused on the most represented and well-known forms (COLVI-RD, FCMD, LMNA-RD, LAMA2-RD, SEPN1-MD, FKRP). Only two articles mentioned other forms, although they are grouped in a single analysis category [19,21].
Out of the 16 studies selected, 11 were cross-sectional studies. Only five studies were longitudinal and therefore analyzed the progression of motor function over time [11,15,17,18,22]. The durations of these longitudinal studies varied but were generally short and do not allow broad trend curves to be drawn. Two articles showed results from the same sample of patients to whom different inclusion criteria were applied [18,22]. In fact, 44 of the 48 patients included in the study by Le Goff et al. were included in the study by Jain et al., i.e., all those with a diagnosis of LAMA2-RD or COL6-RD and with the completion of at least two MFM-32 evaluations one year apart. The cross-sectional studies provided information on the motor and functional aspects of the different forms of CMD, describing their common characteristics. However, they do not allow the evolution of the disease over time to be delineated.
The age ranges of the patients varied. Only 8 out of 16 studies showed results from exclusively pediatric patients (0–18 years), although with different age ranges. The other 8 studies included samples of pediatric and adult patients, almost always without distinction in the analysis.
Nevertheless, interesting preliminary data emerged regarding the progression of motor function in patients with LAMA2-RD, COL6-RD, SPN1-RM, and FKRP mutations.
In patients with LAMA2-RD, an annual linear reduction of 6.6° for right elbow flexion and 3.1° for knee flexion was described in the article by Zambon et al. [11], and an annual reduction of 3.21° for left elbow extension in the article by Jain et al. [18]. In the study by Jain et al., a decrement of 2.60 points each year emerged on the MFM32 scale [18]. This decrement was confirmed by the study of Le Goff et al., which showed a greater loss of score in the items related to axial and proximal function (−1.59 points per year) [22].
In patients with COL6-RD, an annual decrease of 4.01 points on the MFM32 scale was described in the study by Jain et al. [18] and confirmed by the study of Le Goff [22]. The domain in which there was the greatest loss of function was that of standing position and transfers (−1.99 points per year) [22].
In patients with SPN1-RM, an annual change of −0.55 points on the HFMS scale was estimated [15].
In patients with FKRP mutations, average annual decreases of 23.41 m at 6MWT, 0.65 points on the MHFMS scale, 1.33 on the PEDI scale, and 0.55 on the PUL scale were reported [17].
Additional larger longitudinal studies differentiated by pediatric age and adulthood with planned follow-up would be recommended to study the course of the diseases over time, better understand the characteristics of the different forms, and trace natural history trajectories. This would also be useful for identifying the outcome measures for trial readiness, as well as implementing innovative care and rehabilitation techniques in clinical practice. This incitement has been partly received by scientific and patient communities. Some disease registries have recently been proposed, such as the Global Registry for COL6-related dystrophies (NCT04020159) or the Global FKRP Registry (NCT04001595), and some natural history studies for clinical trial readiness have been registered such as the study of dystroglycanopathies (NCT00313677) or the study of patients with SEPN1 or LAMA2 mutations (NCT04478981).
Another feature that emerged is the heterogeneity of the assessment tools used in the studies. In 10 out of 16 papers, motor assessment instruments, such as functional scales and time tests, were used (see Table 8). In the other six papers, anamnestic data and information acquired during clinical assessments were analyzed.
Table 8.
Assessment tools used in the various selected studies (MFM32: Motor Function Measure; GMFM: Gross Motor Function Measure; HMFS: Hammersmith Motor Function; MHFMS: Modified Hammersmith Motor Function; PEDI: Pediatric Evaluation of Disability Inventory; NSAA: North Star Ambulatory Assessment; TFTs: timed function tests; 6MWT: 6 min walk test; Jebsen: Jebsen–Taylor Hand Function Test; PUL: Performance of Upper Limb; Brooke scale; QUEST: Quality of Upper Extremity Skills Test).
The absence of evaluation instruments specifically validated for these diseases should be underlined. The functional assessment scales reported have been validated for the assessment of patients with neuromuscular disease or other specific neuromuscular diseases or other motor disorders. The scales used were the Motor Function Measure (MFM32), Gross Motor Function Measure (GMFM), Hammersmith Motor Function (HFMS), Modified Hammersmith Motor Function (MHFMS), Pediatric Evaluation of Disability Inventory (PEDI), North Star Ambulatory Assessment (NSAA), timed function tests (TFTs), NM-Score classification, 6 min walk test (6MWT), Jebsen Taylor Hand Function Test (Jebsen), Performance of Upper Limb (PUL), Brooke scale, Quality of Upper Extremity Skills Test (QUEST), and Upper Limb Function Measure of Muscular Dystrophy. The tools cited allow the different motor and functional aspects to be assessed. According to the scale, it is possible to obtain results relating to gross motor functions, fine motor functions, upper limb abilities, fatigability, and speed of execution. In addition, some articles included instruments for measuring joint range and strength.
As can be seen in Table 8, the MFM32 scale was the most used instrument for the assessment of patients with CMD. The subdivision into domains (D1, standing and transfers; D2, axial and proximal motor function; and D3, distal motor function) allows for a comprehensive assessment of motor function [22]. It should be noted that patients with CMD were also included in the validation study of this scale [24].
The study by Meilleur et al. on patients with COL6-RD and LAMA2-RD showed a high association between the MFM32 scale and HFMS. The MFM32 provides more information, so it may be more convenient for the assessment of patients with CMD. However, it should be pointed out that some difficulties related to muscle contractures interfered with some tasks, either due to the inability to reach the initial position or the inability to complete the items [23].
The NSAA scale, which has been validated for patients with Duchenne muscular dystrophy [25] and spinal muscular atrophy type 3 [26], includes the assessment of more complex skills, which is useful for patients with better motor functioning who show ceiling effects on the MFM32 scale. It could, therefore, be a complementary tool [23].
With respect to upper limb function, the QUEST showed associations with the total score from the MFM32 in patients with COL6-RD and LAMA2-RD. However, further investigations are ongoing to understand which of these tools is more appropriate [23].
There were no studies describing the use of functional assessment scales in CMD patients in the neonatal period. However, it would be interesting to identify the most appropriate tool among the Chop Intend scale, the HFMS [27], or the recently proposed module of the Hammersmith Neonatal Neurological Examination (HNEE) for neonates with SMA [28].
Finally, the presence of contractures should be taken into consideration. The most frequently described muscle contractures are at the level of the long flexors of the fingers, elbows, knees, hips, and Achilles tendons. The interference reported in the MFM32 scale scores [23] can be expanded to all functional assessment scales, as the presence of contractures, regardless of the body area, limits functionality. Moreover, in some scales, not reaching the initial position corresponds to not scoring. This could represent a bias for those patients who, although limited by contractures, manage to complete the task anyway by implementing compensation strategies. The contractures may also highlight strength deficits when strength is assessed with the MRC scale or myometry [27].
As already highlighted at the 173rd ENMC international workshop on CMD, there is a need to identify the best motor and functional outcome tools for the assessment of patients with CMD [27].
The scientific community has made efforts in recent years, although studies only concern some of the forms and are still quantitatively limited.
It would be useful to refine research by focusing on additional parameters, such as age, stage of disease, residual motor function, form, and neurophysiological assessments, even if in the related literature, data are limited because of lower levels of compliance in pediatric age. This would make studies conducted in different centres comparable and aggregable.
5. Conclusions
In recent decades, the implementation of standards of care has changed the survival of patients suffering from congenital muscular dystrophies. Moreover, new genetic diagnostic techniques have made it possible to identify an increasing number of different forms.
There is an increasing need for larger longitudinal natural history studies, which are essential to identifying motor outcome measures for trial readiness, as well as implementing innovative treatment and rehabilitation techniques in clinical practice.
As can be seen from this scoping review, the scientific community, especially in recent years, has begun to respond to this challenge. However, it is essential to further expand the knowledge of the forms that have already been studied and proceed with new studies for the forms that are still underinvestigated. Finally, it would be advisable to define the most appropriate evaluation tools so that they can be used in a more homogeneous and, therefore, comparable manner.
Author Contributions
Conceptualization, I.C., T.M., and F.S.R.; methodology, I.C., R.D., C.B., E.P., T.M. and F.S.R.; validation, I.C., R.D., C.B., E.P., F.R, E.R. and F.S.R.; formal analysis, I.C., R.D., C.B. and F.S.R.; investigation, I.C., R.D. and F.S.R.; resources, I.C.; data curation, I.C.; writing—original draft preparation, I.C.; writing—review and editing, I.C., R.D., C.B., E.P., E.R., F.R., T.M. and F.S.R.; visualization, T.M. and F.R.; supervision, T.M. and F.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are openly available in publicly accessible repositories (Pubmed/MEDLINE, Embase).
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A
Pubmed search string
(((“Muscular Dystrophies”[Mesh] OR “muscular dystroph*”[tiab]) AND (“congenital”[Subheading] OR congenit*[tiab] OR connatal[tiab] OR CMD[tiab] OR Autosomal[tiab] OR inherit*[tiab] OR inborn[tiab] OR heredit*[tiab])) OR "Muscular Dystrophy, Congenital, due to Partial LAMA2 Deficiency"[Supplementary Concept] OR "Muscular dystrophy congenital, merosin negative”[Supplementary Concept] OR “Bethlem myopathy”[Supplementary Concept] OR “Walker-Warburg Syndrome”[Mesh] OR “Muscular Dystrophy, Emery-Dreifuss”[Mesh] OR alpha-dystroglycanopath*[tiab] OR alpha-DGP[tiab] OR “Walker-Warburg”[tiab] OR “Warburg syndrom*”[tiab] OR “Muscle-Eye-Brain Disease*”[tiab] OR COD-MD[tiab] OR “Emery Dreifuss”[tiab] OR laminopath*[tiab] OR "Bethlem myopath*”[tiab] OR ((Ulrich*[tiab] OR Ullrich*[tiab] OR “laminin alpha 2”[Supplementary Concept] OR laminin-alpha2[tiab] OR LAMA2[tiab] OR “laminin alpha 2”[tiab] OR "Laminin”[Mesh] OR merosin[tiab] “OR Collagen Type VI"[Mesh] OR “collagen VI"[tiab] OR "collagen type VI”[tiab] OR Col6[tiab] OR col6A*[tiab] OR Col-6[tiab] OR col-6A*[tiab] OR "Dystroglycans”[Mesh] OR alpha-dystroglycan*[tiab] OR a-dystroglycan*[tiab] OR FKTN[tiab] OR POMT*[tiab] OR FKRP[tiab] OR POMGNT*[tiab] OR ISPD[tiab] OR B3GNT*[tiab] OR GMPPB[tiab] OR DPM1[tiab] OR DPM2[tiab] OR ALG13[tiab] OR B3GALNT*[tiab] OR RXYLT*[tiab] OR "Lamin Type A"[Mesh] OR “lamin A-C”[tiab] OR “lamin A/C”[tiab] OR LMNA[tiab]) AND dystroph*[tiab])) AND ("Motor Skills"[Mesh] OR "Range of Motion, Articular"[Mesh] OR “Movement”[Mesh] OR motion[tiab] OR motor[tiab] OR movement*[tiab] OR locomot*[tiab] OR mobility[tiab] OR ambulat*[tiab] OR walking*[tiab] OR "Scoliosis"[Mesh] OR scolios*[tiab] OR "Natural History"[Mesh] OR "natural history"[tiab]).
Embase search string
(‘motor performance’/exp OR ‘motor skills in infancy and childhood’/exp OR ‘movement (physiology)’/exp OR ‘scoliosis’/exp OR ‘history’/exp OR motion:ab,ti OR motor:ab,ti OR movement*:ab,ti OR locomot*:ab,ti OR mobility:ab,ti OR ambulat*:ab,ti OR walking*:ab,ti OR ‘natural hystory’:ab,ti OR scolios*:ab,ti) AND ((‘muscular dystrophy’/exp AND (congenit*:ab,ti OR connatal:ab,ti OR cmd:ab,ti OR autosomal:ab,ti OR inherit*:ab,ti OR inborn*:ab,ti OR heredit*:ab,ti) OR ‘congenital muscular dystrophy type 1a’/exp OR ‘merosin deficient congenital muscular dystrophy’/exp OR ‘merosin’/exp OR ‘bethlem myopathy’/exp OR ‘walker warburg syndrome’/exp OR ‘emery dreifuss muscular dystrophy’/exp OR ‘alpha dystroglycanopathy’/exp OR ‘muscle eye brain disease’/exp OR ‘laminopathy’/exp OR ‘ullrich congenital muscular dystrophy’/exp OR ‘laminin’/exp OR ‘laminin alpha2’/exp OR ‘collagen type 6’/exp OR ‘dystroglycan’/exp OR ‘alpha dystroglycan’/exp OR ‘lamin a’/exp OR ‘alpha dystroglycanopath*’:ab,ti OR ‘alpha dgp’:ab,ti OR ‘walker-warburg’:ab,ti OR ‘warburg syndrom*’:ab,ti OR ‘muscle-eye-brain disease*’:ab,ti OR ‘cod md’:ab,ti OR ‘emery dreifuss’:ab,ti OR laminopath*:ab,ti OR ‘bethlem myopath*’:ab,ti OR ulrich*:ab,ti OR ullrich*:ab OR ‘laminin alpha2’:ab,ti OR lama2:ab,ti OR ‘laminin alpha 2’:ab,ti OR merosin:ab,ti OR ‘collagen vi’:ab,ti OR ‘collagen type vi’:ab,ti OR col6:ab,ti OR col6a*:ab,ti OR ‘col 6’:ab,ti OR ‘col 6a*’:ab,ti OR ‘alpha dystroglycan*’:ab,ti OR ‘a dystroglycan*’:ab,ti OR fktn:ab,ti OR pomt*:ab,ti OR fkrp:ab,ti OR pomgnt*:ab,ti OR ispd:ab,ti OR b3gnt*:ab,ti OR gmppb:ab,ti OR dpm1:ab,ti OR dpm2:ab,ti OR alg13:ab,ti OR b3galnt*:ab,ti OR rxylt*:ab,ti OR ‘lamin a-c’:ab,ti OR ‘lamin a/c’:ab,ti OR lmna:ab,ti) AND dystroph*:ab,ti).
References
- Pasrija, D.; Tadi, P. Congenital Muscular Dystrophy. In Treasure Island; StatPearls Publishing: Tampa, FL, USA, 2022. Available online: http://www.ncbi.nlm.nih.gov/books/NBK558956/ (accessed on 20 October 2022).
- Bertini, E.; D’Amico, A.; Gualandi, F.; Petrini, S. Congenital Muscular Dystrophies: A Brief Review. Semin. Pediatr. Neurol. 2011, 18, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, R.J. Congenital Muscular Dystrophy and Congenital Myopathy. Contin. Lifelong Learn. Neurol. 2019, 25, 1640–1661. [Google Scholar] [CrossRef]
- Graziano, A.; Bianco, F.; D’Amico, A.; Moroni, I.; Messina, S.; Bruno, C.; Pegoraro, E.; Mora, M.; Astrea, G.; Magri, F.; et al. Prevalence of congenital muscular dystrophy in Italy: A population study. Neurology 2015, 84, 904–911. [Google Scholar] [CrossRef]
- Zambon, A.A.; Muntoni, F. Congenital muscular dystrophies: What is new? Neuromuscul. Disord. 2021, 31, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Tricco, A.C.; Lillie, E.; Zarin, W.; O’Brien, K.K.; Colquhoun, H.; Levac, D.; Moher, D.; Peters, M.D.J.; Horsley, T.; Weeks, L.; et al. PRISMA Extension for Scoping Reviews (PRISMA-ScR): Checklist and Explanation. Ann. Intern. Med. 2018, 169, 467–473. [Google Scholar] [CrossRef] [PubMed]
- The Joanna Briggs Institute. Joanna Briggs Institute Reviewers’ Manual: 2015 Edition/Supplement Adelaide; The Joanna Briggs Institute: Adelaide, Australia, 2015. [Google Scholar]
- Yonekawa, T.; Komaki, H.; Okada, M.; Hayashi, Y.K.; Nonaka, I.; Sugai, K.; Sasaki, M.; Nishino, I. Rapidly progressive scoliosis and respiratory deterioration in Ullrich congenital muscular dystrophy. J. Neurol. Neurosurg. Psychiatry 2013, 84, 982–988. [Google Scholar] [CrossRef]
- Benito, D.N.-D.; Foley, A.R.; Domínguez-González, C.; Ortez, C.; Jain, M.; Mebrahtu, A.; Donkervoort, S.; Hu, Y.; Fink, M.; Yun, P.; et al. Association of Initial Maximal Motor Ability With Long-term Functional Outcome in Patients With COL6-Related Dystrophies. Neurology 2021, 96, e1413–e1424. [Google Scholar] [CrossRef]
- Fan, Y.; Tan, D.; Song, D.; Zhang, X.; Chang, X.; Wang, Z.; Zhang, C.; Chan, S.H.-S.; Wu, Q.; Wu, L.; et al. Clinical spectrum and genetic variations of LMNA-related muscular dystrophies in a large cohort of Chinese patients. J. Med. Genet. 2020, 58, 326–333. [Google Scholar] [CrossRef]
- Zambon, A.A.; Ridout, D.; Main, M.; Mein, R.; Phadke, R.; Muntoni, F.; Sarkozy, A. LAMA2-related muscular dystrophy: Natural history of a large pediatric cohort. Ann. Clin. Transl. Neurol. 2020, 7, 1870–1882. [Google Scholar] [CrossRef]
- Tan, D.; Ge, L.; Fan, Y.; Chang, X.; Wang, S.; Wei, C.; Ding, J.; Liu, A.; Wang, S.; Li, X.; et al. Natural history and genetic study of LAMA2-related muscular dystrophy in a large Chinese cohort. Orphanet J. Rare Dis. 2021, 16, 319. [Google Scholar] [CrossRef]
- Harada, R.; Taniguchi-Ikeda, M.; Nagasaka, M.; Nishii, T.; Inui, A.; Yamamoto, T.; Morioka, I.; Kuroda, R.; Iijima, K.; Nozu, K.; et al. Assessment of the upper limb muscles in patients with Fukuyama muscular dystrophy: Noninvasive assessment using visual ultrasound muscle analysis and shear wave elastography. Neuromuscul. Disord. 2022, 32, 754–762. [Google Scholar] [CrossRef]
- Ishigaki, K.; Ihara, C.; Nakamura, H.; Mori-Yoshimura, M.; Maruo, K.; Taniguchi-Ikeda, M.; Kimura, E.; Murakami, T.; Sato, T.; Toda, T.; et al. National registry of patients with Fukuyama congenital muscular dystrophy in Japan. Neuromuscul. Disord. 2018, 28, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Silwal, A.; Sarkozy, A.; Scoto, M.; Ridout, D.; Schmidt, A.; Laverty, A.; Henriques, M.; D’Argenzio, L.; Main, M.; Mein, R.; et al. Selenoprotein N-related myopathy: A retrospective natural history study to guide clinical trials. Ann. Clin. Transl. Neurol. 2020, 7, 2288–2296. [Google Scholar] [CrossRef] [PubMed]
- Villar-Quiles, R.N.; von der Hagen, M.; Métay, C.; Gonzalez, V.; Donkervoort, S.; Bertini, E.; Castiglioni, C.; Chaigne, D.; Colomer, J.; Cuadrado, M.L.; et al. The clinical, histologic, and genotypic spectrum of SEPN1-related myopathy. Neurology 2020, 95, e1512–e1527. [Google Scholar] [CrossRef]
- Gedlinske, A.M.; Stephan, C.M.; Mockler, S.R.; Laubscher, K.M.; Laubenthal, K.S.; Crockett, C.D.; Zimmerman, M.B.; Mathews, K.D. Motor outcome measures in patients with FKRP mutations. Neurology 2020, 95, e2131–e2139. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.S.; Meilleur, K.; Kim, E.; Norato, G.; Waite, M.; Nelson, L.; McGuire, M.; Duong, T.; Keller, K.; Lott, D.J.; et al. Longitudinal changes in clinical outcome measures in COL6-related dystrophies and LAMA2-related dystrophies. Neurology 2019, 93, e1932–e1943. [Google Scholar] [CrossRef] [PubMed]
- Vuillerot, C.; Meilleur, K.G.; Jain, M.; Waite, M.; Wu, T.; Linton, M.; Datsgir, J.; Donkervoort, S.; Leach, M.E.; Rutkowski, A.; et al. English Cross-Cultural Translation and Validation of the Neuromuscular Score: A System for Motor Function Classification in Patients With Neuromuscular Diseases. Arch. Phys. Med. Rehabilitation 2014, 95, 2064–2070.e1. [Google Scholar] [CrossRef]
- Bendixen, R.M.; Butrum, J.; Jain, M.S.; Parks, R.; Hodsdon, B.; Nichols, C.; Hsia, M.; Nelson, L.; Keller, K.C.; McGuire, M.; et al. Upper extremity outcome measures for collagen VI-related myopathy and LAMA2-related muscular dystrophy. Neuromuscul. Disord. 2016, 27, 278–285. [Google Scholar] [CrossRef]
- Vuillerot, C.; Rippert, P.; Kinet, V.; Renders, A.; Jain, M.; Waite, M.; Glanzman, A.M.; Girardot, F.; Hamroun, D.; Iwaz, J.; et al. Rasch Analysis of the Motor Function Measure in Patients With Congenital Muscle Dystrophy and Congenital Myopathy. Arch. Phys. Med. Rehabilitation 2014, 95, 2086–2095. [Google Scholar] [CrossRef]
- Le Goff, L.; Meilleur, K.G.; Norato, G.; Rippert, P.; Jain, M.; Fink, M.; Foley, A.R.; Waite, M.; Donkervoort, S.; Bönnemann, C.G.; et al. Responsiveness and Minimal Clinically Important Difference of the Motor Function Measure in Collagen VI-Related Dystrophies and Laminin Alpha2-Related Muscular Dystrophy. Arch. Phys. Med. Rehabilitation 2020, 102, 604–610. [Google Scholar] [CrossRef]
- Meilleur, K.G.; Jain, M.S.; Hynan, L.S.; Shieh, C.-Y.; Kim, E.; Waite, M.; McGuire, M.; Fiorini, C.; Glanzman, A.M.; Main, M.; et al. Results of a two-year pilot study of clinical outcome measures in collagen VI- and laminin alpha2-related congenital muscular dystrophies. Neuromuscul. Disord. 2014, 25, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Bérard, C.; Payan, C.; Hodgkinson, I.; Fermanian, J.; The MFM Collaborative Study Group A Motor Function Measure Scale for Neuromuscular Diseases. Construction and validation study. Neuromuscul. Disord. 2005, 15, 463–470. [Google Scholar] [CrossRef]
- Scott, E.; Eagle, M.; Mayhew, A.; Freeman, J.; Main, M.; Sheehan, J.; Manzur, A.; Muntoni, F.; Disease, T.N.S.C.N.F.P.N. Development of a Functional Assessment Scale for Ambulatory Boys with Duchenne Muscular Dystrophy. Physiother. Res. Int. 2011, 17, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Tiziano, F.D.; Lomastro, R.; Di Pietro, L.; Pasanisi, M.B.; Fiori, S.; Angelozzi, C.; Abiusi, E.; Angelini, C.; Sorarù, G.; Gaiani, A.; et al. Clinical and molecular cross-sectional study of a cohort of adult type III spinal muscular atrophy patients: Clues from a biomarker study. Eur. J. Hum. Genet. 2012, 21, 630–636. [Google Scholar] [CrossRef] [PubMed]
- Bönnemann, C.; Rutkowski, A.; Mercuri, E.; Muntoni, F. 173rd ENMC international workshop: Congenital muscular dystrophy outcome measures 5–7 March 2010, Naarden, The Netherlands. Neuromuscul. Disord. 2011, 21, 513–522. [Google Scholar] [CrossRef]
- Pane, M.; Donati, M.A.; Cutrona, C.; De Sanctis, R.; Pirinu, M.; Coratti, G.; Ricci, M.; Palermo, C.; Berti, B.; Leone, D.; et al. Neurological assessment of newborns with spinal muscular atrophy identified through neonatal screening. Eur. J. Pediatr. 2022, 181, 2821–2829. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).