
Cholesterol Metabolism Pathways Disturbances in Atherosclerosis—Analyses Using Stochastic Petri Net-Based Model



Abstract
1. Introduction
1.1. Cholesterol Significance
1.2. HMG-CoA and Mevalonate Synthesis
1.3. Cholesterol Synthesis
1.4. Cholesterol Transport
- Cholesterol derived from the remnants of CM, HDL, and LDL particles inhibits the synthesis of endogenous cholesterol through the concentration and activity of HMG-CoA;
- Excess cholesterol exceeding the current metabolic needs of the cell causes it to be esterified by acyl-CoA: cholesterol acyltransferase (ACAT). The activity of this enzyme increases with increasing cholesterol content inside the cell. The ACAT transfers a fatty acid residue from the acyl-CoA to the -OH group of cholesterol, forming a cholesterol ester that can be stored in the cell.
- High cholesterol content in the cell causes a decrease in the synthesis of the LDL receptor (LDL-R) by reducing the transcription of the corresponding gene. This helps to reduce the uptake of LDL-C into the cell.
1.5. Regulation of Cholesterol Synthesis
- (a)
- Expression of the sterol-dependent gene, such as sterol-regulatory element-binding protein (SREBP-2). When there is a low cholesterol level, the reductase gene is upregulated by a mechanism involving the transcription factor SREBP-2 protein [12]. The combination of SREBP-2 in response to the DNA’s sterol regulatory element (SRE) leads to an increase in enzyme levels and, thus, to enhanced cholesterol synthesis;
- (b)
- Degradation of the enzyme intensified by sterols (if the concentration of sterols in the SER is high—the reductase is transferred to the cytosol and undergoes ubiquitination and degradation in proteasomes);
- (c)
- Covalent modification catalyzed by AMP-activated protein kinase (AMPK), an enzyme sensitive to the AMP: ATP ratio, and protein phosphatase; a phosphorylated form of AMPK, converts HMGCR to its phosphorylated and inactive form, whereas HMGCR phosphatase reverses this process and produces a dephosphorylated and an active form of HMGCR;
- (d)
- HMGCR activity hormonal control via phosphorylation-dephosphorylation mechanisms —on the one hand, an increase in insulin concentration or in thyroid hormones promotes reductase dephosphorylation (activation). On the other hand, an increase in glucagon, epinephrine, or glucocorticosteroid concentration causes the opposite effect. Moreover, HMGCR can be ubiquitylated by the intermediates of mevalonate and cholesterol derivatives, such as lanosterol, 24,25-dihydro-lanosterol, and 25- and 27-hydroxycholesterol;
- (e)
- Drugs inhibiting selected pathways (statins) are structural HMG-CoA analogs; they are reversible and competitive inhibitors of HMGCR.
1.6. Cholesterol Degradation
1.7. Cholesterol and Atherosclerosis
1.8. Aim of the Study
2. Materials and Methods
2.1. Petri Nets and Stochastic Petri Nets
2.2. Knockout Analysis
3. Results and Discussion
3.1. Stochastic Petri Net-Based Model
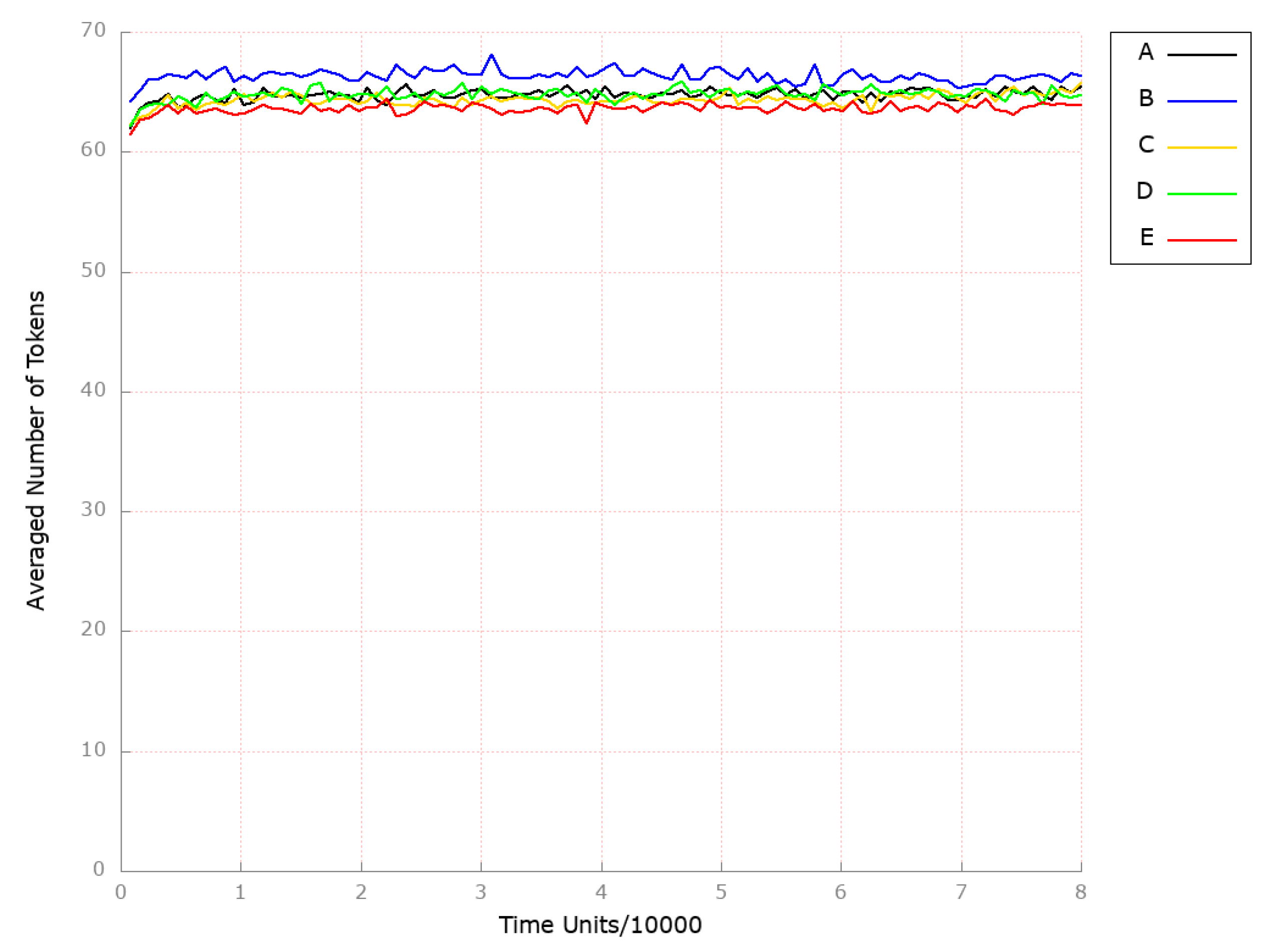
3.2. Stochastic Simulation and Knockout Based Analysis
3.3. Limitations of the Study
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Place | Biological Meaning | Place | Biological Meaning |
|---|---|---|---|
| LIPC (hepatic triacylglycerol lipase) | high unesterified cholesterol pool in the liver | ||
| ApoE | lanosterol | ||
| LDL-R-LDL complex | 2,3 oxidosqualene | ||
| low free cholesterol pool in intestine and the peripheral tissues | squalene | ||
| CaMKKbeta | cAMP-PKA activated | ||
| intermediate density lipoprotein (IDL) | phosphoprotein phosphatase with an increased activity | ||
| LDL-C as CE in endosome | free fatty acids (FFA) in intestinal lumen in micelles | ||
| high expression of LDL-R on cell membrane | hormone-sensitive lipase (HSL) | ||
| HMG-CoA reductase (HMGCR) active | hormone-sensitive lipase (HSL) phosphorylated | ||
| acetyl-CoA carboxylase (ACC) activated | phosphoprotein phosphatase inhibitor 1 (PPI-1) with an increase activity | ||
| serine-threonine kinase 1 (LKB1) | HDL-3 CE in blood | ||
| phosphoprotein phosphatase with a decrease activity | hydrolase of cholesterol esters | ||
| free fatty acids (FFA) | apical sodium bile acid transporter (ASBT) | ||
| LDL-C in serum | free fatty acids (FFA) in adipose tissues | ||
| lysosomal lipases | ACAT in the intestinum | ||
| acetyl-CoA | stored TAG | ||
| malonyl-CoA decarboxylase (MCD) | LCAT | ||
| AMP-activated protein kinase (AMPK-OH) inactive | ABCA1 cholesterol efflux regulatory protein CERP | ||
| AMP-activated protein kinase (AMPK) active | microsomal triglyceride transfer protein (MTTP) | ||
| lipoprotein lipase (LPL) | nascent chylomicrons (CM) with ApoB-48 | ||
| free cholesterol in endosome in intestine | HDL cholesterol non-CE | ||
| acetoacetyl CoA | Niemann–Pick C1-Like 1 (NPC1L1) | ||
| mevalonate | low cholesterol in diet | ||
| malonyl-CoA increase | nascent CM in the blood | ||
| HMGCR phosphatase with a decrease activity | TAG in enterocytes | ||
| nascent VLDL reach in TAG secreted from the liver into the blood | cholesterol from enterocytes and peripheral tissues transported to the blood | ||
| high free cholesterol pool in intestine | ApoC-II | ||
| thiolase | remnant CM receptors in the liver | ||
| HMG-CoA | bile acids | ||
| 2C protein phosphatase with an increase activity | mature CM with ApoB-48, ApoC-II and ApoE | ||
| HMGCR phosphorylated inactive | MAG in intestinal lumen in micelles | ||
| HDL-2 | biliary cholesterol | ||
| HMG-CoA synthase | free fatty acids (FFA) and monoglyceride (MAG) in enterocytes | ||
| isopentenyl PP | ACAT in the liver | ||
| cAMP-PKA low activate AMP-activated | ApoB-100 | ||
| CE transfer protein CETP in blood | remnant CM with ApoB-48 and ApoE | ||
| CoA | LDL receptor-related protein | ||
| farnesyl PP | nascent HDL | ||
| low cAMP | MTTP-ApoB-100 complex | ||
| HMGCR phosphatase | cholesterol stored as cholesteryl esters in the liver | ||
| LRP1 | TAG synthesized in the liver | ||
| enzymes in ER membranes | foamy cells | ||
| increased FA in the liver | macrophages | ||
| geranyl PP | small dense LDL | ||
| cAMP | SRB1 | ||
| inorganic pyrophosphate (PPI)1 OH |
| Transition | Biological Meaning | Transition | Biological Meaning |
|---|---|---|---|
| Hepatic lipase (LIPC) activation | high inorganic pyrophosphate (PPI) OH phosphorylation | ||
| endocytosis via RME | processes increasing inorganic pyrophosphate (PPI) 1 activity | ||
| binding LDL and LDL-R | conversion from CE found in HDL into free cholesterol pool | ||
| LDL-R expression on cell membrane | hydrolase of cholesterol esters activation | ||
| increasing activity by SREBP2 | remaining cholesterol removed by fecal sterols | ||
| LKB1 activation | reabsorption in the intestine and return to the liver | ||
| processes increasing intracellular calcium | apical sodium-dependent bile acid transporter (ASBT) activation | ||
| conversion into LDL | HSL activation | ||
| receptor being returned stimulated by lower pH | hydrolysation of stored TAG | ||
| acetyl CoA synthesis from glucose in the liver | free fatty acids (FFA) pool in adipose tissue increases | ||
| processes lowering free cholesterol pool in the intestine and the peripheral tissues | LCAT activation in serum | ||
| AMP-activated protein kinase AMPK phosphorylation | processes catalyzed by ACAT | ||
| processes decreasing phosphoprotein phosphatase | steroid synthesis | ||
| conversion VLDL into IDL, TAG hydrolysis | TAG storage in adipocytes | ||
| CE hydrolysis | ACAT activation in the intestine | ||
| lysosomal lipases activation | diet and hypertension | ||
| carboxylation catalysed by acetyl CoA carboxylase (ACC) | efflux of cholesterol to ApoA-1 and ApoE catalysed by ABCA1 | ||
| decarboxylation | HDL synthesis in the Liver | ||
| processes increasing AMP-activated protein kinase (AMPK-OH) inactive | nascent CM synthesis in enterocytes | ||
| pancreatic synthesis | HDL secreted by enterocytes and by the liver | ||
| free cholesterol effluxes endosome | cholesterol transport from the lumen to the intestinum | ||
| reaction catalysed by thiolase | NPC1L1 activtion | ||
| mevalonate synthesis | processes lowering cholesterol | ||
| malonyl CoA decarboxylase (MCD) activation | expression remnant CE receptors in the liver when the intestinal pool is high | ||
| dephosphorylation by protein phosphatase 2C | ApoC-2 returned to HDL cholesterol | ||
| protein phosphatase activation | exchanging HDL components in blood | ||
| HMGCR inactivation by phosphorylation | nascent CM exchange components with HDL | ||
| processes decreasing HMGCR phosphatase activity | transport mainly TAG within nascent chylomicrons from the intestine to the blood | ||
| CE transfer from LDL | conversion cholesterol into CE | ||
| thiolase activation | transport by ABCA1 | ||
| acetyl-CoAs conversion | LPL activation | ||
| reaction phosphorylation catalysed by ATP | FFA absorption in enterocyte | ||
| fatty acids (FA) synthesis in the liver | ABCA1 synthesis | ||
| dephosphorylation of ACC and its activation | cholesterol pool increases in the intestine because of biliary cholesterol | ||
| dephosphorylation | formation of the biliary cholesterol | ||
| CE transfer from HDL-2 | bile acids synthesis | ||
| HMG-CoA synthase activation in cytoplasm | reaction increasing cholesterol pool in the liver via RME | ||
| reaction forming farnesyl PP | TG distribution from CM | ||
| decreased PKA activation | binding with glycerol albumins | ||
| HMGCR activation | ACAT activation in the liver | ||
| CETP secretion from the liver | re-esterification of cholesterol by ACAT in the liver | ||
| beta oxidation | APOB100 synthesis in the liver and secreted into circulation | ||
| reaction condensation | forming complex | ||
| processes decreasing cAMP | CM endocytosis in the liver | ||
| PPI 1 OH activation | LDL-R synthesis | ||
| HMGCR phosphatase activation | efflux of free cholesterol from peripheral tissues | ||
| reaction catalyzed by HMG-CoA reductase phosphatase | conversion nascent HDL into HDL-3 | ||
| LRP1 synthesis | reaction forming nascent VLDL reach in TAG in the liver | ||
| reactions 19 leading to cholesterol synthesis in liver | TAG synthesis in the liver | ||
| enzymes activation | MTTP synthesis | ||
| reaction catalysed by squalene synthase | atherosclerosis | ||
| hormonal processes increasing cAMP | transport into peripheral tissue | ||
| activation by LRP1 | conversion HDL-3 into nascent LDL | ||
| reaction catalysed by squalene epoxidase | conversion HDL-3 into HDL-2 | ||
| reaction catalysed by squalene monooxygenase | influx of macrophages | ||
| internalized from blood by the liver | conversion HDL-2 into HDL-3 | ||
| increased PKA activation | oxidation | ||
| reaction catalysed by phosphoprotein phosphatase | binding with macrophage scavenger receptor type A (SRA) I and II | ||
| phosphoprotein phosphatase activation | degradation | ||
| conversion HDL into IDL | cholesterol CE transport to the liver | ||
| phosphorylation by PKA | SRB1 expression |
References
- Wang, Y.; Yutuc, E.; Griffiths, W.J. Cholesterol metabolism pathways—Are the intermediates more important than the products? FEBS J. 2021, 288, 3727–3745. [Google Scholar] [CrossRef] [PubMed]
- Bivona, G.; Gambino, C.M.; Lo Sasso, B.; Scazzone, C.; Giglio, R.V.; Agnello, L.; Ciaccio, M. Serum Vitamin D as a Biomarker in Autoimmune, Psychiatric and Neurodegenerative Diseases. Diagnostics 2022, 12, 130. [Google Scholar] [CrossRef]
- Afonso, M.S.; Machado, R.M.; Lavrador, M.S.; Quintao, E.C.R.; Moore, K.J.; Lottenberg, A.M. Molecular Pathways Underlying Cholesterol Homeostasis. Nutrients 2018, 10, 760. [Google Scholar] [CrossRef]
- Howe, V.; Sharpe, L.J.; Alexopoulos, S.J.; Kunze, S.V.; Chua, K.N.; Li, D.; Brown, A.J. Cholesterol homeostasis: How do cells sense sterol excess? Chem. Phys. Lipids 2016, 199, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Formanowicz, D.; Kozak, A.; Formanowicz, P. A Petri net based model of oxidative stress in atherosclerosis. Found. Comput. Decis. Sci. 2012, 37, 59–78. [Google Scholar] [CrossRef]
- Luo, J.; Yang, H.; Song, B. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 225–245. [Google Scholar] [CrossRef] [PubMed]
- Le Novere, N.; Hucka, M.; Mi, H.; Moodie, S.; Schreiber, F.; Sorokin, A.; Demir, E.; Wegner, K.; Aladjem, M.; Wimalaratne, S.; et al. The Systems Biology Graphical Notation. Nat. Biotechnol. 2009, 27, 735–741. [Google Scholar] [CrossRef]
- Caponio, G.R.; Wang, D.Q.H.; Di Ciaula, A.; De Angelis, M.; Portincasa, P. Regulation of Cholesterol Metabolism by Bioactive Components of Soy Proteins: Novel Translational Evidence. Int. J. Mol. Sci. 2021, 22, 227. [Google Scholar] [CrossRef]
- Desvergne, B.; Michalik, L.; Wahli, W. Transcriptional Regulation of Metabolism. Physiol. Rev. 2006, 86, 465–514. [Google Scholar] [CrossRef]
- Citrin, K.M.; Fernández-Hernando, C.; Suárez, Y. MicroRNA regulation of cholesterol metabolism. Ann. N. Y. Acad. Sci. 2021, 1495, 55–77. [Google Scholar] [CrossRef]
- Asmita, B.; Eviania, M.L.; Crystal, M.W.; Girish, C.S. Regulation of cholesterol biosynthesis and lipid metabolism: A microRNA management perspective. Steroids 2021, 173, 108878. [Google Scholar] [CrossRef]
- Jun, I.; Ryuichiro, S. New insights into the activation of sterol regulatory element-binding proteins by proteolytic processing. Biomol. Concepts 2013, 4, 417–423. [Google Scholar] [CrossRef]
- Woollard, K.J.; Lumsden, N.G.; Andrews, K.L.; Aprico, A.; Harris, E.; Irvine, J.C.; Jefferis, A.m.; Fang, L.; Kanellakis, P.; Bobik, A.; et al. Raised Soluble P-Selectin Moderately Accelerates Atherosclerotic Plaque Progression. PLoS ONE 2014, 9, e97422. [Google Scholar] [CrossRef] [PubMed]
- Márquez, A.B.; van der Vorst, E.P.C.; Maas, S.L. Key Chemokine Pathways in Atherosclerosis and Their Therapeutic Potential. J. Clin. Med. 2021, 10, 3825. [Google Scholar] [CrossRef] [PubMed]
- Harrington, J.R. The Role of MCP-1 in Atherosclerosis. Stem Cells 2000, 18, 65–66. [Google Scholar] [CrossRef]
- Jebari-Benslaiman, S.; Galicia-García, U.; Larrea-Sebal, A.; Olaetxea, J.R.; Alloza, I.; Vandenbroeck, K.; Benito-Vicente, A.; Martín, C. Pathophysiology of Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 3346. [Google Scholar] [CrossRef]
- Mudau, M.; Genis, A.; Lochner, A.; Strijdom, H. Endothelial dysfunction: The early predictor of atherosclerosis: Review article. Cardiovasc. J. Afr. 2012, 23, 222–231. [Google Scholar] [CrossRef]
- Rżosińska, K.; Formanowicz, D.; Formanowicz, P. The study of the influence of micro-environmental signals on macrophage differentiation using a quantitative Petri net based model. Arch. Control Sci. 2017, 27, 331–349. [Google Scholar] [CrossRef]
- Yao, Y.; Xu, X.H.; Jin, L. Macrophage Polarization in Physiological and Pathological Pregnancy. Front. Immunol. 2019, 10, 792. [Google Scholar] [CrossRef]
- Pokhrel, S.; Gudneppanavar, R.; Teegala, L.R.; Duah, E.; Thodeti, C.K.; Paruchuri, S. Leukotriene D4 Upregulates Oxidized Low-Density Lipoprotein Receptor 1 and CD36 to Enhance Oxidized LDL Uptake and Phagocytosis in Macrophages Through Cysteinyl Leukotriene Receptor 1. Front. Physiol. 2021, 12, 756450. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Nikiforov, N.G.; Markin, A.M.; Kashirskikh, D.A.; Myasoedova, V.A.; Gerasimova, E.V.; Orekhov, A.N. Overview of OxLDL and Its Impact on Cardiovascular Health: Focus on Atherosclerosis. Front. Pharmacol. 2021, 11, 613780. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, M. ABCA1 and ABCG1 as potential therapeutic targets for the prevention of atherosclerosis. J. Pharmacol. Sci. 2022, 148, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.H.; Zheng, X.L.; Tang, C.K. Chapter One—Nuclear Factor-κB Activation as a Pathological Mechanism of Lipid Metabolism and Atherosclerosis. In Advances in Clinical Chemistry; Elsevier: Amsterdam, The Netherlands, 2015; Volume 70, pp. 1–30. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Bobryshev, Y.V.; Orekhov, A.N. Macrophage-mediated cholesterol handling in atherosclerosis. J. Cell. Mol. Med. 2016, 20, 17–28. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Scott, M.J. Caspase-1 as a multifunctional inflammatory mediator: Noncytokine maturation roles. J. Leukoc. Biol. 2016, 100, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Mundi, S.; Massaro, M.; Scoditti, E.; Carluccio, M.A.; van Hinsbergh, V.W.M.; Iruela-Arispe, M.L.; De Caterina, R. Endothelial permeability, LDL deposition, and cardiovascular risk factors—A review. Cardiovasc. Res. 2017, 114, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Yin, C.; Luo, S.; Habenicht, A.J.R.; Mohanta, S.K. Vascular Smooth Muscle Cells Contribute to Atherosclerosis Immunity. Front. Immunol. 2019, 10, 1101. [Google Scholar] [CrossRef]
- Churov, A.; Summerhill, V.; Grechko, A.; Orekhova, V.; Orekhov, A. MicroRNAs as Potential Biomarkers in Atherosclerosis. Int. J. Mol. Sci. 2019, 20, 5547. [Google Scholar] [CrossRef]
- Yao, L.; Tanuja, T.; Wenduo, G.; Jingjing, C.; Qingbo, X. Impact of miRNA in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, e159–e170. [Google Scholar] [CrossRef]
- Mireille, O.; Elizabeth, J.H.; Coen, V.S.; Graeme, J.K.; Maryem, A.H.; Bhama, R.; Rayner, K.J.; Ryan, E.T.; Ljubica, P.; Ulf, H.; et al. miRNA Targeting of Oxysterol-Binding Protein-Like 6 Regulates Cholesterol Trafficking and Efflux. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 942–951. [Google Scholar] [CrossRef]
- Bargieł, W.; Cierpiszewska, K.; Maruszczak, K.; Pakuła, A.; Szwankowska, D.; Wrzesińska, A.; Gutowski, Ł.; Formanowicz, D. Recognized and Potentially New Biomarkers—Their Role in Diagnosis and Prognosis of Cardiovascular Disease. Medicina 2021, 57, 701. [Google Scholar] [CrossRef] [PubMed]
- Domanski, M.J.; Tian, X.; Wu, C.O.; Reis, J.P.; Dey, A.K.; Gu, Y.; Zhao, L.; Bae, S.; Liu, K.; Hasan, A.A.; et al. Time Course of LDL Cholesterol Exposure and Cardiovascular Disease Event Risk. J. Am. Coll. Cardiol. 2020, 76, 1507–1516. [Google Scholar] [CrossRef]
- David, R.; Alla, H. Discrete, Continuous and Hybrid Petri Nets; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Koch, I.; Reisig, W.; Schreiber, F. (Eds.) Modeling in Systems Biology. The Petri Net Approach; Springer: London, UK, 2011. [Google Scholar]
- Murata, T. Petri nets: Properties, analysis and applications. Proc. IEEE 1989, 77, 541–580. [Google Scholar] [CrossRef]
- Formanowicz, D.; Kozak, A.; Głowacki, T.; Radom, M.; Formanowicz, P. Hemojuvelin–hepcidin axis modeled and analyzed using Petri nets. J. Biomed. Inform. 2013, 46, 1030–1043. [Google Scholar] [CrossRef] [PubMed]
- Sackmann, A.; Heiner, M.; Koch, I. Application of Petri net based analysis techniques to signal transduction pathway. BMC Bioinform. 2006, 7, 482. [Google Scholar] [CrossRef] [PubMed]
- Marsan, M. Stochastic Petri nets: An elementary introduction. Lect. Notes Comput. Sci. 1989, 424, 1–29. [Google Scholar]
- Bause, F.; Kritzinger, P. Stochastic Petri Nets—An Introduction to the Theory; Vieveg: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Formanowicz, D.; Rybarczyk, A.; Formanowicz, P. Factors influencing essential hypertension and cardiovascular disease modeled and analyzed using stochastic Petri nets. Fundam. Informaticae 2018, 160, 143–165. [Google Scholar] [CrossRef]
- Blazewicz, J.; Formanowicz, D.; Formanowicz, P.; Sackmann, A.; Sajkowski, M. Modeling the process of human body iron homeostasis using a variant of timed Petri nets. Discret. Appl. Math. 2009, 157, 2221–2231. [Google Scholar] [CrossRef]
- Gillespie, D. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 1977, 81, 2340–2361. [Google Scholar] [CrossRef]
- Heiner, M.; Herajy, M.; Liu, F.; Rohr, C.; Schwarick, M. Snoopy—A unifying Petri net tool. Lect. Notes Comput. Sci. 2012, 7347, 398–407. [Google Scholar]
- Formanowicz, D.; Rybarczyk, A.; Radom, M.; Formanowicz, P. A role of inflammation and immunity in essential hypertension—Modeled and analyzed using Petri nets. Int. J. Mol. Sci. 2020, 21, 3348. [Google Scholar] [CrossRef] [PubMed]
- Grunwald, S.; Speer, A.; Ackermann, J.; Koch, I. Petri net modelling of gene regulation of the Duchenne muscular dystrophy. Biosystems 2008, 92, 189–205. [Google Scholar] [CrossRef] [PubMed]
- Formanowicz, D.; Radom, M.; Rybarczyk, A.; Tanas, K.; Formanowicz, P. Control of Cholesterol Metabolism Using a Systems Approach. Biology 2022, 11, 430. [Google Scholar] [CrossRef] [PubMed]
- Atluri, P.; Karakousis, G.; Porrett, P.; Kaiser, L. The Surgical Review: An Integrated Basic and Clinical Science Study Guide, 2nd ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2006. [Google Scholar]
- Scheidel, J.; Lindauer, K.; Ackermann, J.; Koch, I. Quasi-Steady-State Analysis based on Structural Modules and Timed Petri Net Predict System’s Dynamics: The Life Cycle of the Insulin Receptor. Metabolites 2015, 5, 766–793. [Google Scholar] [CrossRef]
- Formanowicz, D.; Rybarczyk, A.; Radom, M.; Tanas, K.; Formanowicz, P. A Stochastic Petri Net-Based Model of the Involvement of Interleukin 18 in Atherosclerosis. Int. J. Mol. Sci. 2020, 21, 8574. [Google Scholar] [CrossRef]
- Paul, G.; Marchelletta, R.; McCole, D.; Barrett, K. Interferon-γ alters downstream signaling originating from epidermal growth factor receptor in intestinal epithelial cells: Functional consequences for ion transport. J. Biol. Chem. 2012, 287, 2144–2155. [Google Scholar] [CrossRef]
- Watson, R. DHEA in Human Health and Aging; CRC Press: Boca Raton, FL, USA, 2011. [Google Scholar] [CrossRef]
- Janeway, C.; Travers, J.; Walport, M.; Shlomchik, M. Immunobiology: The Immune System in Health and Disease, 5th ed.; Garland Science: New York, NY, USA, 2001. [Google Scholar]
- Palsson, S.; Hickling, T.; Bradshaw-Pierce, E.; Zager, M.; Jooss, K.; Brien, P.; Spilker, M.; Palsson, B.; Vicini, P. The development of a fully-integrated immune response model (FIRM) simulator of the immune response through integration of multiple subset models. BMC Syst. Biol. 2013, 7, 95. [Google Scholar] [CrossRef]
- MacEwan, D. TNF ligands and receptors—A matter of life and death. Br. J. Pharmacol. 2002, 135, 855–875. [Google Scholar] [CrossRef]
- Sungjin, P.; Krist, D.; Statsyuk, A. Protein ubiquitination and formation of polyubiquitin chains without ATP, E1 and E2 enzymes. Chem. Sci. 2015, 6, 1770–1779. [Google Scholar] [CrossRef]
- Takahashi, K.; Takeya, M.; Sakashita, N. Multifunctional roles of macrophages in the development and progression of atherosclerosis in humans and experimental animals. Med. Electron Microsc. 2002, 35, 179–203. [Google Scholar] [CrossRef]
- Martin-Valmaseda, E.; Sanchez-Yague, J.; Rodriguez, M.; Gomez, F.; Llanillo, M. Comparison between in vitro lipid peroxidation in fresh sheep platelets and peroxidative processes during sheep platelet ageing under storage at 4 °C. J. Leukoc. Biol. 2010, 87, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Yang, G.; Li, G. Inflammatory mechanisms in ischemic stroke: Role of inflammatory cells. J. Leukoc. Biol. 2010, 87, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Heiner, M.; Lehrack, S.; Gilbert, D.; Marwan, W. Extended Stochastic Petri Nets for Model-Based Design of Wetlab Experiments. Trans. Comput. Syst. Biol. XI 2009, 5750, 138–163. [Google Scholar]
- Memon, S.; Ganga, H.; Masrur, S.; Thompson, P. The Effect of HMG CoA Reductase Inhibitors on the Progression of Aortic Sclerosis: Review Article. Conn. Med. 2016, 80, 169–174. [Google Scholar]
- Bjorkegren, J.; Lusis, A. Atherosclerosis: Recent developments. Cell 2022, 185, 1630–1645. [Google Scholar] [CrossRef]
- Formanowicz, D.; Krawczyk, J. Controlling the thickness of the atherosclerotic plaque by statin medication. PLoS ONE 2020, 10, e0239953. [Google Scholar] [CrossRef]
- Wang, F.; Wang, X.; Ye, P.; Cao, R.; Zhang, Y.; Qi, Y.; Zhao, D. High-density lipoprotein 3 cholesterol is a predictive factor for arterial stiffness: A community-based 4.8-year prospective study. Lipids Health Dis. 2018, 17, 5. [Google Scholar] [CrossRef]
- Schulze, P.; Lee, R. Oxidative stress and atherosclerosis. Curr. Atheroscler. Rep. 2005, 7, 242–248. [Google Scholar] [CrossRef]
- Jiang, F.; Qian, J.; Chen, S.; Zhang, W.; Liu, C. Ligustrazine improves atherosclerosis in rat via attenuation of oxidative stress. Pharm. Biol. 2011, 49, 856–863. [Google Scholar] [CrossRef]
- Kozak, A.; Formanowicz, D.; Formanowicz, P. Structural analysis of a Petri net model of oxidative stress in atherosclerosis. IET Syst. Biol. 2018, 12, 108–117. [Google Scholar] [CrossRef]
- Kim, T. Protein phosphatase inhibitor-1 (PPI-1) has protective activities in stress conditions in E. coli. Int. J. Biol. Macromol. 2006, 38, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Eto, M. Regulation of cellular protein phosphatase-1 (PP1) by phosphorylation of the CPI-17 family, C-kinase-activated PP1 inhibitors. J. Biol. Chem. 2009, 284, 35273–35277. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Bakillah, A. New approaches to target microsomal triglyceride transfer protein. Curr. Opin. Lipidol. 2008, 19, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Hewing, B.; Parathath, S.; Mai, C.; Fiel, M.; Guo, L.; Fisher, E. Rapid regression of atherosclerosis with MTP inhibitor treatment. Atherosclerosis 2013, 227, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Parks, J. ATP-binding cassette transporter AI and its role in HDL formation. Curr. Opin. Lipidol. 2005, 16, 19–25. [Google Scholar] [CrossRef]
- Rust, S.; Rosier, M.; Funke, H.; Real, J.; Amoura, Z.; Piette, J.; Deleuze, J.; Brewer, H.; Duverger, N.; Denefle, P.; et al. Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat. Genet. 1999, 22, 352–355. [Google Scholar] [CrossRef]
- Münch, G.; Bültmann, A.; Li, Z.; Holthoff, H.; Ullrich, J.; Wagner, S.; Ungerer, M. Overexpression of ABCG1 protein attenuates arteriosclerosis and endothelial dysfunction in atherosclerotic rabbits. Heart Int. 2012, 7, e12. [Google Scholar] [CrossRef]
- Yang, N.; Dong, Y.Q.; Jia, G.X.; Fan, S.M.; Li, S.Z.; Yang, S.S.; Li, Y.B. ASBT(SLC10A2): A promising target for treatment of diseases and drug discovery. Biomed. Pharmacother. 2020, 132, 110835. [Google Scholar] [CrossRef]
- Li, M.; Wang, Q.; Li, Y.; Cao, S.; Zhang, Y.; Wang, Z.; Liu, G.; Li, J.; Gu, B. Apical sodium-dependent bile acid transporter, drug target for bile acid related diseases and delivery target for prodrugs: Current and future challenges. Pharmacol. Ther. 2020, 212, 107539. [Google Scholar] [CrossRef]
- Bhat, B.; Rapp, S.; Beaudry, J.; Napawan, N.; Butteiger, D.; Hall, K.; Null, C.; Luo, Y.; Keller, B. Inhibition of ileal bile acid transport and reduced atherosclerosis in apoE-/- mice by SC-435. J. Lipid Res. 2003, 44, 1614–1621. [Google Scholar] [CrossRef]
- van de Peppel, I.; Bertolini, A.; van Dijk, T.; Groen, A.; Jonker, J.; Verkade, H. Efficient reabsorption of transintestinally excreted cholesterol is a strong determinant for cholesterol disposal in mice. J. Lipid Res. 2019, 60, 1562–1572. [Google Scholar] [CrossRef] [PubMed]
- Tiessen, R.; Kennedy, C.; Keller, B.; Levin, N.; Acevedo, L.; Gedulin, B.; van Vliet, A.; Dorenbaum, A.; Palmer, M. Safety, tolerability and pharmacodynamics of apical sodium-dependent bile acid transporter inhibition with volixibat in healthy adults and patients with type 2 diabetes mellitus: A randomised placebo-controlled trial. BMC Gastroenterol. 2018, 18, 3. [Google Scholar] [CrossRef] [PubMed]
















| Place | Initial Number of Tokens | Place | Initial Number of Tokens | Place | Initial Number of Tokens |
|---|---|---|---|---|---|
| 100 | 10 | 100 | |||
| 100 | 0 | 10 | |||
| 0 | 0 | 100 | |||
| 0 | 0 | 0 | |||
| 10 | 0 | 0 | |||
| 10 | 10 | 10 | |||
| 0 | 0 | 0 | |||
| 10 | 0 | 0 | |||
| 0 | 100 | 0 | |||
| 0 | 100 | 0 | |||
| 10 | 10 | 100 | |||
| 0 | 0 | 0 | |||
| 10 | 0 | 10 | |||
| 0 | 100 | 0 | |||
| 10 | 10 | 10 | |||
| 0 | 0 | 0 | |||
| 100 | 0 | 10 | |||
| 0 | 0 | 100 | |||
| 0 | 0 | 100 | |||
| 100 | 0 | 0 | |||
| 10 | 10 | 10 | |||
| 10 | 10 | 0 | |||
| 0 | 10 | 0 | |||
| 0 | 0 | 0 | |||
| 0 | 0 | 0 | |||
| 0 | 10 | 0 | |||
| 10 | 10 | 0 | |||
| 100 | 10 | 10 | |||
| 0 | 0 | 100 | |||
| 0 | 100 | ||||
| 0 | 0 |
| Process | Duration | Time Data Derived from the Literature Sources | Time Interval |
|---|---|---|---|
| binding of ligands and proteins, the influence of molecules, activation/inactivation of enzymes, proteins and processes | 1 | seconds [53,55] | Very, very Short |
| phosphorylation, modulation of the activation of proteins and processes, secretion of molecules, cholesterol efflux | 10 | seconds to a minute [48,56] | Very short |
| Binding for larger complexes (ligands and receptors), larger complexes formation, carboxylation, decarboxylation | 20 | up to a minute [51,52] | Short |
| reduction in the availability of molecules, dissociation of protein complexes, conversion, hydrolysis, degradation | 40 | approximately half as long as the synthesis and expression [52,57] | Medium |
| biomolecule synthesis, formation of molecules, expression, increase in molecules availability/production, transport of molecules, triglycerides storage in adipocytes | 80 | minutes–hours [48,52,58] | Long |
| atherosclerosis | 500 | days [53,54,57,59] | Very Long |
| Transition | Kinetic Parameter | Rate Function | Transition | Kinetic Parameter | Rate Function |
|---|---|---|---|---|---|
| 1.0 | (1.0) | 0.1 | |||
| 0.1 | 0.1 | ||||
| 0.1 | 0.025 | ||||
| 0.0125 | 1.0 | ||||
| 0.1 | 0.025 | ||||
| 1.0 | 0.0125 | ||||
| 0.1 | 1.0 | ||||
| 0.05 | 1.0 | ||||
| 0.1 | 0.1 | ||||
| 1.0 | 1.0 | ||||
| 1.0 | 1.0 | ||||
| 1.0 | 0.025 | ||||
| 0.1 | 0.0125 | ||||
| 0.05 | 0.0125 | ||||
| 0.025 | 1.0 | ||||
| 1.0 | 0.05 | ||||
| 0.05 | 0.1 | ||||
| 0.05 | 1.0 | ||||
| 0.1 | 0.05 | ||||
| 0.1 | 0.1 | ||||
| 0.1 | 0.0125 | ||||
| 0.1 | 1.0 | ||||
| 0.05 | 0.1 | ||||
| 1.0 | 0.1 | ||||
| 0.1 | 0.1 | ||||
| 1.0 | 0.1 | ||||
| 1.0 | 0.1 | ||||
| 0.1 | 0.05 | ||||
| 0.0125 | 0.1 | ||||
| 1.0 | 0.05 | ||||
| 0.1 | 1.0 | ||||
| 1.0 | 0.0125 | ||||
| 0.05 | 0.05 | ||||
| 1.0 | 0.1 | ||||
| 0.1 | 0.1 | ||||
| 0.025 | 0.05 | ||||
| 1.0 | 0.05 | ||||
| 0.1 | 0.1 | ||||
| 1.0 | 1.0 | ||||
| 1.0 | 1.0 | ||||
| 0.1 | 0.05 | ||||
| 0.1 | 0.1 | ||||
| 0.1 | 0.05 | ||||
| 0.1 | 0.1 | ||||
| 0.1 | 0.05 | ||||
| 1.0 | 0.1 | ||||
| 0.1 | 0.025 | ||||
| 0.05 | 0.0125 | ||||
| 0.1 | 0.05 | ||||
| 1.0 | 0.1 | ||||
| 0.1 | 0.002 | ||||
| 0.025 | 0.0125 | ||||
| 1.0 | 0.025 | ||||
| 0.1 | 0.025 | ||||
| 0.1 | 0.1 | ||||
| 0.0125 | 0.025 | ||||
| 1.0 | 0.1 | ||||
| 0.1 | 0.05 | ||||
| 1.0 | 0.025 | ||||
| 0.05 | 0.0125 | ||||
| 0.1 | 0.0125 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rybarczyk, A.; Formanowicz, D.; Radom, M.; Formanowicz, P. Cholesterol Metabolism Pathways Disturbances in Atherosclerosis—Analyses Using Stochastic Petri Net-Based Model. Appl. Sci. 2023, 13, 6149. https://doi.org/10.3390/app13106149
Rybarczyk A, Formanowicz D, Radom M, Formanowicz P. Cholesterol Metabolism Pathways Disturbances in Atherosclerosis—Analyses Using Stochastic Petri Net-Based Model. Applied Sciences. 2023; 13(10):6149. https://doi.org/10.3390/app13106149
Chicago/Turabian StyleRybarczyk, Agnieszka, Dorota Formanowicz, Marcin Radom, and Piotr Formanowicz. 2023. "Cholesterol Metabolism Pathways Disturbances in Atherosclerosis—Analyses Using Stochastic Petri Net-Based Model" Applied Sciences 13, no. 10: 6149. https://doi.org/10.3390/app13106149
APA StyleRybarczyk, A., Formanowicz, D., Radom, M., & Formanowicz, P. (2023). Cholesterol Metabolism Pathways Disturbances in Atherosclerosis—Analyses Using Stochastic Petri Net-Based Model. Applied Sciences, 13(10), 6149. https://doi.org/10.3390/app13106149