
Effect of Reverse Micelles Size on the Electron Transfer Reaction within the Ion Pair of Co (III)/Fe (II) Complexes




Abstract
:1. Introduction
2. Experimental Section
2.1. Reagents
2.2. Synthesis of Metal Complexes
2.2.1. Synthesis of [Co(NH3)5CO3]NO3
2.2.2. Synthesis of [Co(NH3)5Cl]Cl2
2.2.3. Synthesis of [Co(NH3)5trf]trf2
2.3. Preparation of Standard Solutions
2.3.1. Preparation of Standard Solution of [Co(NH3)5OH2]3+
2.3.2. Preparation of Standard Solution of [Fe(CN)6]4−
2.4. Kinetic Studies
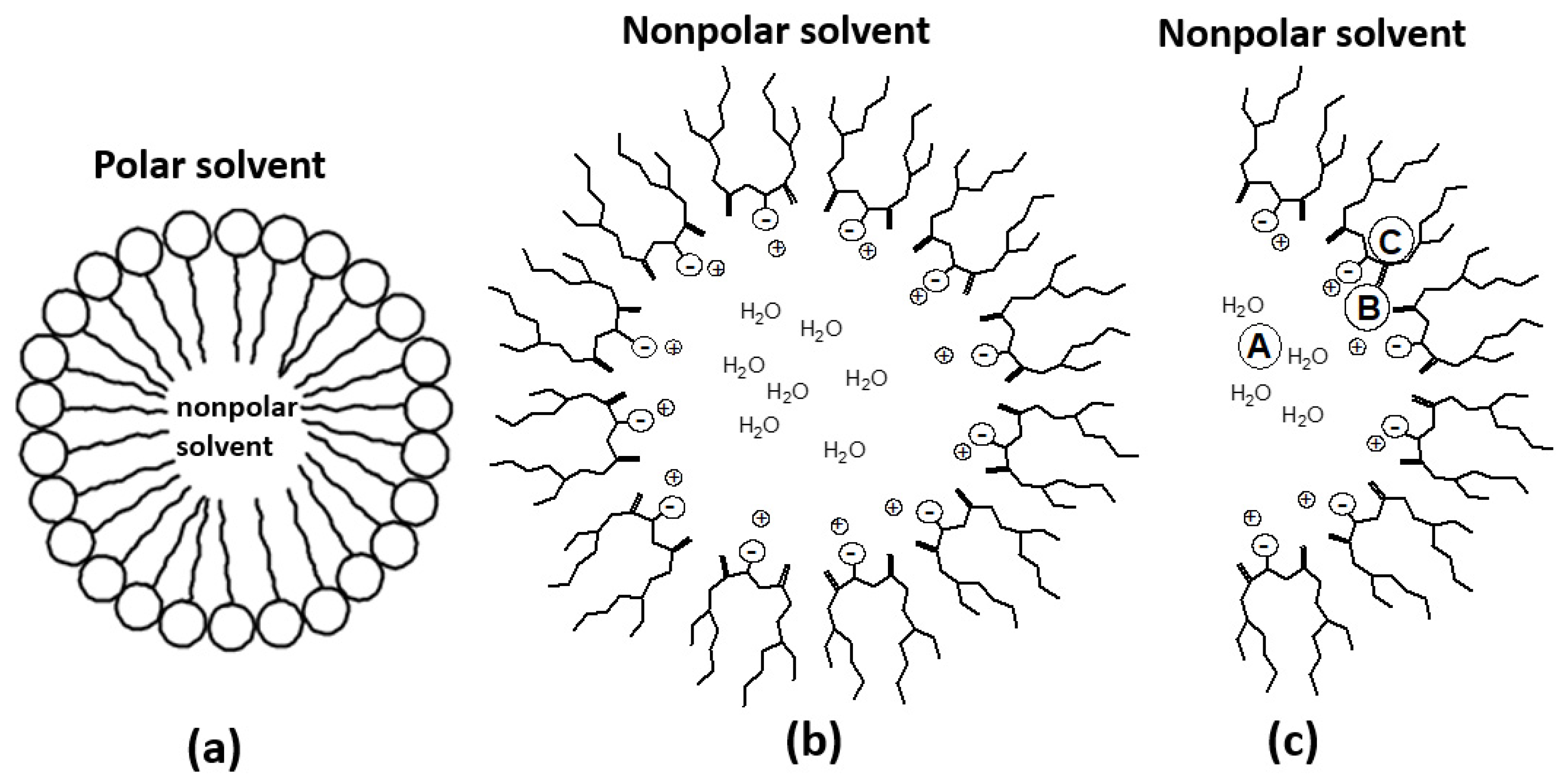
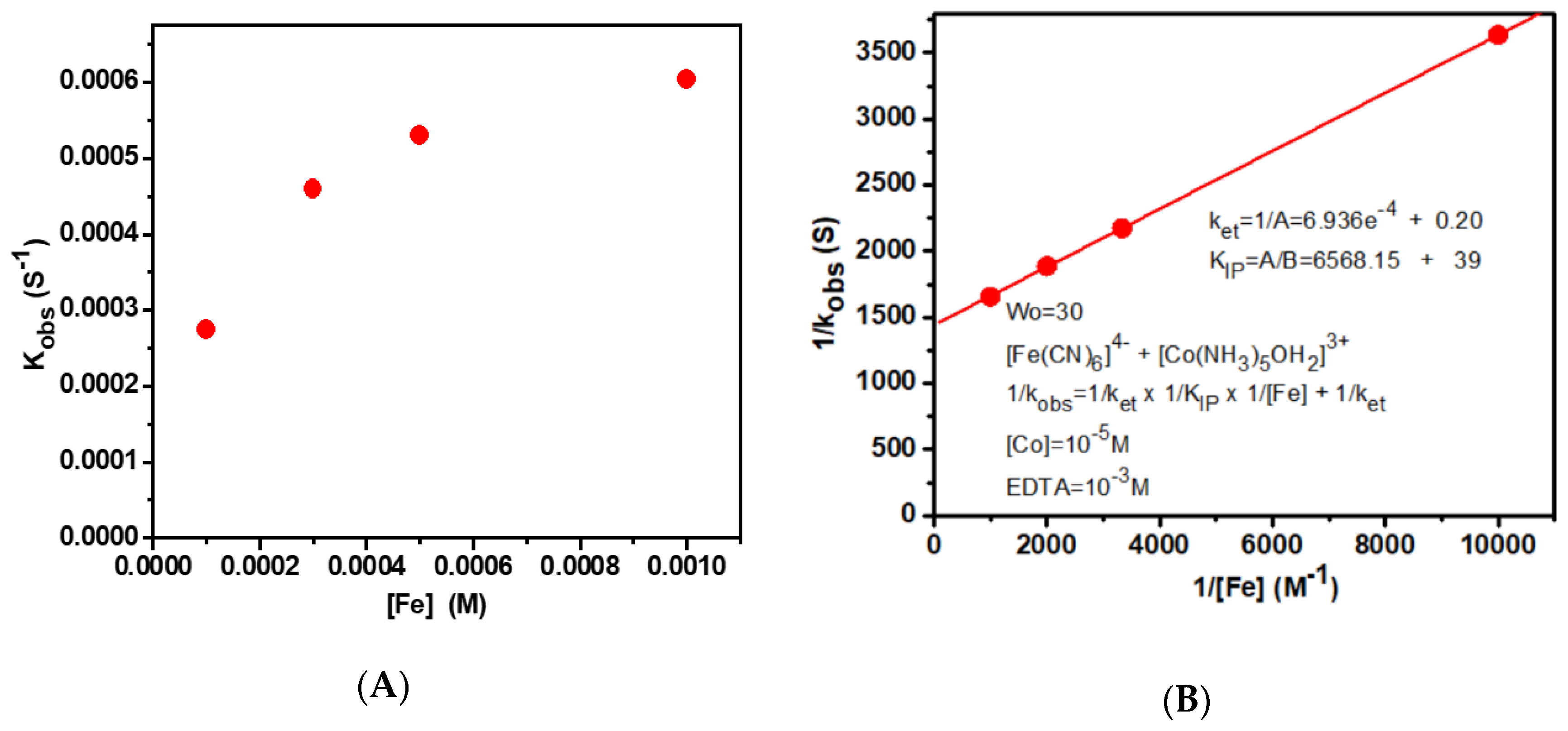
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tonova, K.; Lazarova, Z. Reversed micelle solvents as tools of enzyme purification and enzyme-catalyzed conversion. Biotechnol. Adv. 2008, 26, 516–532. [Google Scholar] [CrossRef]
- Jones, M.C.; Gao, H.; Leroux, J.C. Reverse polymeric micelles for pharmaceutical applications. J. Control. Release 2008, 132, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Dong, X.; Sun, Y. New Development of Reverse Micelles and Applications in Protein Separation and Refolding. Chin. J. Chem. Eng. 2008, 16, 949–955. [Google Scholar] [CrossRef]
- Eastoe, J.; Hollamby, M.J.; Hudson, L. Recent advances in nanoparticle synthesis with reversed micelles. Adv. Colloid Interface Sci. 2006, 128–130, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.L.; Gunderson, W.A.; Weitz, A.C.; Hendrich, M.P.; Ryabov, A.D.; Collins, T.J. Activation of Dioxygen by a TAML Activator in Reverse Micelles: Characterization of an FeIIIFeIV Dimer and Associated Catalytic Chemistry. J. Am. Chem. Soc. 2015, 137, 9704–9715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silber, J.J.; Biasutti, A.; Abuin, E.; Lissi, E. Interactions of small molecules with reverse micelles. Adv. Colloid Interface Sci. 1999, 82, 189–252. [Google Scholar] [CrossRef]
- Volkova, O.I.; Kuleshova, A.A.; Saletsky, A.M. The Effect of Nanoscale Reverse Micelles on the Photophysical Properties of Fluorescein Molecules. Mosc. Univ. Phys. Bull. 2020, 75, 605–610. [Google Scholar] [CrossRef]
- Moulik, S.P.; Paul, B.K. Structure, dynamics and transport properties of microemulsions. Adv. Colloid Interface Sci. 1998, 78, 99–195. [Google Scholar] [CrossRef]
- Maitra, A. Determination of size parameters of water-Aerosol OT-oil reverse micelles from their nuclear magnetic resonance data. J. Phys. Chem. 1984, 88, 5122–5125. [Google Scholar] [CrossRef]
- Pang, Y.; Deàk, J.C.; Huang, W.; Lagutchev, A.; Pakoulev, A.; Patterson, J.E.; Sechler, T.D.; Wang, Z.; Dlott, D.D. Vibrational energy in molecules probed with high time and space resolution. Intl. Rev. Phys. Chem. 2007, 26, 223–248. [Google Scholar] [CrossRef]
- Gebicki, J.L.; Gebicka, L. Radical reactions in reverse micelles studied by pulse radiolysis. Radiat. Phys. Chem. 2020, 177, 109105. [Google Scholar] [CrossRef]
- Jordan, R.B. Reaction Mechanisms of Inorganic and Organometallic Systems; Oxford University Press: Oxford, UK, 2007. [Google Scholar]
- Atkins, P.; de Paula, J. Physical Chemistry, 7th ed.; Oxford University Press: Oxford, UK, 2002; pp. 1015–1018. [Google Scholar]
- Gaus, P.L.; Villanueva, J.L. Direct determinations of the ion pairing equilibrium constant and first-order rate constant for outer sphere electron transfer reactions. J. Am. Chem. Soc. 1980, 102, 1934–1938. [Google Scholar] [CrossRef]
- Bjerrum, N.; Danske, V.S. Mat. Fys. Medd.; Selected Papers; Einar Munksgaard: Copenhagen, Denmark, 1949. [Google Scholar]
- Schellman, J.A. The Application of the Bjerrum Ion Association Theory to the Binding of Anions by Proteins. J. Phys. Chem. 1953, 57, 472–475. [Google Scholar] [CrossRef]
- Nielson, R.M.; Wherland, S. Electron self-exchange by hexakis (cyclohexyl isocyanide) manganese (I/II): Solvent, electrolyte and temperature dependences. Inorg. Chem. 1984, 23, 1338–1344. [Google Scholar] [CrossRef]
- Jassal, V.; Shanker, U.; Shankar, S. Synthesis, Characterization and Applications of Nano-structured Metal Hexacyanoferrates: A Review. J. Environ. Anal. Chem. 2015, 2, 1–14. [Google Scholar] [CrossRef]
- Schlessinger, G.G.; Britton, D.; Rhodes, T.; Ng, E. Chloropentaamminecobalt(III) Chloride. Inorg. Synth. 1967, 9, 160–163. [Google Scholar]
- Van Eldik, R.; Kelm, H. Concerning the pressure dependence of outer-sphere electron-transfer reactions: The reduction of Co(NH3)5OH3+2 by Fe(CN)4−6 in aqueous acidic solution. Inorg. Chim. Acta 1983, 73, 91–94. [Google Scholar] [CrossRef]
- Bera, A.K.; Pal, B.; Sen Gupta, K.K. Outer-sphere reduction of hexacyanoferrate(III) by enolizable and nonenolizable aldehydes in alkaline medium. Int. J. Chem. Kinet. 2012, 44, 494–505. [Google Scholar] [CrossRef]
- Rajchel-Mieldzioć, P.; Tymkiewicz, R.; Sołek, J.; Secomski, W.; Litniewski, J.; Fita, P. Reaction kinetics of sonochemical oxidation of potassium hexacyanoferrate(II) in aqueous solutions. Ultrason. Sonochem. 2020, 63, 104912. [Google Scholar] [CrossRef]
- Marcus, R.A. Transfer reactions in chemistry. Theory and experiment. Pure Appl. Chem. 1997, 69, 13–30. [Google Scholar] [CrossRef]
- Pelizzetti, E.; Mentasti, E.; Pramauro, E. Outer-sphere oxidation of ascorbic acid. Inorg. Chem. 1978, 17, 1181–1186. [Google Scholar] [CrossRef]
- Belletete, M.; Lachapelle, M.; Durocher, G. Polarity of AOT micellar interfaces: Use of the preferential solvation concepts in the evaluation of the effective dielectric constants. J. Phys. Chem. 1990, 94, 5337–5341. [Google Scholar] [CrossRef]
- Chamorovsky, S.K.; Cherepanov, D.A.; Chamorovsky, C.S.; Semenov, A.Y. Correlation of electron transfer rate in photosynthetic reaction centers with intraprotein dielectric properties. Biochim. Biophys. Acta 2007, 1767, 441–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Entry | Wo | ket (s−1) | KIP |
|---|---|---|---|
| 1. | aqueous | 6.88 ± 0.20 | 100 |
| 2. | 30 | [6.94 ± 0.20] × 10−4 | 6550 |
| 3. | 20 | [1.06 ± 0.41] × 10−3 | 6450 |
| 4. | 15 | [1.18 ± 0.35] × 10−3 | 6400 |
| 5. | 10 | [2.43 ± 0.41] × 10−3 | 4500 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hona, R.K.; Thapa, R.; Dhaliwal, G.S. Effect of Reverse Micelles Size on the Electron Transfer Reaction within the Ion Pair of Co (III)/Fe (II) Complexes. Appl. Sci. 2022, 12, 4552. https://doi.org/10.3390/app12094552
Hona RK, Thapa R, Dhaliwal GS. Effect of Reverse Micelles Size on the Electron Transfer Reaction within the Ion Pair of Co (III)/Fe (II) Complexes. Applied Sciences. 2022; 12(9):4552. https://doi.org/10.3390/app12094552
Chicago/Turabian StyleHona, Ram Krishna, Rajesh Thapa, and Gurjot S. Dhaliwal. 2022. "Effect of Reverse Micelles Size on the Electron Transfer Reaction within the Ion Pair of Co (III)/Fe (II) Complexes" Applied Sciences 12, no. 9: 4552. https://doi.org/10.3390/app12094552
APA StyleHona, R. K., Thapa, R., & Dhaliwal, G. S. (2022). Effect of Reverse Micelles Size on the Electron Transfer Reaction within the Ion Pair of Co (III)/Fe (II) Complexes. Applied Sciences, 12(9), 4552. https://doi.org/10.3390/app12094552