
ErbB4 Is a Potential Key Regulator of the Pathways Activated by NTRK-Fusions in Thyroid Cancer

 ,
,  ,
,
Abstract
:Featured Application
Abstract
1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Data Analysis
2.2.1. Identification of Differentially Expressed Genes (DEGs)
2.2.2. Gene Set Enrichment Analysis
2.2.3. Master Regulator Analysis and Upstream Analysis
2.3. Experimental Validation
3. Results
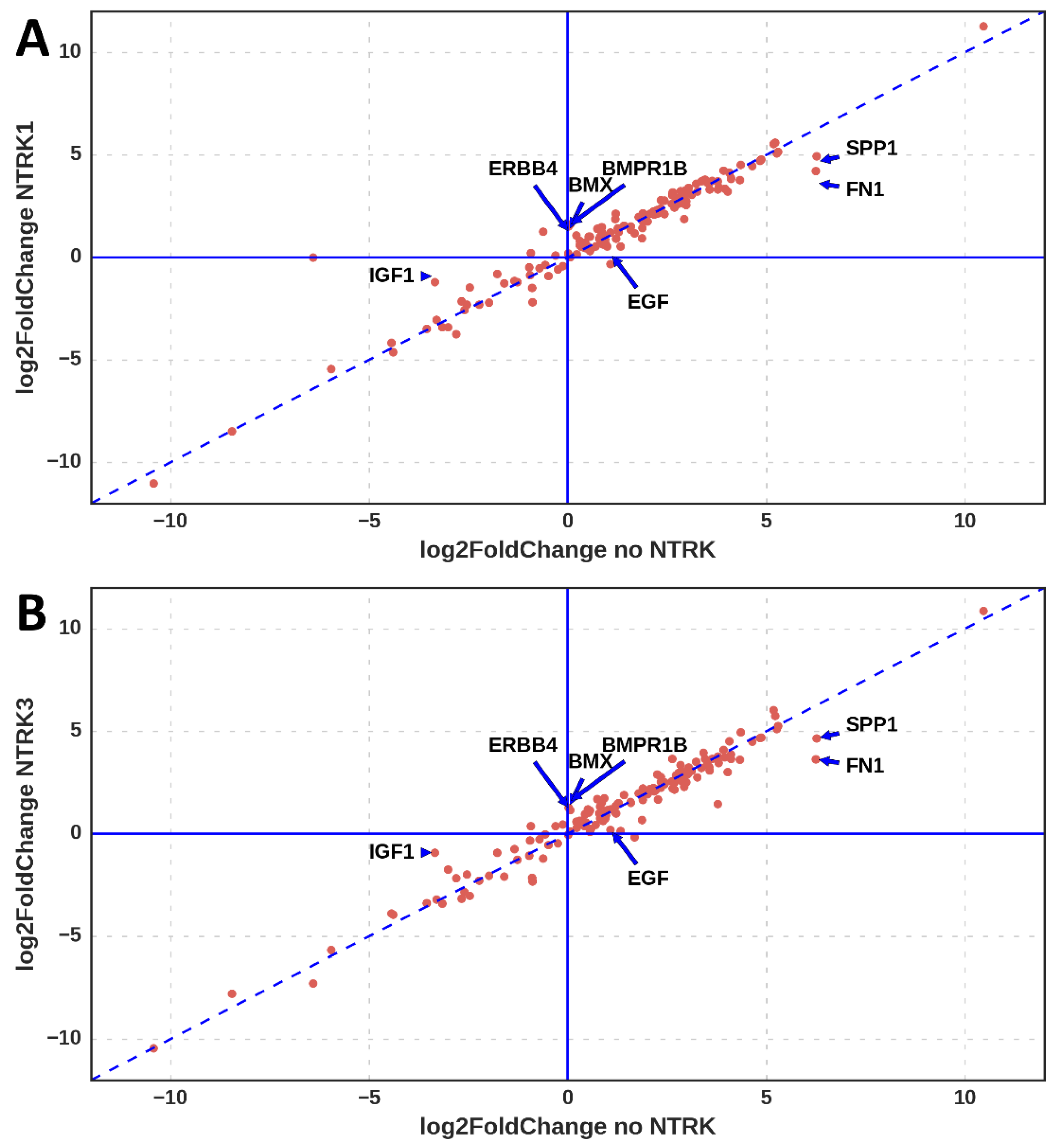
3.1. Differentially Expressed Genes
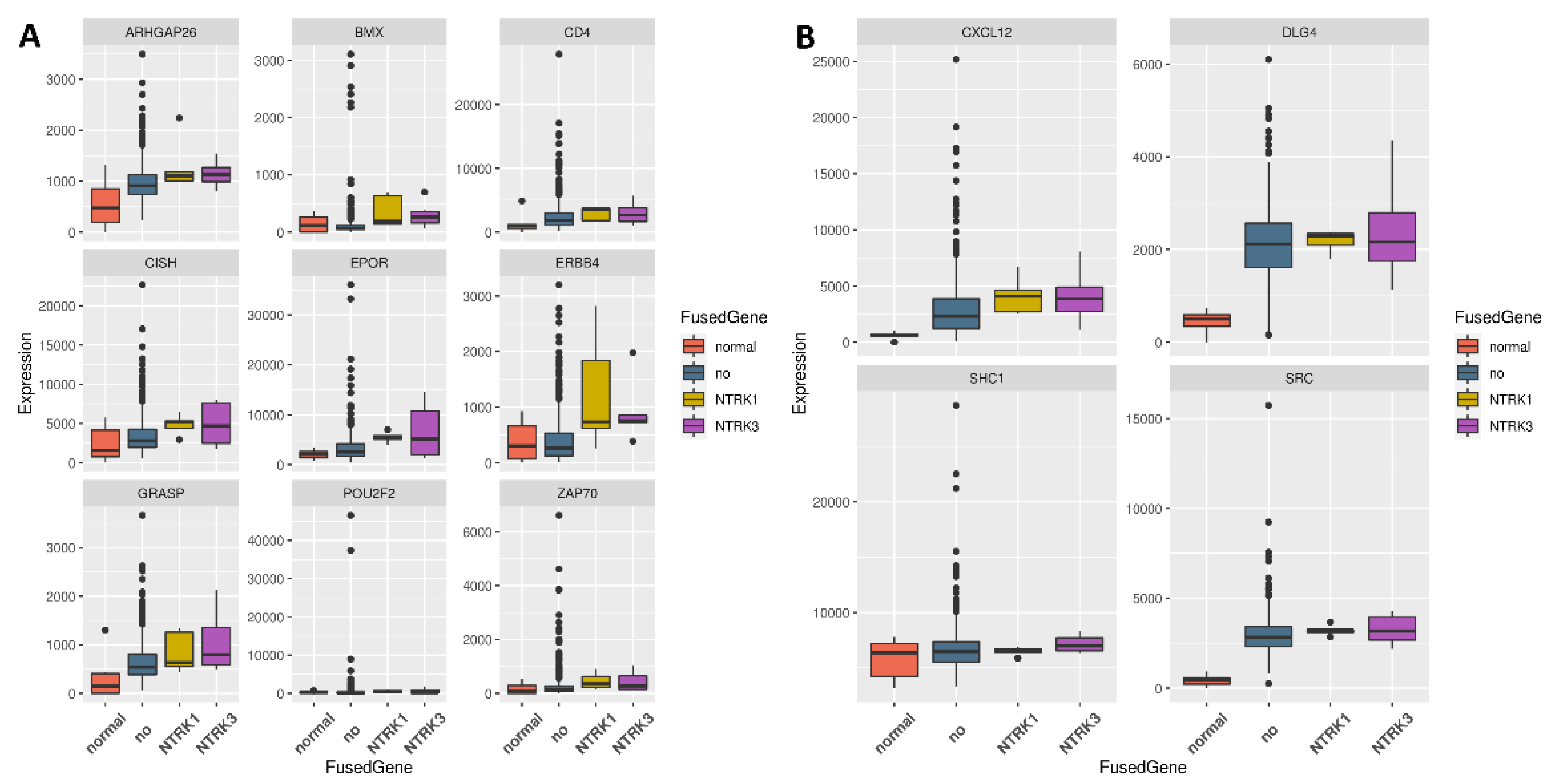
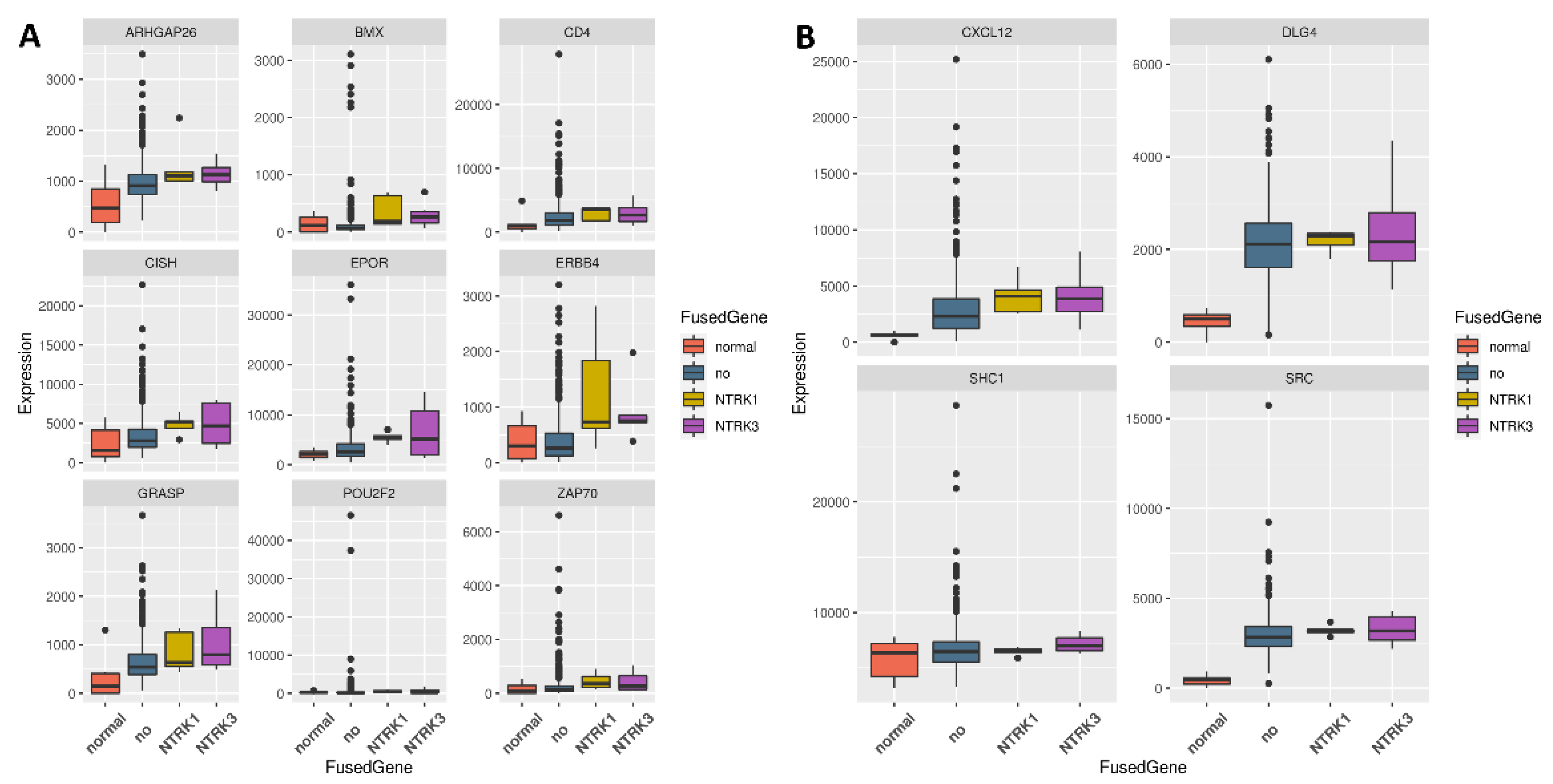
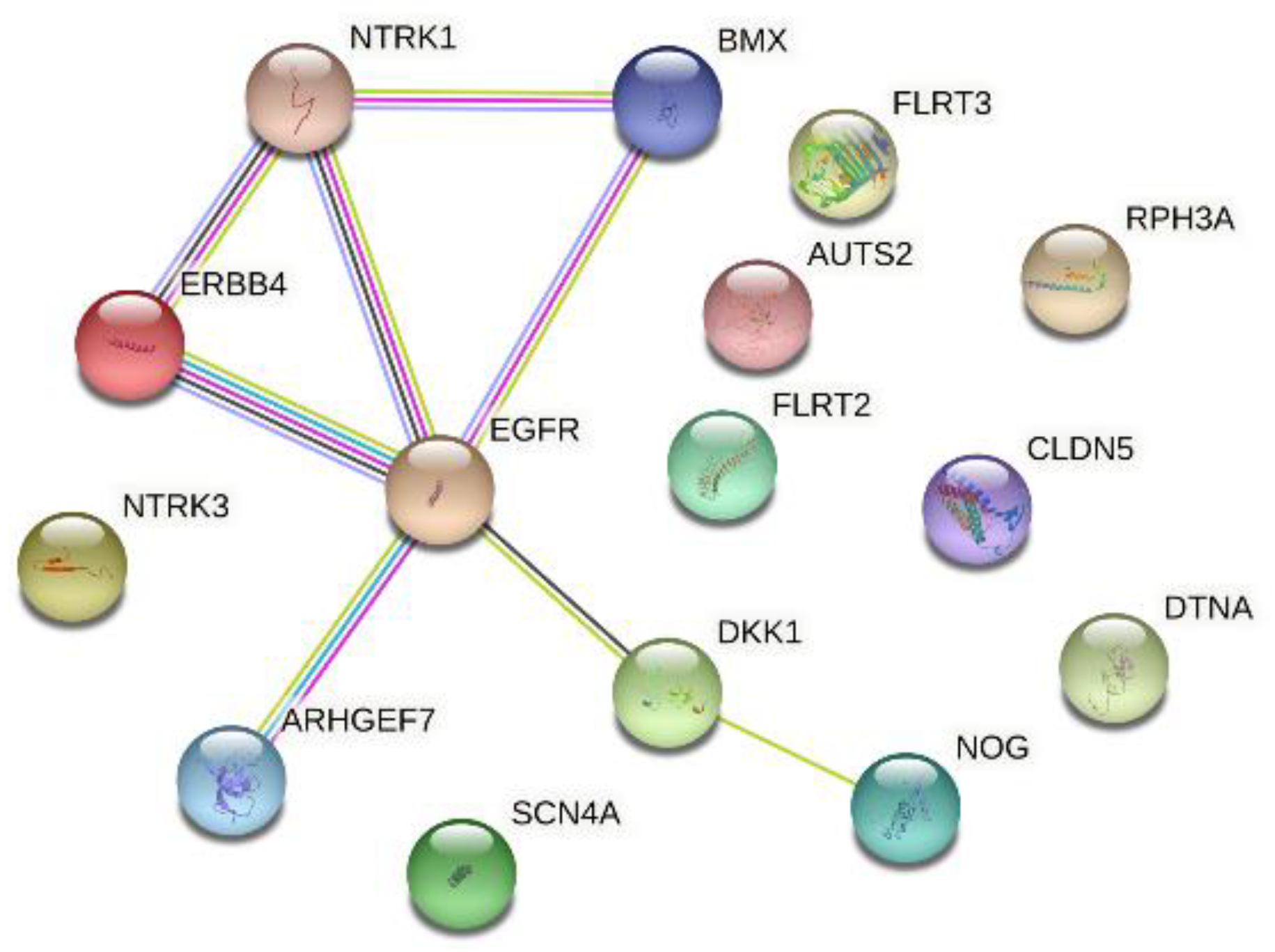
3.2. Genes Upregulated Only in Samples with NTRK1 and NTRK3 Gene Fusions
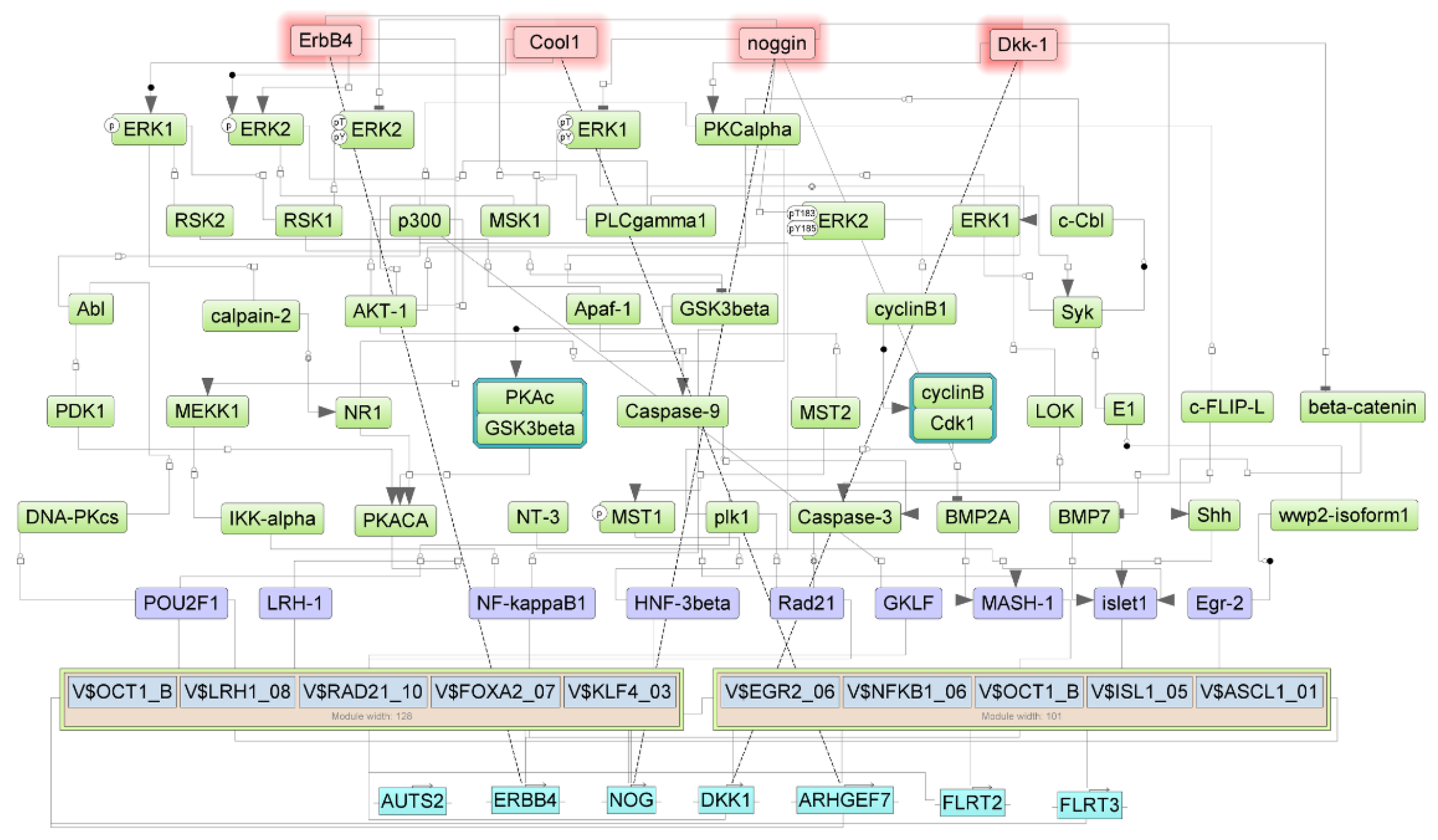
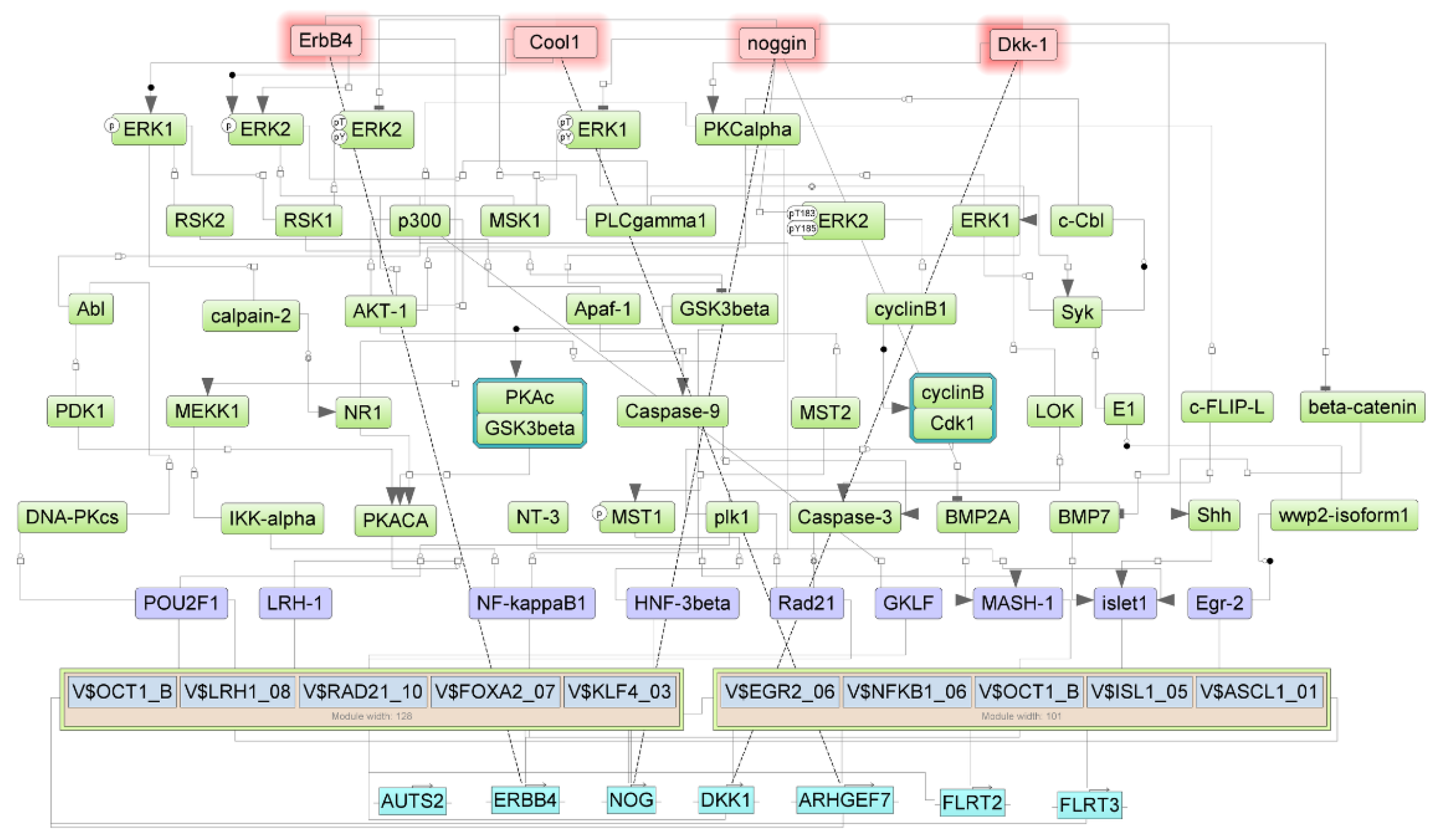
3.3. Regulators for the Genes Upregulated in Samples with NTRK1 and NTRK3 Gene Fusions
3.4. Regulators of Upregulated Genes Associated with the Nervous System
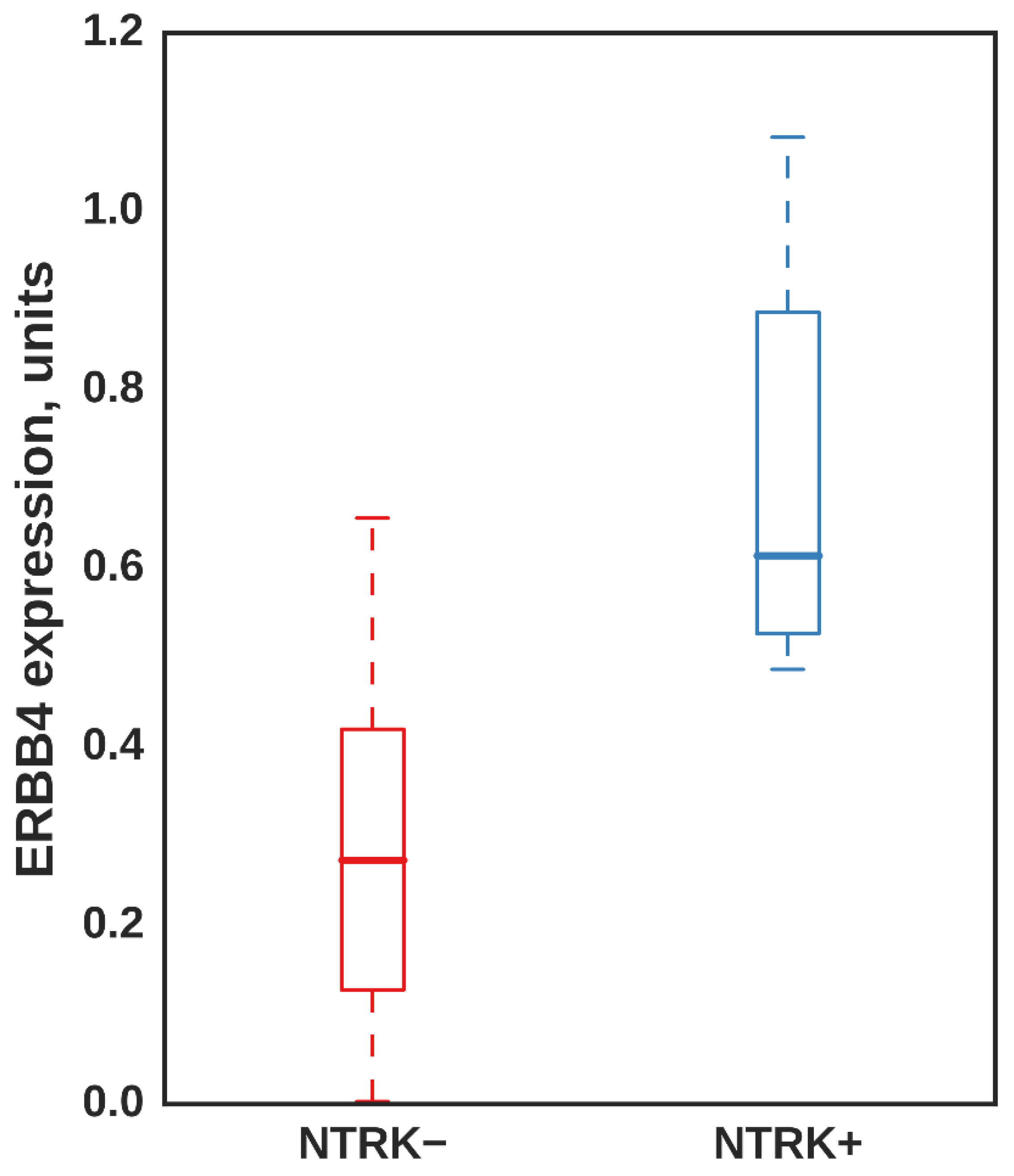
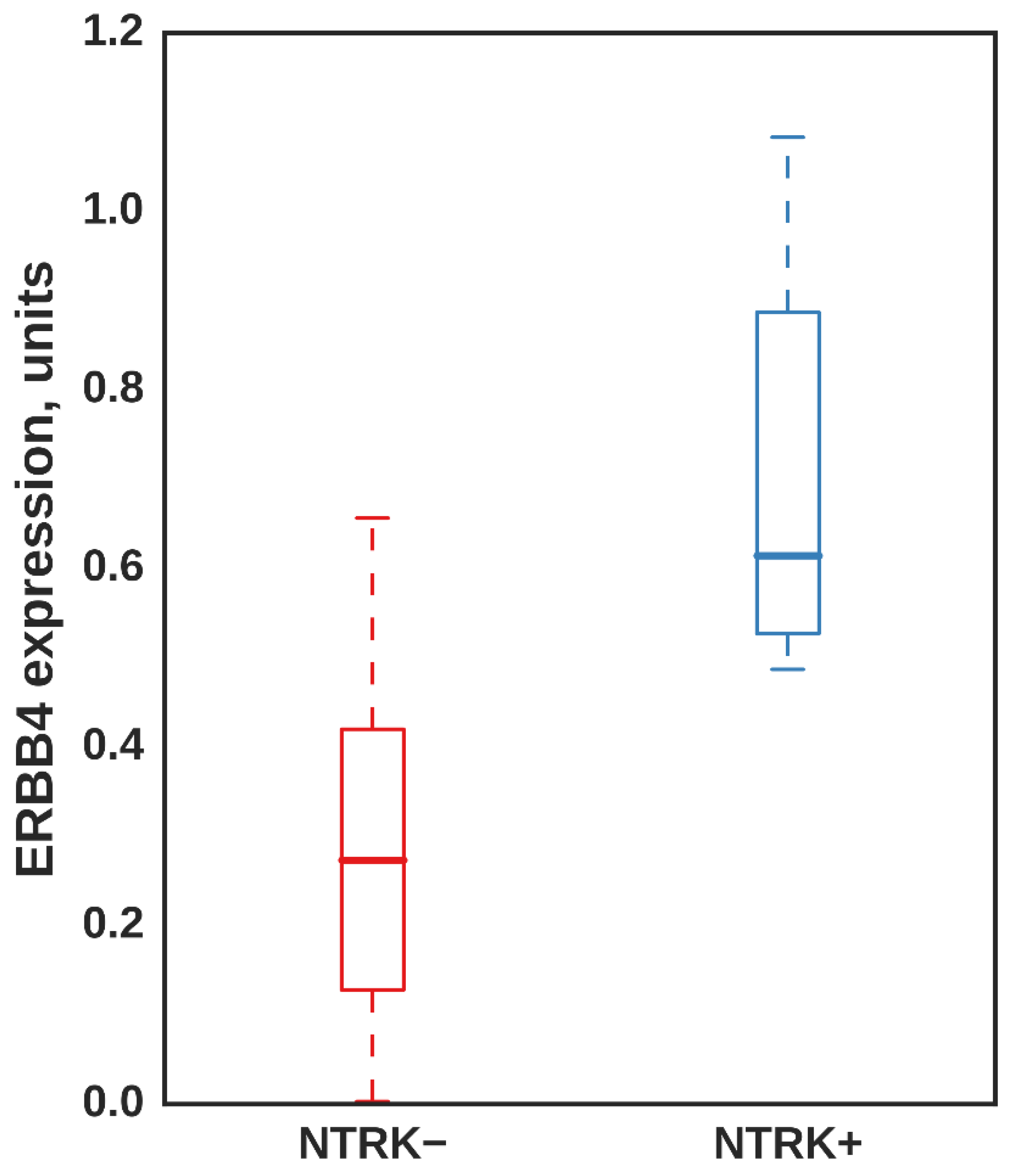
3.5. ERBB4 Was Significantly Upregulated in Independent Samples with NTRK3 Fusion
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Blondy, S.; Christou, N.; David, V.; Verdier, M.; Jauberteau, M.-O.; Mathonnet, M.; Perraud, A. Neurotrophins and Their Involvement in Digestive Cancers. Cell Death Dis. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Marino, F.Z.; Pagliuca, F.; Ronchi, A.; Cozzolino, I.; Montella, M.; Berretta, M.; Errico, M.E.; Donofrio, V.; Bianco, R.; Franco, R. NTRK Fusions, from the Diagnostic Algorithm to Innovative Treatment in the Era of Precision Medicine. Int. J. Mol. Sci. 2020, 21, 3718. [Google Scholar] [CrossRef] [PubMed]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK Fusion-Positive Cancers and TRK Inhibitor Therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef] [PubMed]
- Westphalen, C.B.; Krebs, M.G.; le Tourneau, C.; Sokol, E.S.; Maund, S.L.; Wilson, T.R.; Jin, D.X.; Newberg, J.Y.; Fabrizio, D.; Veronese, L.; et al. Genomic Context of NTRK1/2/3 Fusion-Positive Tumours from a Large Real-World Population. npj Precis. Oncol. 2021, 5, 1–9. [Google Scholar] [CrossRef]
- Bormann Chung, C.; Lee, J.; Barritault, M.; Bringuier, P.P.; Xu, Z.; Huang, W.Y.; Beharry, A.; Castillo, J.; Christiansen, J.; Lin, J.C.; et al. Evaluating Targeted Next-Generation Sequencing Assays and Reference Materials for NTRK Fusion Detection. J. Mol. Diagn. 2022, 24, 18–32. [Google Scholar] [CrossRef]
- Solomon, J.P.; Linkov, I.; Rosado, A.; Mullaney, K.; Rosen, E.Y.; Frosina, D.; Jungbluth, A.A.; Zehir, A.; Benayed, R.; Drilon, A.; et al. NTRK Fusion Detection across Multiple Assays and 33,997 Cases: Diagnostic Implications and Pitfalls. Mod. Pathol. 2019, 33, 38–46. [Google Scholar] [CrossRef]
- Meier, T.; Timm, M.; Montani, M.; Wilkens, L. Gene Networks and Transcriptional Regulators Associated with Liver Cancer Development and Progression. BMC Med. Genom. 2021, 14, 1–23. [Google Scholar] [CrossRef]
- Pan, Z.; Li, L.; Fang, Q.; Qian, Y.; Zhang, Y.; Zhu, J.; Ge, M.; Huang, P. Integrated Bioinformatics Analysis of Master Regulators in Anaplastic Thyroid Carcinoma. BioMed Res. Int. 2019, 1–13. [Google Scholar] [CrossRef]
- Liang, Y.; Wu, H.; Lei, R.; Chong, R.A.; Wei, Y.; Lu, X.; Tagkopoulos, I.; Kung, S.Y.; Yang, Q.; Hu, G.; et al. Transcriptional Network Analysis Identifies BACH1 as a Master Regulator of Breast Cancer Bone Metastasis. J. Biol. Chem. 2012, 287, 33533–33544. [Google Scholar] [CrossRef] [Green Version]
- Schoeberl, B.; Pace, E.A.; Fitzgerald, J.B.; Harms, B.D.; Xu, L.; Nie, L.; Linggi, B.; Kalra, A.; Paragas, V.; Bukhalid, R.; et al. Therapeutically Targeting ErbB3: A Key Node in Ligand-Induced Activation of the ErbB Receptor-PI3K Axis. Sci. Signal. 2009, 2, ra31. [Google Scholar] [CrossRef]
- Liang, Y.; Tsoi, L.C.; Xing, X.; Beamer, M.A.; Swindell, W.R.; Sarkar, M.K.; Berthier, C.C.; Stuart, P.E.; Harms, P.W.; Nair, R.P.; et al. A Gene Network Regulated by the Transcription Factor VGLL3 as a Promoter of Sex-Biased Autoimmune Diseases. Nat. Immunol. 2017, 18, 152–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smestad, J.A.; Maher, J.L. Master Regulator Analysis of Paragangliomas Carrying SDHx, VHL, or MAML3 Genetic Alterations. BMC Cancer 2019, 19, 1–19. [Google Scholar] [CrossRef]
- Suntsova, M.; Gaifullin, N.; Allina, D.; Reshetun, A.; Li, X.; Mendeleeva, L.; Surin, V.; Sergeeva, A.; Spirin, P.; Prassolov, V.; et al. Atlas of RNA Sequencing Profiles for Normal Human Tissues. Sci. Data 2019, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Wang, Q.; Tang, M.; Barthel, F.; Amin, S.; Yoshihara, K.; Lang, F.M.; Martinez-Ledesma, E.; Lee, S.H.; Zheng, S.; et al. TumorFusions: An Integrative Resource for Cancer-Associated Transcript Fusions. Nucleic Acids Res. 2018, 46, D1144. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Rzhetsky, A.; Iossifov, I.; Koike, T.; Krauthammer, M.; Kra, P.; Morris, M.; Yu, H.; Duboué, P.; Weng, W.; Wilbur, W.; et al. GeneWays: A System for Extracting, Analyzing, Visualizing, and Integrating Molecular Pathway Data. J. Biomed. Inform. 2004, 37, 43–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kel, A.; Boyarskikh, U.; Stegmaier, P.; Leskov, L.S.; Sokolov, A.V.; Yevshin, I.; Mandrik, N.; Stelmashenko, D.; Koschmann, J.; Kel-Margoulis, O.; et al. Walking Pathways with Positive Feedback Loops Reveal DNA Methylation Biomarkers of Colorectal Cancer. BMC Bioinform. 2019, 20, 1–20. [Google Scholar] [CrossRef]
- Kel, A.; Voss, N.; Jauregui, R.; Kel-Margoulis, O.; Wingender, E. Beyond Microarrays: Finding Key Transcription Factors Controlling Signal Transduction Pathways. BMC Bioinform. 2006, 7, S13. [Google Scholar] [CrossRef] [Green Version]
- Koschmann, J.; Bhar, A.; Stegmaier, P.; Kel, A.; Wingender, E. “Upstream Analysis”: An Integrated Promoter-Pathway Analysis Approach to Causal Interpretation of Microarray Data. Microarrays 2015, 4, 270–286. [Google Scholar] [CrossRef]
- Boyarskikh, U.; Pintus, S.; Mandrik, N.; Stelmashenko, D.; Kiselev, I.; Evshin, I.; Sharipov, R.; Stegmaier, P.; Kolpakov, F.; Filipenko, M.; et al. Computational Master-Regulator Search Reveals MTOR and PI3K Pathways Responsible for Low Sensitivity of NCI-H292 and A427 Lung Cancer Cell Lines to Cytotoxic Action of P53 Activator Nutlin-3. BMC Med. Genom. 2018, 11, S12018–S12920. [Google Scholar] [CrossRef] [Green Version]
- Matys, V.; Kel-Margoulis, O.V.; Fricke, E.; Liebich, I.; Land, S.; Barre-Dirrie, A.; Reuter, I.; Chekmenev, D.; Krull, M.; Hornischer, K.; et al. TRANSFAC and Its Module TRANSCompel: Transcriptional Gene Regulation in Eukaryotes. Nucleic Acids Res. 2006, 34, D108–D110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krull, M. TRANSPATH(R): An integrated database on signal transduction and a tool for array analysis. Nucleic Acids Res. 2003, 31, 97–100. [Google Scholar] [CrossRef] [Green Version]
- Waleev, T.; Shtokalo, D.; Konovalova, T.; Voss, N.; Cheremushkin, E.; Stegmaier, P.; Kel-Margoulis, O.; Wingender, E.; Kel, A. Composite Module Analyst: Identification of Transcription Factor Binding Site Combinations Using Genetic Algorithm. Nucleic Acids Res. 2006, 34, W541–W545. [Google Scholar] [CrossRef]
- Wingender, E.; Dietze, P.; Karas, H.; Knüppel, R. TRANSFAC: A Database on Transcription Factors and Their DNA Binding Sites. Nucleic Acids Res. 1996, 24, 238–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deyneko, I.V.; Kel, A.E.; Kel-Margoulis, O.V.; Deineko, E.V.; Wingender, E.; Weiss, S. MatrixCatch—A Novel Tool for the Recognition of Composite Regulatory Elements in Promoters. BMC Bioinform. 2013, 14, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meckbach, C.; Tacke, R.; Hua, X.; Waack, S.; Wingender, E.; Gültas, M. PC-TraFF: Identification of Potentially Collaborating Transcription Factors Using Pointwise Mutual Information. BMC Bioinform. 2015, 16, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meckbach, C.; Wingender, E.; Gültas, M. Removing Background Co-Occurrences of Transcription Factor Binding Sites Greatly Improves the Prediction of Specific Transcription Factor Cooperations. Front. Genet. 2018, 9, 189. [Google Scholar] [CrossRef]
- Segers, V.F.M.; Dugaucquier, L.; Feyen, E.; Shakeri, H.; De Keulenaer, G.W. The Role of ErbB4 in Cancer. Cell. Oncol. 2020, 43, 335–352. [Google Scholar] [CrossRef]
- Luo, B.; Liu, Z.; Lin, D.; Chen, W.; Ren, D.; Yu, Z.; Xiong, M.; Zhao, C.; Fei, E.; Li, B. ErbB4 Promotes Inhibitory Synapse Formation by Cell Adhesion, Independent of Its Kinase Activity. Transl. Psychiatry 2021, 11, 1–11. [Google Scholar] [CrossRef]
- Cao, S.X.; Zhang, Y.; Hu, X.Y.; Hong, B.; Sun, P.; He, H.Y.; Geng, H.Y.; Bao, A.M.; Duan, S.M.; Yang, J.M.; et al. ErbB4 Deletion in Noradrenergic Neurons in the Locus Coeruleus Induces Mania-like Behavior via Elevated Catecholamines. eLife 2018, 7, e39907. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Jung, H.H.; Do, I.-G.; Bae, S.; Lee, S.K.; Kim, S.W.; Lee, J.E.; Nam, S.J.; Ahn, J.S.; Park, Y.H.; et al. Prognostic Value of ERBB4 Expression in Patients with Triple Negative Breast Cancer. BMC Cancer 2016, 16, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.S.; Bernard, J.K.; Demory Beckler, M.; Almohazey, D.; Washington, M.K.; Smith, J.J.; Frey, M.R. ERBB4 Is Over-Expressed in Human Colon Cancer and Enhances Cellular Transformation. Carcinogenesis 2015, 36, 710–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauf, F.; Festa, F.; Park, J.G.; Magee, M.; Eaton, S.; Rinaldi, C.; Betanzos, C.M.; Gonzalez-Malerva, L.; LaBaer, J. Ibrutinib Inhibition of ERBB4 Reduces Cell Growth in a WNT5A-Dependent Manner. Oncogene 2018, 37, 2237–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayati, A.; Moghimi, S.; Salarinejad, S.; Safavi, M.; Pouramiri, B.; Foroumadi, A. A Review on Progression of Epidermal Growth Factor Receptor (EGFR) Inhibitors as an Efficient Approach in Cancer Targeted Therapy. Bioorg. Chem. 2020, 99, 103811. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.; Weihua, Z. Rethink of EGFR in Cancer With Its Kinase Independent Function on Board. Front. Oncol. 2019, 9, 800. [Google Scholar] [CrossRef] [PubMed]
- de Farias, C.B.; Heinen, T.E.; dos Santos, R.P.; Abujamra, A.L.; Schwartsmann, G.; Roesler, R. BDNF/TrkB Signaling Protects HT-29 Human Colon Cancer Cells from EGFR Inhibition. Biochem. Biophys. Res. Commun. 2012, 425, 328–332. [Google Scholar] [CrossRef] [Green Version]
- Ekman, N.; Lymboussaki, A.; Västrik, I.; Sarvas, K.; Kaipainen, A.; Alitalo, K. Bmx Tyrosine Kinase Is Specifically Expressed in the Endocardium and the Endothelium of Large Arteries. Circulation 1997, 96, 1729–1732. [Google Scholar] [CrossRef]
- Holopainen, T.; Räsänen, M.; Anisimov, A.; Tuomainen, T.; Zheng, W.; Tvorogov, D.; Hulmi, J.J.; Andersson, L.C.; Cenni, B.; Tavi, P.; et al. Endothelial Bmx Tyrosine Kinase Activity Is Essential for Myocardial Hypertrophy and Remodeling. Proc. Natl. Acad. Sci. USA 2015, 112, 13063–13068. [Google Scholar] [CrossRef] [Green Version]
- Qiu, L.; Wang, F.; Liu, S.; Chen, X.-L. Current Understanding of Tyrosine Kinase BMX in Inflammation and Its Inhibitors. Burn. Trauma 2014, 2, 121. [Google Scholar] [CrossRef] [Green Version]
- Paavonen, K.; Ekman, N.; Wirzenius, M.; Rajantie, I.; Poutanen, M.; Alitalo, K. Bmx Tyrosine Kinase Transgene Induces Skin Hyperplasia, Inflammatory Angiogenesis, and Accelerated Wound Healing. Mol. Biol. Cell 2004, 15, 4226–4233. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Cai, C.; Sowalsky, A.G.; Ye, H.; Ma, F.; Yuan, X.; Simon, N.I.; Gray, N.S.; Balk, S.P. BMX-Mediated Regulation of Multiple Tyrosine Kinases Contributes to Castration Resistance in Prostate Cancer. Cancer Res. 2018, 78, 5203–5215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.-F.; Gao, F.; Wang, J.-J.; Long, C.; Chen, X.; Tao, L.; Yang, L.; Ding, L.; Ji, Y. BMX-ARHGAP Fusion Protein Maintains the Tumorigenicity of Gastric Cancer Stem Cells by Activating the JAK/STAT3 Signaling Pathway. Cancer Cell Int. 2019, 19, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haines, B.P.; Wheldon, L.M.; Summerbell, D.; Heath, J.K.; Rigby, P.W.J. Regulated Expression of FLRT Genes Implies a Functional Role in the Regulation of FGF Signalling during Mouse Development. Dev. Biol. 2006, 297, 14–25. [Google Scholar] [CrossRef]
- Jauhiainen, S.; Laakkonen, J.P.; Ketola, K.; Toivanen, P.I.; Nieminen, T.; Ninchoji, T.; Levonen, A.-L.; Kaikkonen, M.U.; Ylä-Herttuala, S. Axon Guidance-Related Factor FLRT3 Regulates VEGF-Signaling and Endothelial Cell Function. Front. Physiol. 2019, 10, 224. [Google Scholar] [CrossRef] [Green Version]
- Oksenberg, N.; Stevison, L.; Wall, J.D.; Ahituv, N. Function and Regulation of AUTS2, a Gene Implicated in Autism and Human Evolution. PLoS Genet. 2013, 9, e1003221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, K.; Yamashiro, K.; Nagai, T.; Shan, W.; Egusa, S.F.; Shimaoka, K.; Kuniishi, H.; Sekiguchi, M.; Go, Y.; Tatsumoto, S.; et al. AUTS2 Regulation of Synapses for Proper Synaptic Inputs and Social Communication. iScience 2020, 23, 101183. [Google Scholar] [CrossRef] [PubMed]
- Oksenberg, N.; Haliburton, G.D.E.; Eckalbar, W.L.; Oren, I.; Nishizaki, S.; Murphy, K.; Pollard, K.S.; Birnbaum, R.Y.; Ahituv, N. Genome-Wide Distribution of Auts2 Binding Localizes with Active Neurodevelopmental Genes. Transl. Psychiatry 2014, 4, e431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molnar, C.; de Celis, J.F. Tay Bridge Is a Negative Regulator of EGFR Signalling and Interacts with Erk and Mkp3 in the Drosophila Melanogaster Wing. PLoS Genet. 2013, 9, e1003982. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-C.; Lin, J.-D.; Chen, J.-T.; Chang, C.-M.; Weng, H.-F.; Hsueh, C.; Chien, H.-P.; Yu, J.-S. Integrated Analysis of Fine-Needle-Aspiration Cystic Fluid Proteome, Cancer Cell Secretome, and Public Transcriptome Datasets for Papillary Thyroid Cancer Biomarker Discovery. Oncotarget 2018, 9, 12079–12100. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.J.; Chung, K.W.; Lee, R.A.; Park, I.A.; Lee, S.H.; Park, D.E.; Noh, D.Y. Overexpression of BetaPix-a in Human Breast Cancer Tissues. Cancer Lett. 2003, 193, 99–107. [Google Scholar] [CrossRef]
- Ishaque, N.; Abba, M.L.; Hauser, C.; Patil, N.; Paramasivam, N.; Huebschmann, D.; Leupold, J.H.; Balasubramanian, G.P.; Kleinheinz, K.; Toprak, U.H.; et al. Whole Genome Sequencing Puts Forward Hypotheses on Metastasis Evolution and Therapy in Colorectal Cancer. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Primers and Probes |
|---|---|
| TBP | TBP-R2 5′-CACTGTGGATACAATATTTTGCAG-3′ TBP-U2 5′-CACTCCTGCCACGCCAGCT-3′ TBP-PC2R 5′-ROX-CGGAGAGTTCTGGGATTGTACCG-BHQ2-3′ |
| ERBB4 | ERBB4-U2 5′-CCAGATCGGGAGTGCCACC-3′ ERBB4-R2 5′-ATGACTAGTGGGACCGTTACAC-3′ ERBB4-PF2 5′-FAM-TGCCATCCAAACTGCACCCAAGG-BHQ1-3′ |
| ETV6-NTRK3 | ETV6-NTRK3-R 5′-CACCCAGTTCTCGCTTCAGC-3′ ETV6-NTRK3-U 5′-GAGCACGCCATGCCCATTG-3′ ETV6-NTRK3-PH 5′-HEX-CAGCACATTAAGAGGAGAGACATCG-BHQ1-3′ |
| Gene | DEG with p-Value < 0.05 | Identified as Regulator | Expression More than in Samples without NTRK-Fusions | Interactions with Trk Found in STRING | Known Engagement in Malignancy |
|---|---|---|---|---|---|
| ERBB4 | yes/no | yes | yes | yes | yes |
| EGFR | yes | yes | no | yes | yes |
| BMX | no | yes | yes/no | yes | yes |
| AUTS2 | yes | no | yes | no | yes |
| DKK1 | yes | yes | yes | yes | yes |
| ARHGEF7 | yes | yes | yes | yes | yes |
| FLRT3 | yes | no | yes | no | no |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kechin, A.; Borobova, V.; Kel, A.; Ivanov, A.; Filipenko, M. ErbB4 Is a Potential Key Regulator of the Pathways Activated by NTRK-Fusions in Thyroid Cancer. Appl. Sci. 2022, 12, 2506. https://doi.org/10.3390/app12052506
Kechin A, Borobova V, Kel A, Ivanov A, Filipenko M. ErbB4 Is a Potential Key Regulator of the Pathways Activated by NTRK-Fusions in Thyroid Cancer. Applied Sciences. 2022; 12(5):2506. https://doi.org/10.3390/app12052506
Chicago/Turabian StyleKechin, Andrey, Viktoriya Borobova, Alexander Kel, Anatoliy Ivanov, and Maxim Filipenko. 2022. "ErbB4 Is a Potential Key Regulator of the Pathways Activated by NTRK-Fusions in Thyroid Cancer" Applied Sciences 12, no. 5: 2506. https://doi.org/10.3390/app12052506
APA StyleKechin, A., Borobova, V., Kel, A., Ivanov, A., & Filipenko, M. (2022). ErbB4 Is a Potential Key Regulator of the Pathways Activated by NTRK-Fusions in Thyroid Cancer. Applied Sciences, 12(5), 2506. https://doi.org/10.3390/app12052506