1. Introduction
Luminescence examinations of bone samples can provide valuable forensic information and archeological insight into the environmental conditions a find was exposed to. Recently, the photoluminescence emission, in particular, of heated bones has been found to be affected by the exposure temperature [
1,
2,
3]. Photoluminescence consists of both quickly decaying fluorescence with lifetimes in the picosecond and nanosecond range and longer prevailing phosphoresce with lifetimes in the microsecond to minute range. In particular, the study of the phosphorescence could aid in the search of human skeletal remains or indicate the maximum temperature a bone has been exposed to, for example during a previous fire as has been proposed by Krap et al. [
1,
3]. In Ref. [
3], the authors have quantified the phosphorescence intensity by visual inspection and observer assessment through a scoring index scheme. The authors found the visibly observable phosphorescence intensity to depend strongly on the maximum exposure temperature, with the highest emissions observed in the range from 450 °C to 800 °C, and to a much lesser degree to depend on exposure duration. Photoluminescence of bones has been observed before [
4] and originally assumed to stem mainly from organic substances like collagen, which is known to fluoresce but does not phosphoresce, and to a lower degree to bone minerals (e.g., apatite). Recent studies have suggested, however, that inorganic material of the bone generates the temperature-dependent phosphorescence, in particular, the main inorganic component hydroxyapatite [
1,
5]. The detailed microscopic origins and the components contributing to phosphorescence in particular in the range of 700 °C to 800 °C are not fully understood and for lower exposure temperatures even contradicting findings on the role of organic and inorganic components have been reported [
3]. So far mainly spectroscopy methods, which have been based on continuous excitation with UV or blue light, have been used to investigate the bone photoluminescence. Krap et al. suggested to distinguish between “short-decay” and “long-decay” phosphorescence with the short-decay phosphoresce being the part which is “visibly not observable” but has a longer decay time than fluorescence [
3].
In life sciences, phosphorescence lifetime microscopy (PLIM) has been used so far mainly for studies in cell biology to investigate the oxygen consumption of single cells during the cell metabolism by recording phosphorescence from exogenous markers (metal complexes) [
6,
7]. The marker phosphorescence emission can be affected by the system under study, e.g., a cell, and thereby indirectly indicates the sample condition. The technique can be combined with fluorescence lifetime imaging (FLIM) [
8,
9,
10]. The novelty of this study lies in the measurement of autophosphorescence, an intrinsic sample property, in contrast to measuring phosphorescence from an externally added marker. Autophosphorescence is usually very weak or absent in biological samples but can be considered as a “direct signal” from the specimen. To the best of our knowledge the autophosphorescence of biological specimens has not been studied (i) with high-resolution microscopy/tomography, (ii) with two-photon excitation, and (iii) with precise time resolution. In this paper, we have used high-resolution femtosecond laser scanning imaging in combination with time-correlated single-photon counting (TCSPC, [
9]) to investigate the spatial and temporal distributions of the two-photon excited autophosphorescence and autofluorescence emission from bones without any additional labeling with phosphorescent markers. The measurements provided the microscopic distribution of autophosphorescent substances in heated bone samples within a temporal measurement range of tens of microseconds as well as the simultaneous measurement of fast decaying fluorescence in the picosecond/nanosecond range. By evaluating the fluorescence-to-phosphorescence intensity ratio, the temperature effect on the bone photoluminescence could be unbiasedly derived and quantified without the need for a scoring index by observers.
3. Results and Discussion
A photographic image of the bones samples after the heating which feature typical heat-induced color changes [
16] is shown in
Figure 2a. The color ranges from black (400 °C) to brown and grey and completely white (900 °C).
The color changes of bones as a result of temperature are well-known and caused by changes of the chemical composition of the organic components [
16].
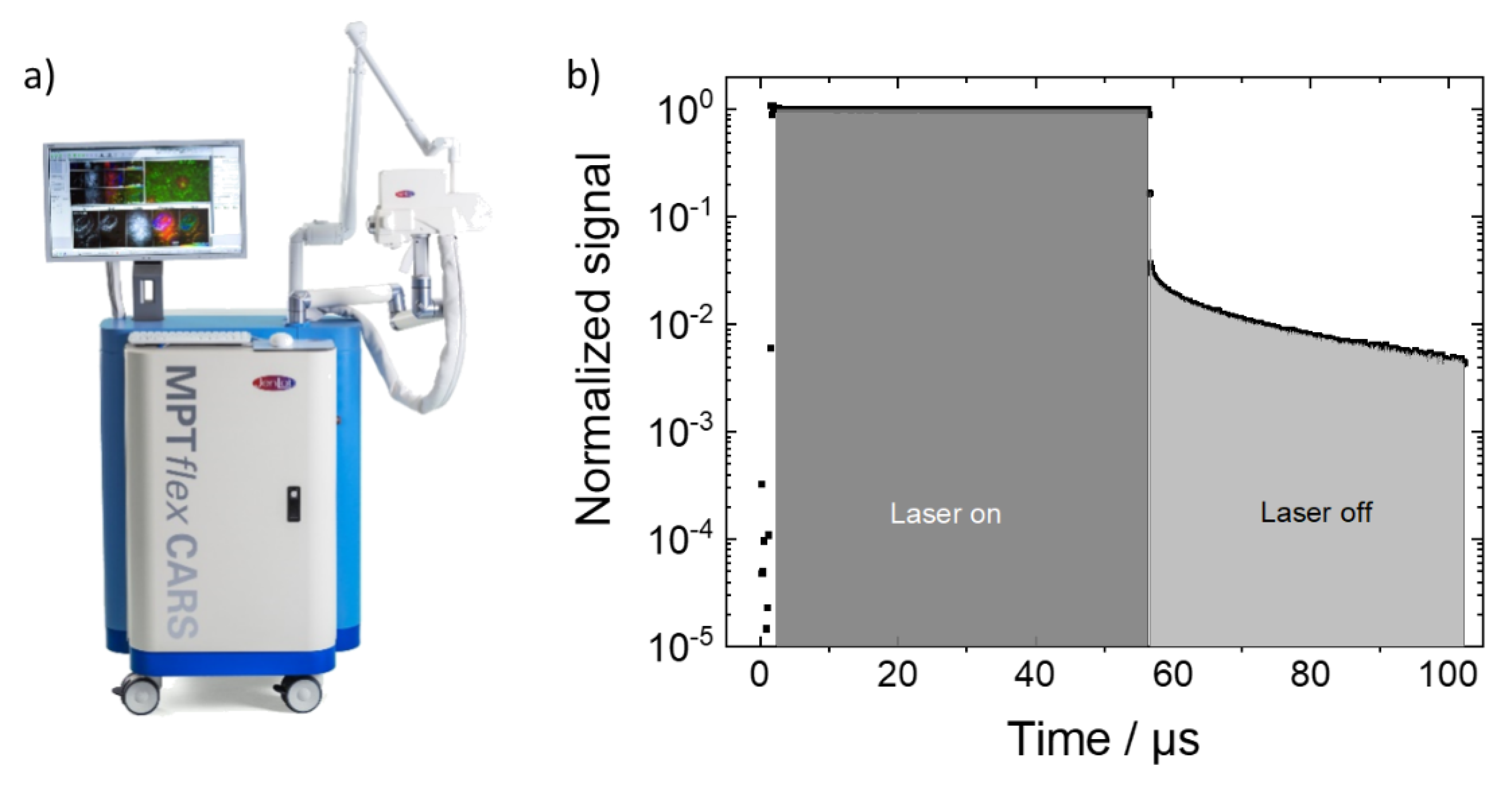
Figure 2b shows microscopic images which were derived from the pixelwise-collected photon counts during “laser on” and “laser off” times. The laser “on-time” images reflect the fluorescence intensity distribution (with negligible phosphorescence contributions) and the “laser-off times” reflect the phosphorescence signal. Microscopic fine structures of the bone samples can be recognized in both the fluorescence and phosphorescence signals, although, the fluoresce intensity is up to two orders of magnitude higher. For example, the total photon counts were 233 × 10
6 and 4 × 10
6 for fluorescence and phosphorescence in the 600 °C images, respectively. For both, the 400 °C- and the 900 °C-heated samples, only fluorescence was detectable. The ratio of the total phosphoresce to fluorescence intensities for the different maximum exposure temperatures is shown in
Figure 3.
The figure indicates a clear dependence of the intensity ratio on the exposure temperature. The bone sample which had been subjected to 400 °C hardly phosphorescents. With increasing exposure temperature, the ratio significantly increases up to a maximum of 600 °C from where it drops to 700 °C and 800 °C until at 900 °C the phosphorescence vanishes again. This general trend has been observed with all measured samples, although, different regions of a sample do exhibit varying phosphorescence and fluorescence emission intensities. The temperature dependence of the data in
Figure 3 is in line with the trend described by Krap et al. for the phosphoresce intensity scores [
3]. The phosphoresce-to-fluorescence ratio is an easy to obtain and more unbiased measure of the phosphorescence, whose absolute intensity is affected by the exact excitation and focusing conditions, without subjective observer evaluation. However, it should be noted that the described temperature dependence of the ratio does not rule out individual temperature dependencies of both the fluorescence as well as the phosphorescence. In fact, our data seems to indicate that absolute intensity values cannot be directly compared for the different samples since they depend on the region of interest and other imaging conditions.
With our measurement setup, the phosphorescence decay can also be analyzed pixelwise. This phosphorescence lifetime microscopy (PLIM) is the phosphorescence analogue of the better-known fluorescent lifetime microscopy (FLIM) [
9,
10,
15]: the time-dependent data are pixelwise color-coded according the to fit parameters. Usually, the day data are fitted by a mono or bi-exponential decay curve. We have used a bi-exponetial fit curve for the decay time tmean = a1 × t1 + a2 × t2 with amplitudes a1,2 (a1 + a2 = 100%) of the short and long decay times t1,2 as fit parameters [
9]. For the sample that was exposed to 700 °C (a photo as well as the fluorescence and phosphorescence intensity images are shown in
Figure 1),
Figure 4 exemplarily shows the corresponding PLIM data with color coding according to values of the decay time components. In the PLIM image in
Figure 4a, the short decay time t1 is pseudo-color coded and the pixel brightness corresponds to the photoluminescence intensity. The color coding covers a range between 2 µs (red) and 12 µs (blue) as indicated by the background colors of the histogram (
Figure 4b). The t1 histogram has two maxima around 4.5 µs and 10 µs. The decay time data of the phosphorescence for a single pixel is shown in
Figure 4c (blue dots) along with the fit curve (solid red line) on a semi-logarithmic scale. The time range covers the “laser off” time between 56 µs and 110 µs as described in
Section 2.1. The progression of the measured decay data indicates a bi-exponential decay which is the reason why a fitting with two decay times was applied. The fit parameters for that pixel are shown in
Figure 4d. They reveal the fast and slow decay times and the corresponding amplitudes t1 = 5.6 µs and a1 = 54% and t2 = 52.2 µs and a2 = 46%, respectively. The PLIM image with pseudo-color coding of t2 and the corresponding t2 histogram are shown in
Figure 4e,f, respectively. The color coding spans the decay-time range from 20 µs (red) up to 60 µs (blue). It exhibits a maximum around 40 µs but is non-zero for the whole covered range. Both PLIM images (
Figure 4a,e) exhibit regions with different decay times (compare blue, green, and reddish regions). This could be an indication that different phosphorescent materials with different decay times are present or the microscopic environment affects the phosphorescence decay time. It should be noted, however, that the fitting results are also affected by the choice of fitting conditions like the fitting boundaries. This concerns in particular the absolute values of the fitting parameters but to a much lesser degree the spatial variation of decay times. Further investigations are necessary, which are outside the scope of this paper, to investigate the possible presence as well as the nature of different phosphorescent substances and the influence of artifacts due to the fit conditions and settings.
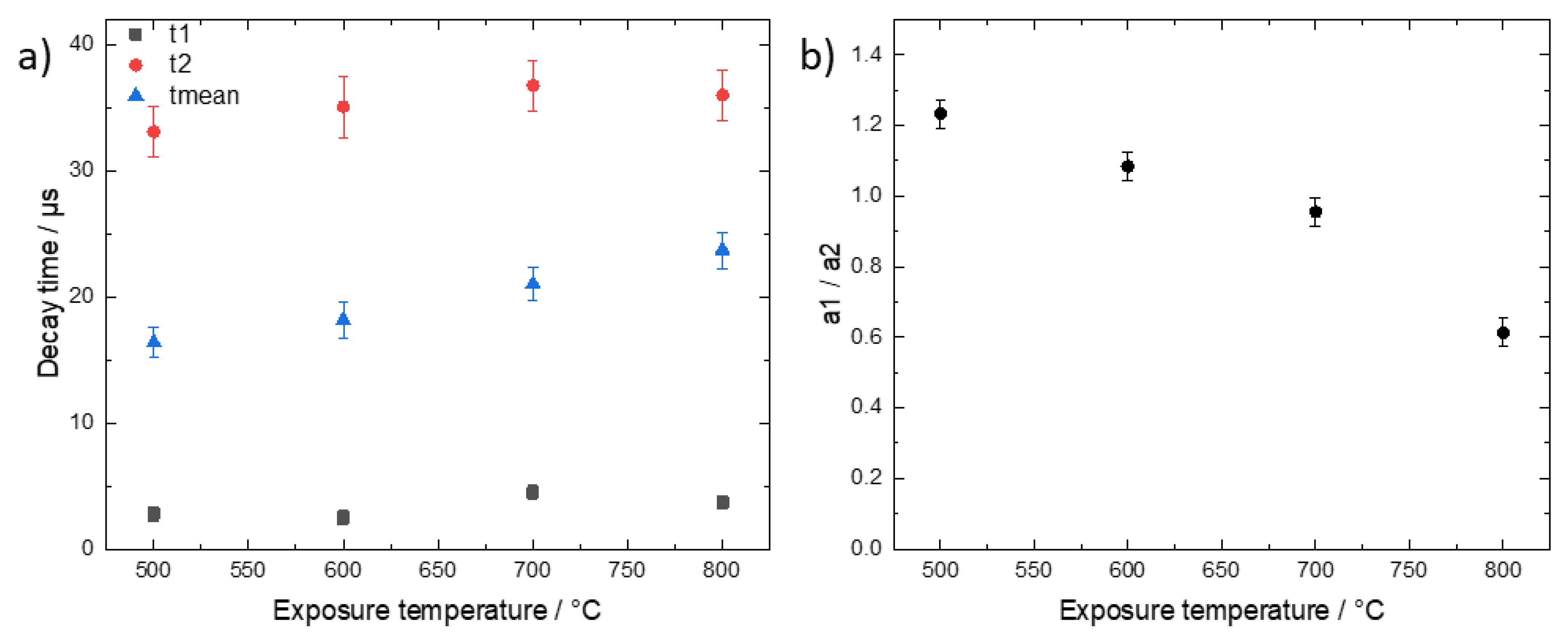
Since we record the time-resolved phosphorescence signals, it is interesting to check for a possible temperature dependence of the decay times. Such a dependence would indicate a change of the type of phosphorescent substance or the microenvironment with exposure temperature.
Figure 5 shows data derived from global fit analysis, i.e., by summing the signals from all image pixels for each time channel and fitting the resulting decay time data.
The decay time data were fitted again with a two-exponential decay function. The phosphorescence intensities of the 400 °C and 900 °C samples were too low for a meaningful decay time analysis and, therefore, are not included in
Figure 5. While the values of both the fast and slow decay times do not significantly change with increasing temperature, the mean decay time increases from 16 µs to 24 µs. This increase reflects mainly a decrease of the contribution of the fast decay component as depicted in
Figure 5b. This could result from a change of the composition of the phosphorescent bone substances. However, more data are needed to clarify and validate this dependence and the possible conclusions which are outside the scope of this paper. The general observation confirms the temperature dependence of the intensity-ratio dependence of
Figure 3 and the findings by Krap et at. in the way that, depending on the exposure temperature, the phosphorescent components which give rise to t1 and t2 are either present or absent but not significantly changed further [
3].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




