
Modelling the Defect Processes of Materials for Energy Applications


 , ,
, , 
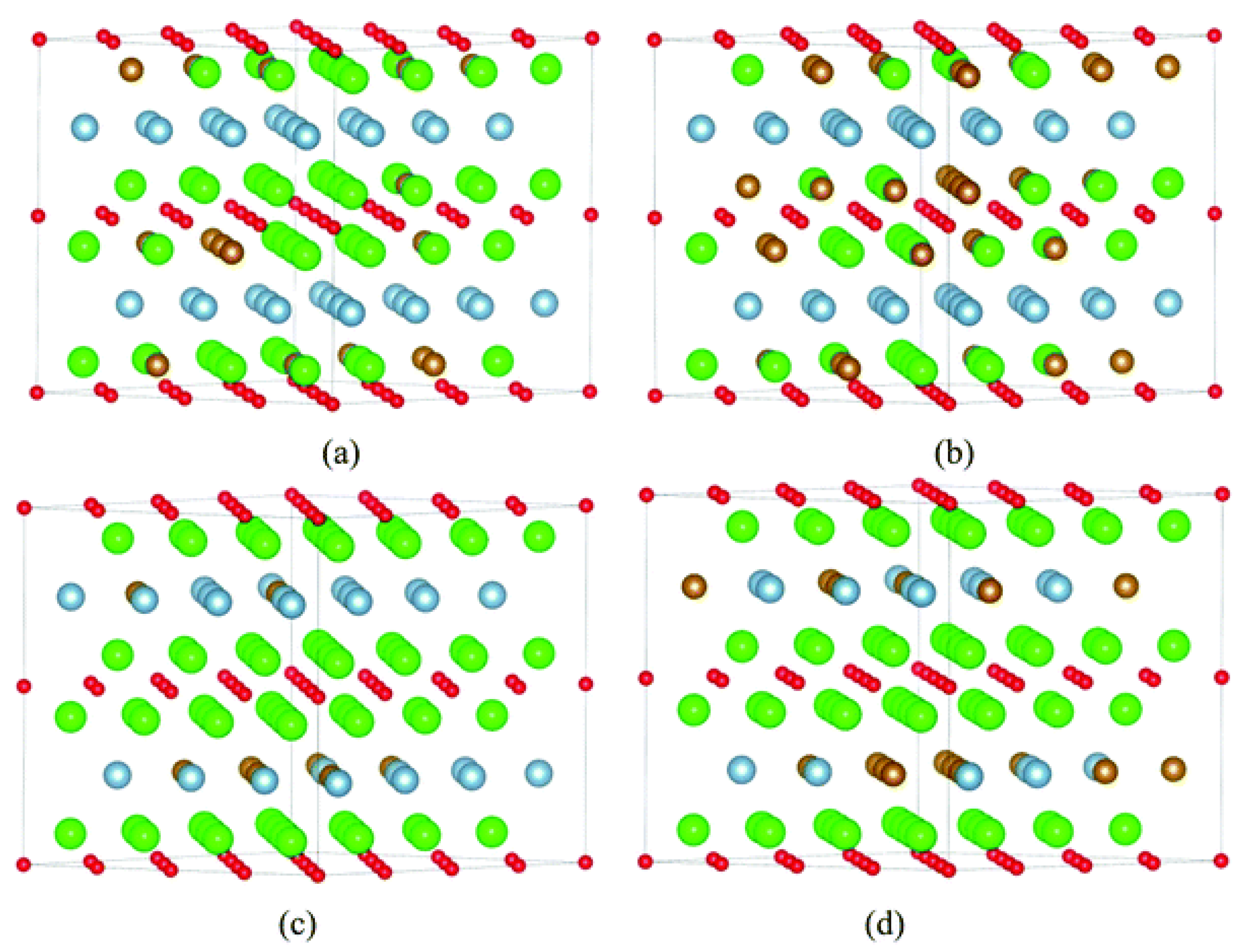{kind=link}
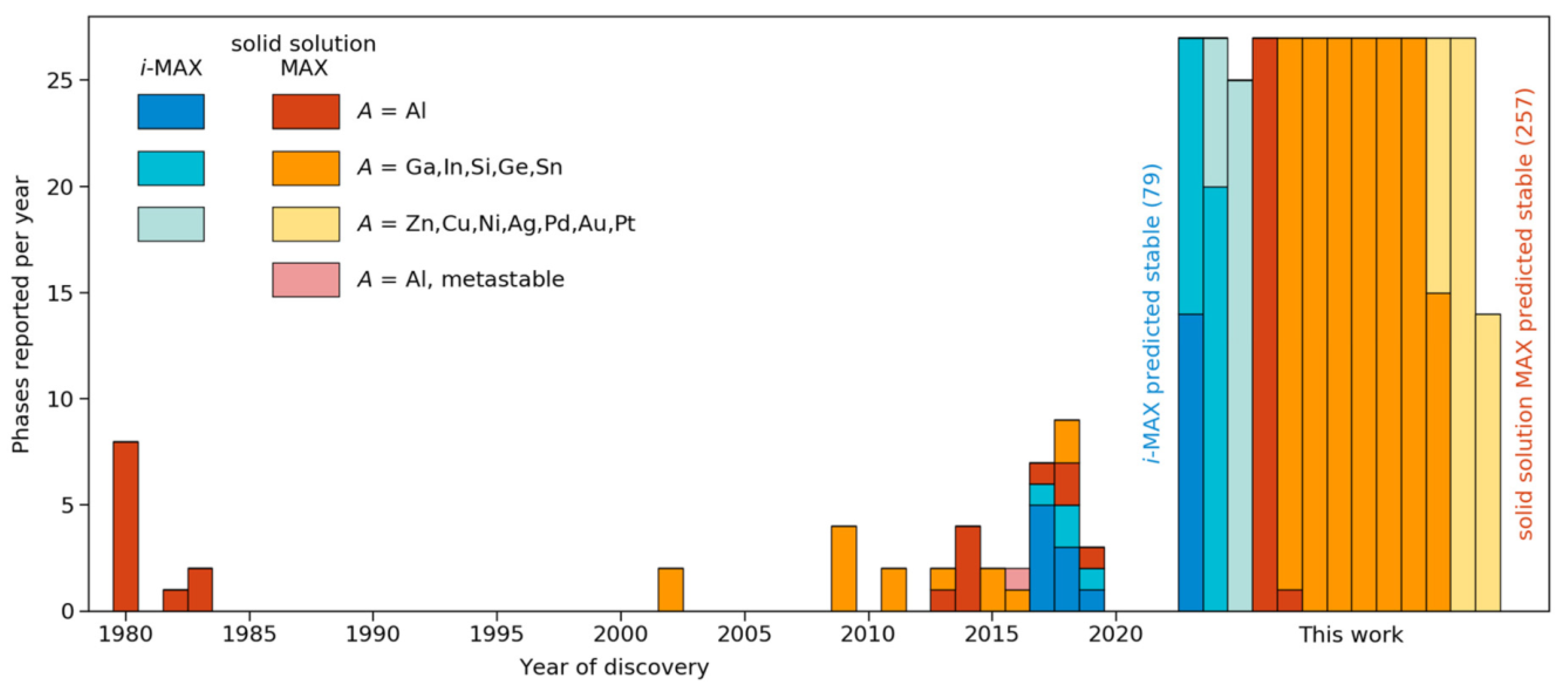{kind=link}
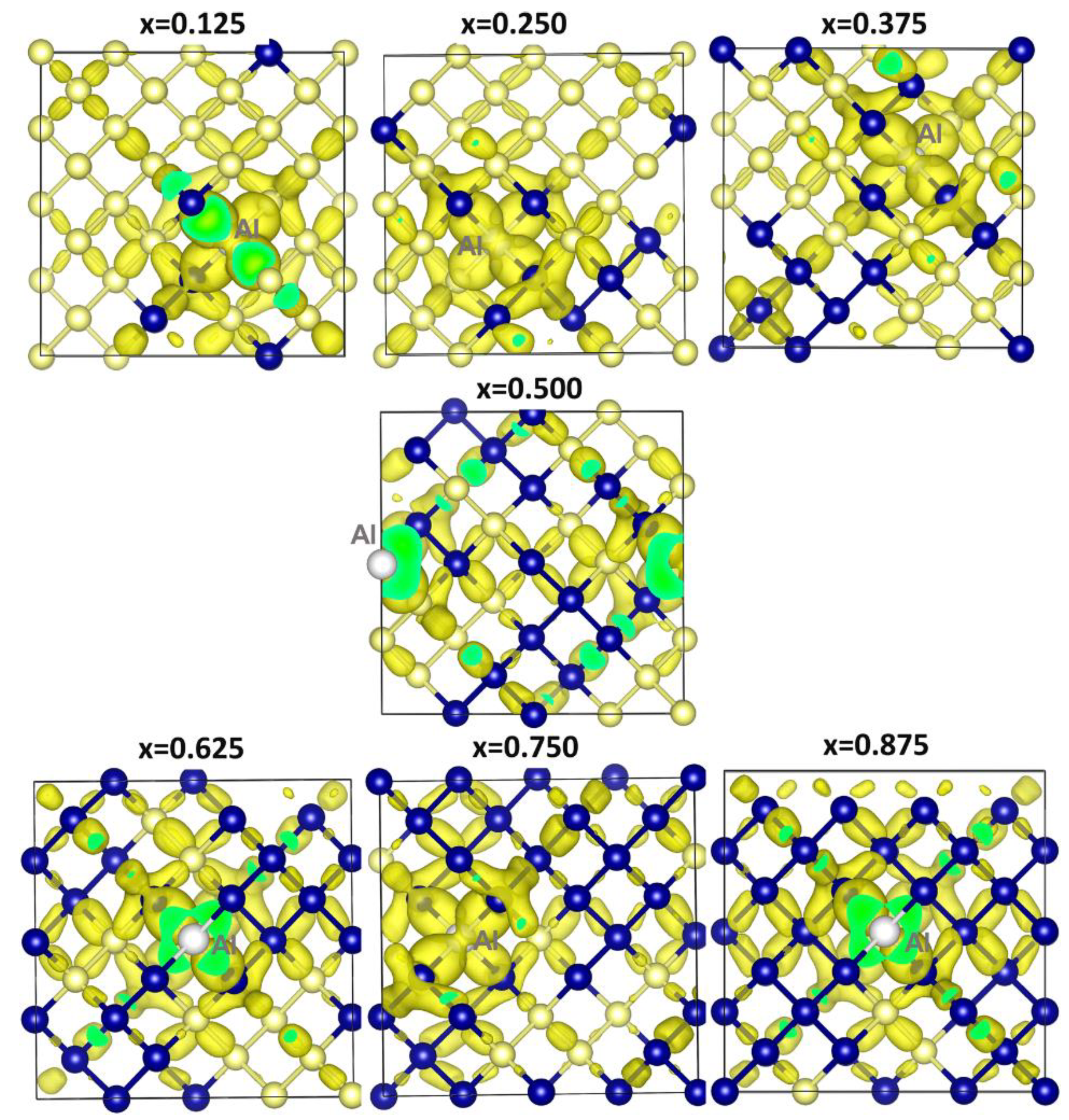{kind=link}
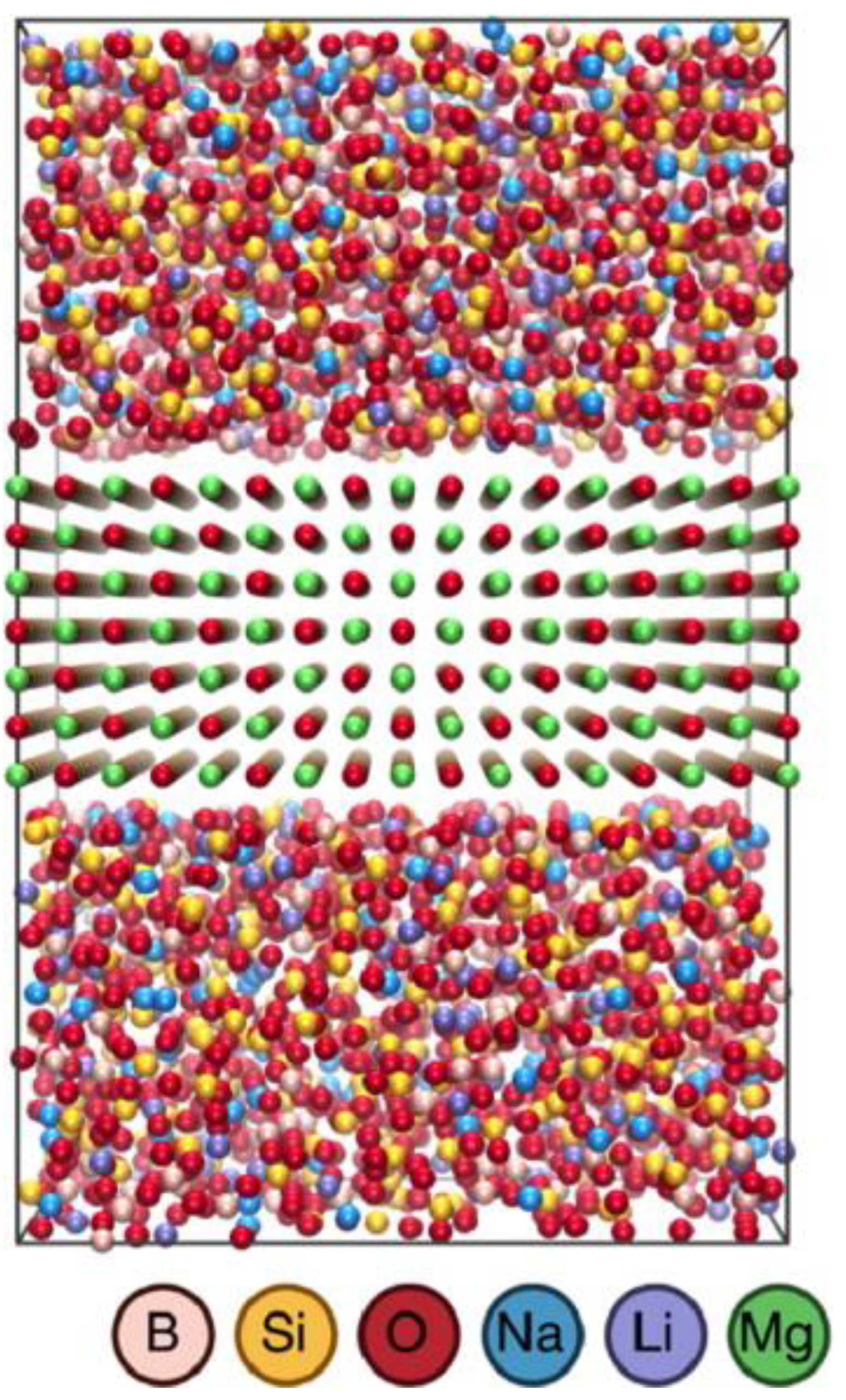{kind=link}
Abstract
1. Introduction
2. Methodology
2.1. Thermodynamic Methodology
2.2. Cluster Expansion and Special Quasirandom Structures
3. Advanced Materials for Energy Applications
4. Oxide Interfaces
5. Machine Learning Approaches for Defect Prediction
6. Summary, Perspective and Future Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Middleburgh, S.C.; Lumpkin, G.R.; Riley, D. Accommodation, accumulation, and migration of defects in Ti3SiC2 and Ti3AlC2 MAX phases. J. Am. Ceram. Soc. 2013, 96, 3196–3201. [Google Scholar] [CrossRef]
- Cooper, M.W.D.; Middleburgh, S.C.; Grimes, R.W. Vacancy mediated cation migration in uranium dioxide: The influence of cluster configuration. Solid State Ionics 2014, 266, 68–72. [Google Scholar] [CrossRef]
- Yang, Y.; Fei, H.; Ruan, G.; Xiang, C.; Tour, J.M. Edge-oriented MoS2 nanoporous films as flexible electrodes for hydrogen evolution reactions and supercapacitor devices. Adv. Mater. 2014, 23, 8163–8168. [Google Scholar] [CrossRef] [PubMed]
- Hadi, M.A.; Vovk, R.V.; Chroneos, A. Physical properties of the recently discovered Zr2(Al1-xBix)C MAX phases. J. Mater. Sci. Mater. Electron. 2016, 27, 11925–11933. [Google Scholar] [CrossRef]
- Duvel, A.; Heitjans, P.; Fedorov, P.; Scholz, G.; Cibin, G.; Chadwick, A.V.; Pickup, D.M.; Ramos, S.; Sayle, L.W.L.; Sayle, E.K.L.; et al. Is geometric frustration-induced disorder a recipe for high ionic conductivity? J. Am. Chem. Soc. 2017, 139, 5842–5848. [Google Scholar] [CrossRef]
- Yun, S.; Zhou, X.; Even, J.; Hagfeldt, A. Theoretical treatment of CH3NH3PbI3 perovskite solar cells. Angew. Chem. 2017, 56, 15806–15817. [Google Scholar] [CrossRef] [PubMed]
- Timerkaeva, D.; Caliste, D.; Deutsch, T.; Pochet, P. Oxygen in silicon: Switch in the diffusion-mediated mechanism. Phys. Rev. B 2017, 96, 195306. [Google Scholar] [CrossRef]
- Dahlqvist, M.; Petruhins, A.; Lu, J.; Hultman, L.; Rosen, J. Origin of chemically ordered atomic laminates (i-MAX): Expanding the elemental space by a theoretical/experimental approach. ACS Nano 2018, 12, 7761–7770. [Google Scholar] [CrossRef]
- Thoda, O.; Xanthopoulou, G.; Vekinis, G.; Chroneos, A. Review of recent studies on solution combustion synthesis of nanostructured catalysts. Adv. Eng. Mater. 2018, 20, 1800047. [Google Scholar] [CrossRef]
- Ning, D.; Baki, A.; Scherb, T.; Song, J.; Fantin, A.; Liu, X.Z.; Schumacher, G.; Banhart, J.; Bouwmeester, H.J.M. Influence of A-site deficiency on structural evolution of Pr2−xNiO4+δ with temperature. Solid State Ionics 2019, 342, 115056. [Google Scholar] [CrossRef]
- Kuganathan, N.; Iyngaran, P.; Vovk, R.; Chroneos, A. Defects, dopants and Mg diffusion in MgTiO3. Sci. Rep. 2019, 9, 4394. [Google Scholar] [CrossRef] [PubMed]
- Grieshammer, S.; Belova, I.V.; Murch, G.E. Thermodiffusion and ion transport in doped ceria by molecular dynamics simulations. Acta Mater. 2021, 210, 116802. [Google Scholar] [CrossRef]
- Shi, J.; Han, C.; Niu, H.; Zhu, Y.; Yun, S. Theoretical investigation of proton diffusion in Dion-Jacobson layered perovskite RbBiNb2O7. Nanomaterials. 2021, 11, 1953. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Xing, Y.; Hu, E.; Wang, J.; Shi, J.; Yun, S.; Zhu, B. PN heterostructure interface-facilitated proton conduction in 3C-SiC/Na0.6CoO2 electrolyte for fuel cell application. ACS Appl. Energy Mater. 2021, 4, 7519–7525. [Google Scholar] [CrossRef]
- Igumbor, E.; Olaniyan, O.; Dongho-Nguimbo, G.M.; Mapasha, R.E.; Ahmad, S. Electronic properties and defect levels induced by n/p-type defect complexes in Ge. Mater. Sci. Semicond. Proc. 2022, 150, 106906. [Google Scholar] [CrossRef]
- Pelenitsyn, V.; Korotaev, P. First-principles study of radiation defects in silicon. Comp. Mater. Sci. 2022, 207, 111273. [Google Scholar] [CrossRef]
- Arshad, A.; Yun, S.; Shi, J.; Sun, M.; Zafar, N.; Hagfeldt, A. N-coordinated bimetallic defect-rich nanocarbons as highly efficient electrocatalysts in advanced energy conversion applications. Chem. Eng. J. 2022, 435, 134913. [Google Scholar] [CrossRef]
- Varley, J.B.; Shen, B.; Higashiwaki, M. Wide bandgap semiconductor materials and devices. J. Appl. Phys. 2022, 131, 230401. [Google Scholar] [CrossRef]
- Hassan, J.Z.; Raza, A.; Qumar, U.; Li, G. Recent advances in engineering strategies of Bi-based photocatalysts for environmental remediation. Sustain. Mater. Technol. 2022, 33, e00478. [Google Scholar] [CrossRef]
- Sivaranjani, P.R.; Janani, B.; Thomas, A.M.; Raju, L.L.; Khan, S.S. Recent development in MoS2 based nano-photocatalyst for the degradation of pharmaceutically active compounds. J. Clean. Prod. 2022, 352, 131506. [Google Scholar] [CrossRef]
- Steele, B.C.H. Appraisal of Ce1-yGdyO2-y/2 electrolytes for IT-SOFC operation at 500 °C. Solid State Ionics 2000, 129, 95–110. [Google Scholar] [CrossRef]
- Singhal, S.C. Advances in solid oxide fuel cell technology. Solid State Ionics 2000, 135, 305–313. [Google Scholar] [CrossRef]
- Sata, N.; Eberman, K.; Eberl, K.; Maier, J. Mesoscopic fast ion conduction in nanometre scale planar heterostructures. Nature 2000, 408, 946–949. [Google Scholar] [CrossRef] [PubMed]
- Steele, B.C.H.; Heinzel, A. Materials for fuel-cell technologies. Nature 2001, 414, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Stanek, C.R.; Sickafus, K.E.; Uberuaga, B.P. First-principles prediction of disordering tendencies in pyrochlore oxides. Phys. Rev. B 2009, 79, 104203. [Google Scholar] [CrossRef]
- Kube, R.; Bracht, H.; Chroneos, A.; Posselt, M.; Schmidt, B. Intrinsic and extrinsic diffusion in germanium. J. Appl. Phys. 2009, 106, 063534. [Google Scholar] [CrossRef]
- Devanathan, R.; Weber, W.J.; Gale, G.D. Radiation tolerance of ceramics- insights from atomistic simulation of damage accumulation in pyrochlores. Energy Environ. Sci. 2010, 3, 1551–1559. [Google Scholar] [CrossRef]
- Horlait, D.; Middleburgh, S.C.; Chroneos, A.; Lee, W.E. Synthesis and DFT investigation of new bismuth-containing MAX phases. Sci. Rep. 2016, 6, 18829. [Google Scholar] [CrossRef]
- Horlait, D.; Grasso, S.; Chroneos, A.; Lee, W.E. Attempts to synthesize quaternary MAX phases (Zr,M)2AlC and Zr2(Al,A)C as a way to approach Zr2AlC. Mater. Res. Lett. 2016, 4, 137–144. [Google Scholar] [CrossRef]
- Zapata-Solvas, E.; Christopoulos, S.R.G.; Ni, N.; Parfitt, D.C.; Horlait, D.; Fitzpatrick, M.E.; Chroneos, A.; Lee, W.E. Experimental synthesis and density functional theory investigation of radiation tolerance of Zr3(Al1−xSix)C2 MAX phases. J. Am. Ceram. Soc. 2017, 100, 1377–1387. [Google Scholar] [CrossRef]
- Zunger, A.; Wei, S.H.; Ferreira, L.G.; Bernard, J.E. Special quasirandom structures. Phys. Rev. Lett. 1990, 65, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Chroneos, A.; Jiang, C.; Grimes, R.W.; Schwingenschlögl, U.; Bracht, H. Defect interactions in Sn1−xGex alloys. Appl. Phys. Lett. 2009, 94, 252104. [Google Scholar] [CrossRef]
- Chroneos, A.; Jiang, C.; Grimes, R.W.; Schwingenschlögl, U.; Bracht, H. E centers in Si1−x−yGexSny alloys. Appl. Phys. Lett. 2009, 95, 112101. [Google Scholar] [CrossRef]
- Christopoulos, S.R.G.; Kuganathan, N.; Chroneos, A. Electronegativity and doping Si1−xGex alloys. Sci. Rep. 2020, 10, 7459. [Google Scholar] [CrossRef]
- Gubaev, K.; Podryabinkin, E.V.; Hart, G.L.W.; Shapeev, A.V. Accelerating high-throughput searches for new alloys with active learning of interatomic potentials. Comp. Mater. Sci. 2019, 156, 148–156. [Google Scholar] [CrossRef]
- Varotsos, P. Calculation of the migration volume of vacancies in ionic solids from macroscopic parameters. Phys. Stat. Sol. 1978, 47, K133–K136. [Google Scholar] [CrossRef]
- Varotsos, P.; Alexopoulos, K. Thermodynamics of Point Defects and Their Relation with the Bulk Properties; Elsevier: Amsterdam, The Netherlands, 1986. [Google Scholar]
- Zhang, B.; Wu, X.; Xu, J.; Zhou, R. Application of the cBΩ model for the calculation of oxygen self-diffusion coefficients in minerals. J. Appl. Phys. 2010, 108, 053505. [Google Scholar] [CrossRef]
- Vallianatos, F.; Saltas, V. Application of the cBΩ model to the calculation of diffusion parameters of He in olivine. Phys. Chem. Miner. 2014, 41, 181–188. [Google Scholar] [CrossRef]
- Cooper, M.W.D.; Grimes, R.W.; Fitzpatrick, M.E.; Chroneos, A. Modeling oxygen self-diffusion in UO2 under pressure. Solid State Ionics 2015, 282, 26–30. [Google Scholar] [CrossRef]
- Zhang, B.; Shan, S. Application of the cBΩ model to the calculation of diffusion parameters of Si in silicates. Geochem. Geophys. Geosyst. 2015, 16, 705–718. [Google Scholar] [CrossRef]
- Chroneos, A.; Vovk, R.V. Modeling self-diffusion in UO2 and ThO2 by connecting point defect parameters with bulk properties. Solid State Ionics 2015, 274, 1–3. [Google Scholar] [CrossRef]
- Parfitt, D.C.; Cooper, M.W.D.; Rushton, M.J.D.; Christopoulos, S.-R.G.; Fitzpatrick, M.E.; Chroneos, A. Thermodynamic calculations of oxygen self-diffusion in mixed-oxide nuclear fuels. RSC Adv. 2016, 6, 74018–74028. [Google Scholar] [CrossRef]
- Saltas, V.; Chroneos, A.; Vallianatos, F.A. A thermodynamic approach of self- and hetero-diffusion in GaAs: Connecting point defect parameters with bulk properties. RSC Adv. 2016, 6, 53324–53330. [Google Scholar] [CrossRef]
- Varotsos, P. Comparison of models that interconnect point defect parameters in solids with bulk properties. J. Appl. Phys. 2007, 101, 123503. [Google Scholar] [CrossRef]
- Varotsos, P. Point defect parameters in β-PbF2 revisited. Solid State Ionics 2008, 179, 438–441. [Google Scholar] [CrossRef]
- Chroneos, A. Connecting point defect parameters with bulk properties to describe diffusion in solids. Appl. Phys. Rev. 2016, 3, 041304. [Google Scholar] [CrossRef]
- Saltas, V.; Chroneos, A.; Vallianatos, F.A. A thermodynamic approach to self-diffusion in silicon: Evidence of a single diffusion mechanism? Mater. Chem. Phys. 2016, 181, 204–208. [Google Scholar] [CrossRef]
- Sarlis, N.V.; Skordas, E.S. Estimating the compressibility of osmium from recent measurements of Ir-Os alloys under high pressure. J. Phys. Chem. A 2016, 120, 1601–1604. [Google Scholar] [CrossRef]
- Saltas, V.; Chroneos, A.; Vallianatos, F.A. Composition and temperature dependence of self-diffusion in Si1−xGex alloys. Sci. Rep. 2017, 7, 1374. [Google Scholar] [CrossRef] [PubMed]
- Skordas, E.S.; Sarlis, N.V.; Varotsos, P.A. Applying the cBΩ thermodynamical model to LiF using its equation of state obtained from high pressure diamond anvil cell measurements. Solid State Ionics 2020, 354, 115404. [Google Scholar] [CrossRef]
- Connolly, J.W.D.; Williams, A.R. Density functional theory applied to phase transformations in transition metal alloys. Phys. Rev. B 1983, 27, 5169–5172. [Google Scholar] [CrossRef]
- Sanchez, J.M.; Ducastelle, F.; Gratias, D. Generalized cluster description of multicomponent systems. Physica A 1984, 128, 334–350. [Google Scholar] [CrossRef]
- Laks, D.B.; Ferreira, L.G.; Froyen, S.; Zunger, A. Efficient cluster expansion for substitutional systems. Phys. Rev. B 1992, 46, 12587–12605. [Google Scholar] [CrossRef]
- Wolverton, C.; Zunger, A. Ising-like description of structurally released ordered and disordered alloys. Phys. Rev. Lett. 1995, 75, 3162–3165. [Google Scholar] [CrossRef]
- Zunger, A.; Wang, L.G.; Hart, G.L.W.; Sanati, M. Obtaining Ising-like expansions for binary alloys from first principles. Model. Simul. Mater. Sci. Eng. 2002, 10, 685–706. [Google Scholar] [CrossRef]
- Jiang, C.; Sordelet, D.J.; Gleeson, B. First-principles study of phase stability in pseudobinary (Ni1−xPtx)3Al. Phys. Rev. B 2005, 72, 184203. [Google Scholar] [CrossRef]
- Chroneos, A.; Rushton, M.J.D.; Jiang, C.; Tsoukalas, L.H. Nuclear wasteform materials: Atomistic simulation case studies. J. Nucl. Mater. 2013, 441, 29–39. [Google Scholar] [CrossRef]
- Wei, S.H.; Ferreira, L.G.; Bernard, J.E.; Zunger, A. Electronic properties of random alloys: Special quasirandom structures. Phys. Rev. B 1990, 42, 9622–9649. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Wolverton, C.; Sofo, J.; Chen, L.Q.; Liu, Z.K. First-principles study of binary bcc alloys using special quasirandom structures. Phys. Rev. B 2004, 69, 214202. [Google Scholar] [CrossRef]
- Murphy, S.T.; Chroneos, A.; Jiang, C.; Schwingenschlögl, U.; Grimes, R.W. Deviations from Vegard’s law in ternary III-V alloys. Phys. Rev. B 2010, 82, 073201. [Google Scholar] [CrossRef]
- Murphy, S.T.; Chroneos, A.; Grimes, R.W.; Jiang, C.; Schwingenschlögl, U. Phase stability and the arsenic vacancy defect in InxGa1−xAs. Phys. Rev. B 2011, 84, 184108. [Google Scholar] [CrossRef]
- Wang, H.; Chroneos, A.; Jiang, C.; Schwingenschlögl, U. Modelling zirconium hydrides using the special quasirandom structure approach. Phys. Chem. Chem. Phys. 2013, 15, 7599–7603. [Google Scholar] [CrossRef] [PubMed]
- Saltas, V.; Vallianatos, F. Silicon self-diffusion in Stishovite: Calculations of point defect parameters based on the cBΩ thermodynamic model. Environ. Sci. Proc. 2021, 6, 6. [Google Scholar]
- Chroneos, A.; Bracht, H.; Jiang, C.; Uberuaga, B.P.; Grimes, R.W. Nonlinear stability of E centers in Si1-xGex: Electronic structure calculations. Phys. Rev. B 2008, 78, 195201. [Google Scholar] [CrossRef]
- Lumley, S.C.; Grimes, R.W.; Murphy, S.T.; Burr, P.A.; Chroneos, A.; Chard-Tuckey, P.R.; Wenman, M.R. The thermodynamics of hydride precipitation: The importance of entropy, enthalpy and disorder. Acta Mater. 2014, 79, 351–362. [Google Scholar] [CrossRef]
- Wang, H.; Chroneos, A.; Jiang, C.; Schwingenschlögl, U. Special quasirandom structures for gadolinia-doped ceria and related materials. Phys. Chem. Chem. Phys. 2012, 14, 11737–11742. [Google Scholar] [CrossRef][Green Version]
- Chroneos, A.; Jiang, C.; Grimes, R.W.; Schwingenschlögl, U. Special quasirandom structures for binary/ternary group IV alloys. Chem. Phys. Lett. 2010, 493, 97–102. [Google Scholar] [CrossRef]
- Sarlis, N.V.; Skordas, E.S. Pressure and temperature dependence of the oxygen self-diffusion activation volume in UO2 by a thermodynamical model. Solid State Ionics 2016, 290, 121–123. [Google Scholar] [CrossRef]
- Saltas, V.; Chroneos, A.; Cooper, M.W.D.; Fitzpatrick, M.E.; Vallianatos, F. Investigation of oxygen self-diffusion in PuO2 by combining molecular dynamics with thermodynamic calculations. RSC Adv. 2016, 6, 103641–103649. [Google Scholar] [CrossRef]
- Saltas, V.; Horlait, D.; Sgourou, E.N.; Vallianatos, F.; Chroneos, A. Modelling solid solutions with cluster expansion, special quasirandom structures, and thermodynamic approaches. Appl. Phys. Rev. 2017, 4, 41301. [Google Scholar] [CrossRef]
- Varotsos, P.; Sarlis, N.; Lazaridou, M. Interconnection of defect parameters and stress-induced electric signals in ionic crystals. Phys. Rev. B 1999, 59, 24–27. [Google Scholar] [CrossRef]
- Zhang, B.; Xu, J. Thermodynamic estimation of the compressibility of ferropericlase under high pressure. AIP Adv. 2016, 6, 115112. [Google Scholar] [CrossRef]
- Magomedov, M. Dependence of the parameters of vacancy formation and self-diffusion in a single-component crystal on temperature and pressure. J. Phys. Chem. Solids 2022, 165, 110653. [Google Scholar] [CrossRef]
- Dahlqvist, M.; Rosen, J. The rise of MAX phase alloys—large-scale theoretical screening for the prediction of chemical order and disorder. Nanoscale 2022, 14, 10958. [Google Scholar] [CrossRef] [PubMed]
- Christopoulos, S.R.G.; Kordatos, A.; Cooper, M.W.D.; Fitzpatrick, M.E.; Chroneos, A. Activation volumes of oxygen self-diffusion in fluorite structured oxides. Mater. Res. Express 2016, 3, 105504. [Google Scholar] [CrossRef]
- Sarlis, N.V.; Skordas, E.S. Bulk moduli of PbSxSe1-x, PbSxTe1-x, and PbSexTe1-x from the combination of the cBΩ model with the modified Born theory compared to generalized gradient approximation. Mod. Phys. Lett. B 2016, 30, 1650409. [Google Scholar] [CrossRef]
- Varotsos, P.A.; Sarlis, N.V.; Skordas, E.S. Thermodynamics of point defects in solids and relation with the bulk properties: Recent results. Crystals 2022, 12, 686. [Google Scholar] [CrossRef]
- Lafaye, P.; Toffolon-Masclet, C.; Crivello, J.C.; Joubert, J.M. Experimental study, first-principles calculation and thermodynamic modelling of the Cr-Fe-Nb-Sn-Zr quinary system for application as cladding materials in nuclear reactors. J. Nucl. Mater. 2021, 544, 152692. [Google Scholar] [CrossRef]
- Sarlis, N.V.; Skordas, E.S. Interconnection of a thermodynamical model for point defect parameters in solids with the dynamical theory of diffusion. Solid State Ionics 2019, 335, 82–85. [Google Scholar] [CrossRef]
- Saltas, V.; Chroneos, A.; Vallianatos, F. Mg diffusion in Si on a thermodynamic basis. J. Mater. Sci. Mater. Electron. 2018, 29, 12022–12027. [Google Scholar] [CrossRef]
- Kumar, V.; Di Stefano, D.; Rignanese, G.M.; Gonze, X. Li diffusion in Si and LiSi: Nuclear quantum effects and anharmonisity. J. Chem. Phys. 2020, 152, 244101. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Bridges, C.A.; Unocic, R.R.; Pitike, K.C.; Cooper, V.R.; Zhang, Y.P.; Lin, D.Y.; Page, K. Probing the local site disorder and distortion in pyrochlore high-entropy oxides. J. Am. Chem. Soc. 2021, 143, 4193–4204. [Google Scholar] [CrossRef] [PubMed]
- Vovk, R.V.; Obolenskii, M.A.; Zavgorodniy, A.A.; Goulatis, I.L.; Beleskii, V.I.; Chroneos, A. Structural relaxation, metal to insulator transition and pseudo-gap in oxygen deficient HoBa2Cu3O7-δ single crystals. Physica C 2009, 469, 203–206. [Google Scholar] [CrossRef]
- Vovk, R.V.; Vovk, N.R.; Shekhovtsov, O.V.; Goulatis, I.L.; Chroneos, A. c-axis hopping conductivity in heavily Pr-doped YBCO single crystals. Semicond. Sci. Technol. 2013, 26, 085017. [Google Scholar] [CrossRef]
- Rushton, M.J.D.; Chroneos, A.; Skinner, S.J.; Kilner, J.A.; Grimes, R.W. Effect of strain on the oxygen diffusion in yttria and gadolinia co-doped ceria. Solid State Ionics 2013, 230, 37–42. [Google Scholar] [CrossRef]
- Rushton, M.J.D.; Chroneos, A. Impact of uniaxial strain and doping on oxygen diffusion in CeO2. Sci. Rep. 2014, 4, 6068. [Google Scholar] [CrossRef]
- Chroneos, A.; Sgourou, E.N.; Londos, C.A.; Schwingenschlögl, U. Oxygen defect processes in silicon and silicon germanium. Appl. Phys. Rev. 2015, 2, 021306. [Google Scholar] [CrossRef]
- Zhu, J.; Vasilopoulou, M.; Davazoglou, D.; Kennou, S.; Chroneos, A.; Schwingenschlögl, U. Intrinsic defects and H doping in WO3. Sci. Rep. 2017, 7, 40882. [Google Scholar] [CrossRef]
- Jiang, C.; Chroneos, A. Ab initio modelling of MAX phase solid solutions using the special quasirandom structure approach. Phys. Chem. Chem. Phys. 2018, 20, 1173–1180. [Google Scholar] [CrossRef]
- Ewing, R.C. Nuclear waste forms for actinides. Proc. Natl. Acad. Sci. USA 1999, 96, 3432–3439. [Google Scholar] [CrossRef]
- Sickafus, K.E.; Minervini, L.; Grimes, R.W.; Valdez, J.A.; Ishimaru, M.; Li, F.; McClellan, K.J.; Hartmann, T. Radiation tolerance of complex oxides. Science 2000, 289, 748–751. [Google Scholar] [CrossRef] [PubMed]
- Grimes, R.W.; Konings, R.J.M.; Edwards, L. Greater tolerance for nuclear materials. Nat. Mater. 2008, 7, 683–685. [Google Scholar] [CrossRef] [PubMed]
- Grimes, R.W.; Nuttal, W.J. Generating the option of a two-stage nuclear renaissance. Science 2010, 329, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Armaroli, N.; Balzani, V. Towards an electricity-powered world. Energy Environ. Sci. 2011, 4, 3193–3322. [Google Scholar] [CrossRef]
- Garcia-Barriocanal, J.; Rivera-Calzada, A.; Varela, M.; Sefrioui, Z.; Iborra, E.; Leon, C.; Pennycook, S.J.; Santamaria, J. Colossal ionic conductivity at interfaces of epitaxial ZrO2:Y2O3/SrTiO3 heterostructures. Science 2008, 321, 676–680. [Google Scholar] [CrossRef]
- Kilner, J.A. Ionic conductors: Feel the strain. Nat. Mater. 2008, 7, 838. [Google Scholar] [CrossRef]
- Guo, X. Comment on “Colossal ionic conductivity at interfaces of epitaxial ZrO2:Y2O3/SrTiO3 heterostructures”. Science 2009, 324, 5926. [Google Scholar] [CrossRef]
- Schichtel, N.; Korte, C.; Hesse, D.; Janek, J. Elastic strain at interfaces and its influence on ionic conductivity in nanoscaled solid electrolyte thin films- theoretical considerations and experimental studies. Phys. Chem. Chem. Phys. 2009, 11, 3043–3048. [Google Scholar] [CrossRef]
- Kushima, A.; Yildiz, B. Oxygen ion diffusivity in strained yttria stabilized zirconia: Where is the fastest strain? J. Mater. Chem. 2010, 20, 4809–4819. [Google Scholar] [CrossRef]
- Chroneos, A.; Yildiz, B.; Tarancόn, A.; Parfitt, D.; Kilner, J.A. Oxygen diffusion in solid oxide fuel cell cathode and electrolyte materials: Mechanistic insights from atomistic simulations. Energy Environ. Sci. 2011, 4, 2774–2789. [Google Scholar] [CrossRef]
- Cavallaro, A.; Burriel, M.; Roqueta, J.; Apostolidis, A.; Bernardi, A.; Tarancόn, A.; Srinivasan, R.; Cook, S.N.; Fraser, H.L.; Kilner, J.A.; et al. Electronic nature of the enhanced conductivity in YSZ-STO multilayers deposited by PLD. Solid State Ionics 2010, 181, 592–601. [Google Scholar] [CrossRef]
- Pennycook, T.J.; Beck, M.J.; Varga, K.; Varela, M.; Pennycook, S.J.; Pantelides, S.T. Origin of colossal ionic conductivity in oxide multilayers: Interface induced sublattice disorder. Phys. Rev. Lett. 2010, 104, 115901. [Google Scholar] [CrossRef] [PubMed]
- De Souza, R.A.; Ramadan, A.; Hörner, S. Modifying the barriers for oxygen-vacancy migration in fluorite-structured CeO2 electrolytes through strain; A computer simulation study. Energy Environ. Sci. 2012, 5, 5445–5453. [Google Scholar] [CrossRef]
- Korte, C.; Schichtel, N.; Hesse, D.; Janek, J. Influence of interface structure on mass transport in phase boundaries between different ionic materials: Experimental studies and formal considerations. Monatsh. Chem. 2009, 140, 1069–1080. [Google Scholar] [CrossRef]
- Chiabrera, F.; Garbayo, I.; Lopez-Conesa, L.; Martin, G.; Ruiz-Caridad, A.; Walls, M.; Ruiz-Gonzalez, L.; Kordatos, A.; Nunez, M.; Morata, A.; et al. Engineering transport in manganites by tuning local nonstoichiometry in grain boundaries. Adv. Mater. 2019, 31, 1805360. [Google Scholar] [CrossRef]
- Acosta, M.; Baiutti, F.; Tarancon, A.; MacManus-Driscoll, J.L. Nanostructured materials and interfaces for advanced ionic electronic conducting oxides. Adv. Mater. Interfaces 2019, 6, 1–15. [Google Scholar] [CrossRef]
- Lee, D.; Gao, X.; Sun, L.; Jee, Y.; Poplawsky, J.; Farmer, T.O.; Fan, L.; Guo, E.-J.; Lu, Q.; Heller, W.T.; et al. Colossal oxygen vacancy formation at a fluorite-bixbyite interface. Nat. Commun. 2021, 11, 1371. [Google Scholar] [CrossRef]
- Baiutti, F.; Chiabrera, F.; Acosta, M.; Diercks, D.; Parfitt, D.; Santiso, J.; Wang, X.; Cavallaro, A.; Morata, A.; Wang, H.; et al. A high-entropy manganite in an ordered nanocomposite for long-term application in solid oxide cells. Nat. Commun. 2021, 12, 2660. [Google Scholar] [CrossRef]
- Kuganathan, N.; Baiutti, F.; Tarancon, A.; Fleig, J.; Chroneos, A. Defects energetics in the SrTiO3-LaCrO3 system. Solid State Ionics 2021, 361, 115570. [Google Scholar] [CrossRef]
- Navickas, E.; Huber, T.M.; Chen, Y.; Hetaba, W.; Holzlechner, G.; Rupp, G.; Stöger-Pollach, M.; Friedbacher, G.; Hutter, H.; Yildiz, B.; et al. Fast oxygen exchange and diffusion kinetics of grain boundaries in Sr-doped LaMnO3 thin films. Phys. Chem. Chem. Phys. 2015, 17, 7659–7669. [Google Scholar] [CrossRef]
- Saranya, A.M.; Pla, D.; Morata, A.; Cavallaro, A.; Canales-Vazquez, J.; Kilner, J.A.; Burriel, M.; Tarancon, A. Engineering mixed ionic electronic conduction in La0.8Sr0.2MnO3+δ nanostructures through fast grain boundary oxygen diffusivity. Adv. Energy Mater. 2015, 5, 1500377. [Google Scholar] [CrossRef]
- Saranya, A.M.; Morata, A.; Pla, D.; Burriel, M.; Chiabrera, F.; Garbayo, I.; Hornes, A.; Kilner, J.A.; Tarancon, A. Unveiling the outstanding oxygen mass transport properties of Mn-rich perovskites in grain boundary-dominated La0.8Sr0.2(Mn1−xCox)0.85O3±δ nanostructures. Chem. Mater. 2018, 30, 5621–5629. [Google Scholar] [CrossRef]
- Sun, L.; Marrocchelli, D.; Yildiz, B. Edge dislocation slows down oxide ion diffusion in doped CeO2 by segregation of charged defects. Nat. Commun. 2015, 6, 6294. [Google Scholar] [CrossRef] [PubMed]
- Marrocchelli, D.; Sun, L.; Yildiz, B. Dislocation in SrTiO3: Easy to reduce but not so fast for oxygen transport. J. Am. Chem. Soc. 2015, 137, 4735–4748. [Google Scholar] [CrossRef] [PubMed]
- Solovjov, A.L.; Tkachenko, M.A.; Vovk, R.V.; Chroneos, A. Fluctuation conductivity and pseudogap in HoBa2Cu3O7-δ single crystals under pressure with transport current flowing under an angle 45° to the twin boundaries. Physica C 2014, 501, 24–31. [Google Scholar] [CrossRef]
- Vovk, R.V.; Nazyrov, Z.F.; Obolenskii, M.A.; Goulatis, I.L.; Chroneos, A.; Simoes, V.M.P. Phase separation in oxygen deficient HoBa2Cu3O7-δ single crystals: Effect of pressure and twin boundaries. Philos. Mag. 2011, 91, 2291–2302. [Google Scholar] [CrossRef]
- Vovk, R.V.; Obolenskii, M.A.; Zavgorodniy, A.A.; Bondarenko, A.V.; Goulatis, I.L.; Samoilov, A.V.; Chroneos, A. Effect of high pressure on the fluctuation conductivity and the charge transfer of YBa2Cu3O7-δ single crystals. J. Alloys Compds. 2008, 453, 69–74. [Google Scholar] [CrossRef]
- Vovk, R.V.; Obolenskii, M.A.; Nazyrov, Z.F.; Goulatis, I.L.; Chroneos, A.; Simoes, V.M.P. Electro-transport and structure of 1-2-3 HTSC single crystals with different plane defect topologies. J. Mater. Sci. Mater. Electron. 2012, 23, 1255–1259. [Google Scholar] [CrossRef]
- Solovjov, A.L.; Petrenko, E.V.; Omelchenko, L.V.; Vovk, R.V.; Goulatis, I.L.; Chroneos, A. Effect of annealing on a pseudogap state in untwinned YBa2Cu3O7-δ single crystals. Sci. Rep. 2019, 9, 9274. [Google Scholar] [CrossRef]
- Seymour, I.D.; Chroneos, A.; Kilner, J.A.; Grimes, R.W. Defect processes in orthorhombic LnBaCo2O5.5 double perovskites. Phys. Chem. Chem. Phys. 2011, 13, 15305–15310. [Google Scholar] [CrossRef]
- Yildiz, B. “Stretching” the energy landscape of oxides- Effects on eletrocatalysis and diffusion. MRS Bull. 2014, 39, 147–156. [Google Scholar] [CrossRef]
- Ma, W.; Kim, J.J.; Tsvetkov, N.; Daio, T.; Kuru, Y.; Cai, Z.; Chen, Y.; Sasaki, K.; Tuller, H.L.; Yildiz, B. Vertically aligned nanocomposite La0.8Sr0.2CoO3/(La0.5Sr0.5)2CoO4 cathodes—electronic structure, surface chemistry and oxygen reduction kinetics. J. Mater. Chem. A 2015, 3, 207–219. [Google Scholar] [CrossRef]
- Jay, E.E.; Rushton, M.J.D.; Chroneos, A.; Grimes, R.W.; Kilner, J.A. Genetics of superionic conductivity in lithium lanthanum titanates. Phys. Chem. Chem. Phys. 2015, 17, 178–183. [Google Scholar] [CrossRef]
- Kuganathan, N.; Rushton, M.J.D.; Grimes, R.W.; Kilner, J.A.; Gkanas, E.I.; Chroneos, A. Self-diffusion in garnet-type Li7La3Zr2O12 solid electrolytes. Sci. Rep. 2021, 11, 451. [Google Scholar] [CrossRef] [PubMed]
- Kuganathan, N.; Chroneos, A. Defects, diffusion, dopants and encapsulation of Na in NaZr2(PO4)3. Materialia 2021, 16, 101039. [Google Scholar] [CrossRef]
- Kuganathan, N.; Chroneos, A. Defects, dopants and sodium mobility in Na2MnSiO4. Sci. Rep. 2018, 8, 14669. [Google Scholar] [CrossRef] [PubMed]
- Tsuruaka, T.; Tsujita, T.; Su, J.; Nishitani, Y.; Hamamura, T.; Inatomi, Y.; Nakura, K.; Terabe, K. Fabrication of a magnesium-ion conducting magnesium phosphate electrolyte film using atomic layer deposition. Jpn. J. Appl. Phys. 2020, 59, SIIG08. [Google Scholar] [CrossRef]
- Kuganathan, N.; Davazoglou, K.; Chroneos, A. Computer modelling investigation of MgV2O4 for Mg-ion batteries. J. Appl. Phys. 2020, 127, 035106. [Google Scholar] [CrossRef]
- Hasa, I.; Mariyappan, S.; Saurel, D.; Adelhelm, P.; Koposov, A.Y.; Masquelier, C.; Croguennec, L.; Casas-Cabanas, M. Challenges of today for Na-based batteries of the future: From materials to cell metrics. J. Power Sources 2021, 482, 228872. [Google Scholar] [CrossRef]
- Liu, R.Q.; Kumar, A.; Chen, Z.Z.; Agrawal, A.; Sundararaghavan, V.; Choudhary, A. A predictive machine learning approach for microstructure optimization and materials design. Sci. Rep. 2015, 5, 22552. [Google Scholar] [CrossRef]
- Bouchard, P.J. Validated residual stress profiles for fracture assessments of stainless steel pipe girth welds. Int. J. Press. Vessels Pip. 2007, 84, 195–222. [Google Scholar] [CrossRef]
- Wei, J.; Chu, X.; Sun, X.Y.; Xu, K.; Deng, H.X.; Chen, J.G.; Wei, Z.M.; Lei, M. Machine learning in materials science. InfoMat 2019, 1, 338–358. [Google Scholar] [CrossRef]
- Gubernatis, J.E.; Lookman, T. Machine learning in materials design and discovery: Examples from the present and suggestions for the future. Phys. Rev. Mater. 2018, 2, 12301–123016. [Google Scholar] [CrossRef]
- Mathew, J.; Parfitt, D.; Wilford, K.; Riddle, N.; Alamaniotis, M.; Chroneos, A.; Fitzpatrick, M.E. Reactor pressure vessel embrittlement: Insights from neural network modelling. J. Nucl. Mater. 2018, 502, 311–322. [Google Scholar] [CrossRef]
- Pace, J.V.; Rosseel, T.M.; Wang, J.A. US NRC Embrittlement Data Base. 1999. Available online: http://www.osti.gov/scitech/biblio/14353 (accessed on 25 August 2022).
- Rosenblatt, F. The perceptron: A probabilistic model for information storage and organization in the brain. Psychol. Rev. 1958, 65, 386–408. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lei, G.; Bramerdorfer, G.; Peng, S.; Sun, X.; Zhu, J. Machine learning for design optimization of electromagnetic devices: Recent developments and future directions. Appl. Sci. 2021, 11, 1627. [Google Scholar] [CrossRef]
- Alamaniotis, M.; Mathew, J.; Chroneos, A.; Fitzpatrick, M.; Tsoukalas, L. Probabilistic kernel machines for predictive monitoring of weld residual stress in energy systems. Eng. Appl. Artif. Intell. 2018, 71, 138–154. [Google Scholar] [CrossRef]
- Odette, G.R.; Lucas, G.E.; Klingensmith, D.; Wirth, B.D.; Gragg, D. The Effects of Composition and Heat Treatment on Hardening and Embrittlement of Reactor Pressure Vessel Steels. [Internet], NUREG/CR-6778; 2003. Available online: http://www.nrc.gov/docs/ML0317/ML031700122.pdf (accessed on 25 August 2022).
- Mathew, J.; Griffin, J.; Alamaniotis, M.; Kanarachos, S.; Fitzpatrick, M.E. Prediction of welding residual stresses using machine learning: Comparison between neural networks and neuro-fuzzy systems. Appl. Soft Comp. 2018, 70, 131–146. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sgourou, E.N.; Daskalopulu, A.; Goulatis, I.; Panayiotatos, Y.; Solovjov, A.L.; Vovk, R.V.; Chroneos, A. Modelling the Defect Processes of Materials for Energy Applications. Appl. Sci. 2022, 12, 9872. https://doi.org/10.3390/app12199872
Sgourou EN, Daskalopulu A, Goulatis I, Panayiotatos Y, Solovjov AL, Vovk RV, Chroneos A. Modelling the Defect Processes of Materials for Energy Applications. Applied Sciences. 2022; 12(19):9872. https://doi.org/10.3390/app12199872
Chicago/Turabian StyleSgourou, Efstratia N., Aspassia Daskalopulu, Ioannis Goulatis, Yerassimos Panayiotatos, Andrei L. Solovjov, Ruslan V. Vovk, and Alexander Chroneos. 2022. "Modelling the Defect Processes of Materials for Energy Applications" Applied Sciences 12, no. 19: 9872. https://doi.org/10.3390/app12199872
APA StyleSgourou, E. N., Daskalopulu, A., Goulatis, I., Panayiotatos, Y., Solovjov, A. L., Vovk, R. V., & Chroneos, A. (2022). Modelling the Defect Processes of Materials for Energy Applications. Applied Sciences, 12(19), 9872. https://doi.org/10.3390/app12199872