
Comparison of Methods to Identify and Monitor Mold Damages in Buildings

 ,
, 
Abstract
Featured Application
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. Morphological Identification of Material-Colonizing Fungi
2.3. Fungal DNA Metabarcoding
2.4. Bioinformatics Analyses
2.5. Statistical Analyses
3. Results
3.1. Mold vs. Dust Mycobiota by DNA Metabarcoding
3.2. Fungi Growing on Building Materials Identified by Microscopy
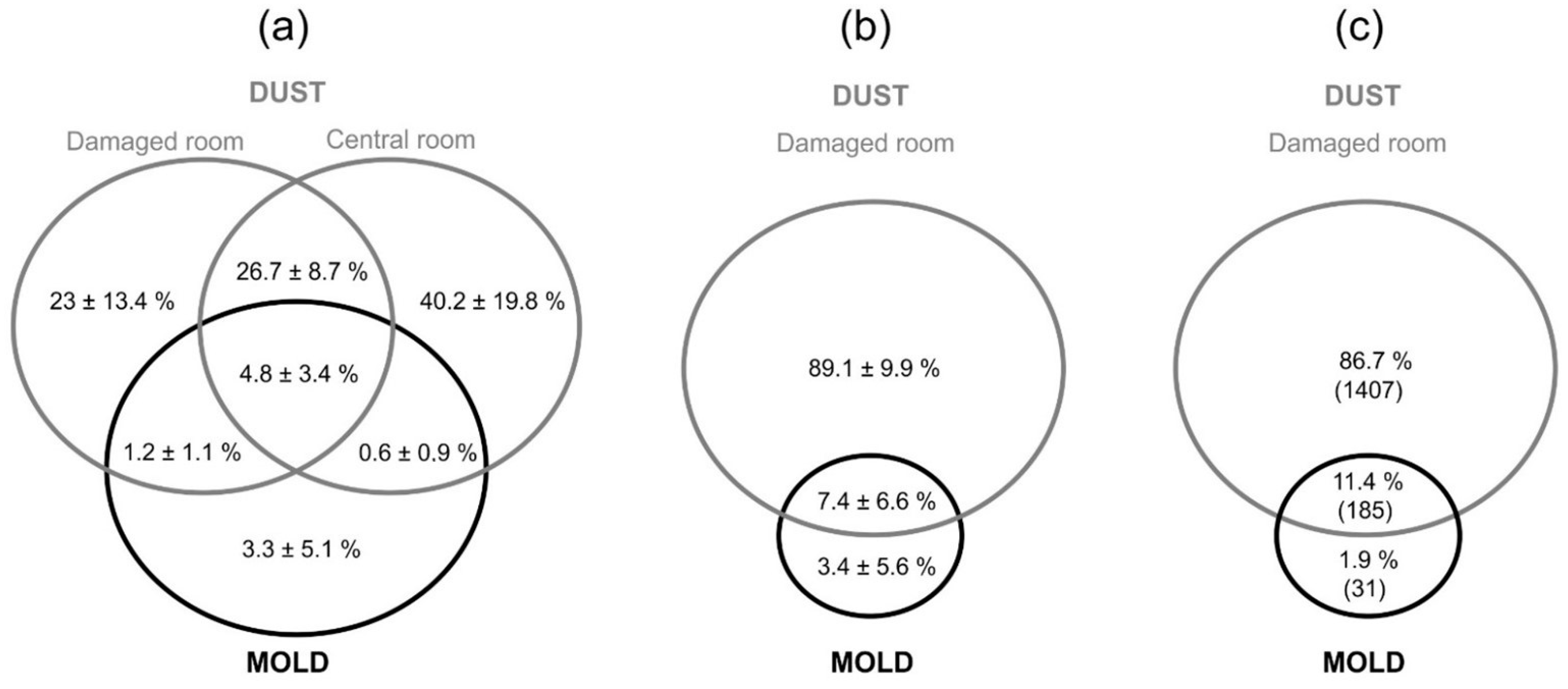
3.3. Dispersal of Fungi in the Apartments as Assesed by DNA Metabarcoding
4. Discussion
4.1. Moisture-Damage Indicator Fungi
4.2. Pros and Cons of Different Methods: Microscopy vs. DNA Metabarcoding
4.3. Contribution of Dust DNA Analyses for Assessment of Indoor Moisture Problems
4.4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization, Regional Office for Europe. WHO Guidelines for Indoor Air Quality: Dampness and Mould; WHO, Regional Office for Europe: Copenhagen, Denmark, 2009; Available online: https://apps.who.int/iris/handle/10665/164348 (accessed on 14 September 2022).
- Mendell, M.J.; Mirer, A.G.; Cheung, K.; Tong, M.; Douwes, J. Respiratory and allergic health effects of dampness, mold, and dampness-related agents: A review of the epidemiologic evidence. Environ. Health Perspect. 2011, 119, 748–756. [Google Scholar] [CrossRef] [PubMed]
- National Academies of Sciences, Engineering, and Medicine. Microbiomes of the Built Environment: A Research Agenda for Indoor Microbiology, Human Health, and Buildings; The National Academies Press: Washington, DC, USA, 2017. [Google Scholar] [CrossRef]
- Hyvärinen, A.; Meklin, T.; Vepsäläinen, A.; Nevalainen, A. Fungi and actinobacteria in moisture-damaged building materials: Concentrations and diversity. Int. Biodeterior. Biodegrad. 2002, 49, 27–37. [Google Scholar] [CrossRef]
- Andersen, B.; Frisvad, J.C.; Søndergaard, I.; Rasmussen, I.S.; Larsen, L.S. Associations between fungal species and water-damaged building materials. Appl. Environ. Microbiol. 2011, 77, 4180–4188. [Google Scholar] [CrossRef]
- Verdier, T.; Coutand, M.; Bertron, A.; Roques, C. A review of indoor microbial growth across building materials and sampling and analysis methods. Build. Environ. 2014, 80, 136–149. [Google Scholar] [CrossRef]
- Dedesko, S.; Siegel, J.A. Moisture parameters and fungal communities associated with gypsum drywall in buildings. Microbiome 2015, 3, 71. [Google Scholar] [CrossRef] [PubMed]
- Coombs, K.; Vesper, S.; Green, B.J.; Yermakov, M.; Reponen, T. Fungal microbiomes associated with green and non-green building materials. Int. Biodeterior. Biodegrad. 2017, 125, 251–257. [Google Scholar] [CrossRef]
- Horisawa, S.; Yoshida, M.; Umezawa, K.; Wada, T.; Abe, H.; Doi, S.; Samejima, M.; Momohara, I. Diversity and community structure of wood-inhabiting fungi found in Japanese wooden houses analyzed by the next-generation sequencing. J. Wood Sci. 2017, 63, 369–378. [Google Scholar] [CrossRef]
- Viitanen, H. Moisture and bio-deterioration risk of building materials and structures. In Mass Transfer-Advanced Aspects; Nakajima, H., Ed.; InTech Europe: Rijeka, Croatia, 2011; pp. 579–594. [Google Scholar] [CrossRef]
- Nunez, M.; Sivertsen, M.A.; Mattsson, J. Growth preferences on substrate, construction, and room location for indoor moulds and Actinomycetes. In Proceedings of the 10th International Conference on Healthy Buildings 2012, Brisbane, Australia, 5–12 July 2012; Morawska, L., Ed.; ISIAQ: Santa Cruz, CA, USA; Volume 1, 5H.3. p. 1312. [Google Scholar]
- Nunez, M.; Hammer, H. Microbial specialists in below-grade foundation walls in Scandinavia. Indoor Air 2014, 24, 543–551. [Google Scholar] [CrossRef]
- Andersen, B.; Frisvad, J.C.; Dunn, R.R.; Thrane, U. A pilot study on baseline fungi and moisture indicator fungi in Danish homes. J. Fungi 2021, 7, 71. [Google Scholar] [CrossRef]
- Prezant, B.; Miller, D.; Weekes, D. Recognition, Evaluation, and Control of Indoor Air Mold; American Industrial Hygiene Association: Falls Church, VA, USA, 2008. [Google Scholar]
- Flannigan, B.; Samson, R.A.; Miller, J.D. Microorganisms in Home and Indoor Work Environments: Diversity, Health Impacts, Investigation and Control; CRC Press: London, UK, 2011. [Google Scholar]
- Mensah-Attipoe, J.; Täubel, M. Analysis Approaches for Fungi in Indoor Environmental Assessments. In Exposure to Microbiological Agents in Indoor and Occupational Environments; Viegas, C., Viegas, S., Gomes, A., Täubel, M., Sabino, R., Eds.; Springer: Cham, Switzerland, 2017; pp. 109–127. [Google Scholar] [CrossRef]
- Reponen, T. Sampling for Microbial Determinations. In Exposure to Microbiological Agents in Indoor and Occupational Environments; Viegas, C., Viegas, S., Gomes, A., Täubel, M., Sabino, R., Eds.; Springer: Cham, Switzerland, 2017; pp. 85–96. [Google Scholar] [CrossRef]
- Wu, B.; Hussain, M.; Zhang, W.; Stadler, M.; Liu, X.; Xiang, M. Current insights into fungal species diversity and perspective on naming the environmental DNA sequences of fungi. Mycology 2019, 10, 127–140. [Google Scholar] [CrossRef]
- Haugland, R.A.; Vesper, S.J. Identification and Quantification of Specific Fungi and Bacteria. U.S. Patent 6,387,652, 14 May 2002. [Google Scholar]
- Gilbert, J.A.; Stephens, B. Microbiology of the built environment. Nat. Rev. Microbiol. 2018, 16, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.H.; Anslan, S.; Bahram, M.; Wurzbacher, C.; Baldrian, P.; Tedersoo, L. Mycobiome diversity: High-throughput sequencing and identification of fungi. Nat. Rev. Microbiol. 2019, 17, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Martin-Sanchez, P.M.; Estensmo, E.L.F.; Morgado, L.N.; Maurice, S.; Engh, I.B.; Skrede, I.; Kauserud, H. Analysing indoor mycobiomes through a large-scale citizen science study in Norway. Mol. Ecol. 2021, 30, 2689–2705. [Google Scholar] [CrossRef] [PubMed]
- Estensmo, E.L.F.; Botnen, S.S.; Maurice, S.; Martin-Sanchez, P.M.; Morgado, L.; Engh, I.B.; Høiland, K.; Skrede, I.; Kauserud, H. The indoor mycobiome of daycare centers is affected by occupancy and climate. Appl. Environ. Microbiol. 2022, 88, e0211321. [Google Scholar] [CrossRef]
- Sylvain, I.A.; Adams, R.I.; Taylor, J.W. A different suite: The assemblage of distinct fungal communities in water-damaged units of a poorly-maintained public housing building. PLoS ONE 2019, 14, e0213355. [Google Scholar] [CrossRef]
- Hegarty, B.; Haverinen-Shaughnessy, U.; Shaughnessy, R.J.; Peccia, J. Spatial gradients of fungal abundance and ecology throughout a damp building. Environ. Sci. Technol. Lett. 2019, 6, 329–333. [Google Scholar] [CrossRef]
- Hegarty, B.; Pan, A.; Haverinen-Shaughnessy, U.; Shaughnessy, R.; Peccia, J. DNA sequence-based approach for classifying the mold status of buildings. Environ. Sci. Technol. 2020, 54, 15968–15975. [Google Scholar] [CrossRef]
- Jayaprakash, B.; Adams, R.I.; Kirjavainen, P.; Karvonen, A.; Vepsäläinen, A.; Valkonen, M.; Järvi, K.; Sulyok, M.; Pekkanen, J.; Hyvärinen, A.; et al. Indoor microbiota in severely moisture damaged homes and the impact of interventions. Microbiome 2017, 5, 138. [Google Scholar] [CrossRef]
- Gams, W. The analysis of communities of saprophytic microfungi with special reference to soil fungi. Handb. Veg. Sci. 1992, 19, 182–223. [Google Scholar]
- de Hoog, G.S.; Guarro, J.; Gené, J.; Figueras, M.J. Atlas of Clinical Fungi, 2nd ed.; Centraalbureau voor Schimmelcultures: Baarn, The Netherlands; Universitat di Rovira i Virgili: Reus, Spain, 2000. [Google Scholar]
- Ihrmark, K.; Bödeker, I.T.M.; Cruz-Martinez, K.; Friberg, H.; Kubartova, A.; Schenck, J.; Strid, Y.; Stenlid, J.; Brandström-Durling, M.; Clemmensen, K.E.; et al. New primers to amplify the fungal ITS2 region—Evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol. Ecol. 2012, 82, 666–677. [Google Scholar] [CrossRef] [PubMed]
- White, T.J.; Bruns, T.D.; Lee, S.B.; Taylor, J.W. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR–Protocols and Applications–A Laboratory Manual; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: New York, NY, USA, 1990; pp. 315–322. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org/ (accessed on 14 September 2022).
- Wickham, H. Tidyverse: Easily Install and Load the ‘Tidyverse’ R Package Version 1.2.1. 2017. Available online: https://CRAN.R-project.org/package=tidyverse (accessed on 14 September 2022).
- Oksanen, J.; Simpson, G.L.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’hara, R.B.; Solymos, P.; Stevens, M.H.H.; Szoecs, E.; et al. Vegan: Community Ecology Package. R Package Version 2.5-6. 2019. Available online: https://CRAN.R-project.org/package=vegan (accessed on 14 September 2022).
- Adams, R.I.; Miletto, M.; Taylor, J.W.; Bruns, T.D. Dispersal in microbes: Fungi in indoor air are dominated by outdoor air and show dispersal limitation at short distances. ISME J. 2013, 7, 1262–1273. [Google Scholar] [CrossRef] [PubMed]
- Barberán, A.; Dunn, R.R.; Reich, B.J.; Pacifici, K.; Laber, E.B.; Menninger, H.L.; Morton, J.M.; Henley, J.B.; Leff, J.W.; Miller, S.L.; et al. The ecology of microscopic life in household dust. Proc. R. Soc. B Boil. Sci. 2015, 282, 20151139. [Google Scholar] [CrossRef] [PubMed]
- Estensmo, E.; Morgado, L.; Maurice, S.; Martin-Sanchez, P.M.; Engh, I.B.; Mattsson, J.; Kauserud, H.; Skrede, I. Spatiotemporal variation of the indoor mycobiome in daycare centers. Microbiome 2021, 9, 220. [Google Scholar] [CrossRef] [PubMed]
- An, C.; Yamamoto, N. Fungal compositions and diversities on indoor surfaces with visible mold growths in residential buildings in the Seoul Capital Area of South Korea. Indoor Air 2016, 26, 714–723. [Google Scholar] [CrossRef]
- Pitkäranta, M.; Meklin, T.; Hyvärinen, A.; Nevalainen, A.; Paulin, L.; Auvinen, P.; Lignell, U.; Rintala, H. Molecular profiling of fungal communities in moisture damaged buildings before and after remediation—A comparison of culture-dependent and culture-independent methods. BMC Microbiol. 2011, 11, 235. [Google Scholar] [CrossRef]
- Mensah-Attipoe, J.; Reponen, T.; Veijalainen, A.M.; Rintala, H.; Täubel, M.; Rantakokko, P.; Ying, J.; Hyvärinen, A.; Pasanen, P. Comparison of methods for assessing temporal variation of growth of fungi on building materials. Microbiology 2016, 162, 1895–1903. [Google Scholar] [CrossRef]
- Shinohara, N.; Woo, C.; Yamamoto, N.; Hashimoto, K.; Yoshida-Ohuchi, H.; Kawakami, Y. Comparison of DNA sequencing and morphological identification techniques to characterize environmental fungal communities. Sci. Rep. 2021, 11, 2633. [Google Scholar] [CrossRef]
- Visagie, C.M.; Hirooka, Y.; Tanney, J.B.; Whitfield, E.; Mwange, K.; Meijer, M.; Amend, A.S.; Seifert, K.A.; Samson, R.A. Aspergillus, Penicillium and Talaromyces isolated from house dust samples collected around the world. Stud. Mycol. 2014, 78, 63–139. [Google Scholar] [CrossRef]
- Woudenberg, J.H.; Groenewald, J.Z.; Binder, M.; Crous, P.W. Alternaria redefined. Stud. Mycol. 2013, 75, 171–212. [Google Scholar] [CrossRef]
- Xu, Y.; Tandon, R.; Ancheta, C.; Arroyo, P.; Gilbert, J.A.; Stephens, B.; Kelley, S.T. Quantitative profiling of built environment bacterial and fungal communities reveals dynamic material dependent growth patterns and microbial interactions. Indoor Air 2021, 31, 188–205. [Google Scholar] [CrossRef]
- Carini, P.; Marsden, P.J.; Leff, J.W.; Morgan, E.E.; Strickland, M.S.; Fierer, N. Relic DNA is abundant in soil and obscures estimates of soil microbial diversity. Nat. Microbiol. 2016, 2, 16242. [Google Scholar] [CrossRef] [PubMed]
- Skrede, I.; Engh, I.B.; Binder, M.; Carlsen, T.; Kauserud, H.; Bendiksby, M. Evolutionary history of Serpulaceae (Basidiomycota): Molecular phylogeny, historical biogeography and evidence for a single transition of nutritional mode. BMC Evol. Biol. 2011, 11, 230. [Google Scholar] [CrossRef] [PubMed]
- Vesper, S.; McKinstry, C.; Haugland, R.; Wymer, L.; Bradham, K.; Ashley, P.; Cox, D.; Dewalt, G.; Friedman, W. Development of an Environmental Relative Moldiness index for US homes. J. Occup. Environ. Med. 2007, 49, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Täubel, M.; Karvonen, A.M.; Reponen, T.; Hyvärinen, A.; Vesper, S.; Pekkanen, J. Application of the Environmental Relative Moldiness index in Finland. Appl. Environ. Microbiol. 2015, 82, 578–584. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials 1 (n) | ||||||
|---|---|---|---|---|---|---|
| Taxa | Gypsum Board (19) | Chipboard (4) | Wood (12) | MDF (3) | Building Paper (3) | Wall Paper (4) |
| Chaetomium | 15.8 | 25 | 16.6 | |||
| Chaetomium globosum | 47.4 | 25 | 66.6 | 25 | ||
| Stachybotrys chartarum | 36.8 | 8.3 | 25 | |||
| Stachybotrys echinata | 5.3 | |||||
| Cladosporium | 31.6 | 25 | 25 | 25 | ||
| Penicillium | 31.6 | 50 | 25 | 33.3 | 75 | |
| Acremonium | 26.3 | 25 | ||||
| Acremonium ovobatum | 8.3 | |||||
| Aspergillus | 15.8 | 25 | 16.6 | |||
| Aspergillus versicolor | 21 | 33.3 | ||||
| Aspergillus niger | 8.3 | |||||
| Aspergillus penicillioides | 8.3 | |||||
| Aspergillus glaucus | 33.3 | |||||
| Ulocladium | 15.8 | 8.3 | ||||
| Ascotricha erinacea | 10.5 | |||||
| Rhizopus | 10.5 | |||||
| Pseudoallescheria | 5.3 | 8.3 | ||||
| Scopulariopsis brevicaulis | 5.3 | |||||
| Tritirachium | 5.3 | 25 | ||||
| Sepedonium | 5.3 | |||||
| Monodictys | 25 | |||||
| Trichoderma | 16.6 | |||||
| Coniophora puteana | 16.6 | |||||
| Wallemia sebi | 25 | |||||
| Bjerkandera adusta | 8.3 | |||||
| Phoma glomerata | 8.3 | |||||
| Niesslia heterophora | 33.3 | |||||
| Black fungi | 5.3 | 8.3 | ||||
| Actinobacteria 2 | 10.5 | 8.3 | 100 | |||
| Microscopy Results | Fungal DNA Metabarcoding Data (Genus Level) | ||||||
|---|---|---|---|---|---|---|---|
| Mold Sample | Apartment | Material; Visual Aspect | Major Fungi in Mold Sample (RA > 1%) | RA (%) | Presence in Dust Damaged Room (%) | Presence in Dust Central Room (%) | |
| M1 | 1 | Building paper; yellowish growth | Penicillium | 66.6 | 10.6 | 4 | |
| Aspergillus | 12.3 | 21.5 | 22 | ||||
| Diplospora | 6.9 | ||||||
| Malassezia | 4.7 | 0.13 | 0.12 | ||||
| Niesslia heterophora | Monocillium (=Niesslia) | 4 | |||||
| Pachnocybe | 1.3 | ||||||
| Acrostalagmus | 1.1 | ||||||
| Cladosporium | 1 | 21.7 | 12.7 | ||||
| M2 | 1 | Building paper; yellowish growth | Aspergillus | 68.9 | |||
| Penicillium | 25.3 | 10.6 | 4 | ||||
| Monocillium | 1.9 | ||||||
| Acrostalagmus | 1.3 | ||||||
| M3 | 1 | Gypsum board; black mold | Stachybotrys echinata Stachybotrys chartarum | Stachybotrys | 85.2 | ||
| Aspergillus | 8.1 | 21.5 | 22 | ||||
| Exophiala | 5 | 0.01 | 0.06 | ||||
| M9 | 3 | Wall paper; dark mold | Penicillium sp. | Penicillium | 69.8 | 43.1 | 14.9 |
| Monocillium | 22.8 | ||||||
| M10 | 3 | Wall paper; dark mold | Mucor | 81.4 | 0.2 | 0.01 | |
| Penicillium sp. | Penicillium | 17.7 | 20 | 14.9 | |||
| M11 | 3 | Floor wood; dark stains | Trichoderma sp. | Trichoderma | 98.4 | nd | 1.6 |
| Coniochaeta | 1.1 | nd | |||||
| M12 | 3 | Floor wood; dark stains | Coniochaeta | 37.5 | nd | ||
| Penicillium | 15.2 | nd | 14.9 | ||||
| Unidentified Ascomycetes | |||||||
| M24 | 8 | Painted concrete wall | Acremonium | 94.2 | 0.3 | ||
| Capronia | 5.6 | 0.06 | |||||
| Unidentified molds | |||||||
| M25 | 8 | Wall wood panel | Aspergillus penicillioides | Aspergillus | 100 | 40 | 3.2 |
| M30 | 13 | Gypsum board | Penicillium | 33.1 | 2.2 | 13.5 | |
| Cladosporium sp. | Cladosporium | 32.6 | 4.7 | 18.1 | |||
| Monocillium | 17.5 | ||||||
| Sarocladium | 10.9 | 0.01 | |||||
| Trichoderma | 1.3 | 0.1 | |||||
| Acremonium sp. | Acremonium | 1.2 | 0.006 | ||||
| Aspergillus | 1.1 | 6.2 | 5 | ||||
| Ulocladium sp. | |||||||
| M36 | 15 1 | Floor wood | Penicillium sp. | Penicillium | 36.3 | 5.1 | 0.8 |
| Talaromyces | 27.5 | 0.01 | |||||
| Fusarium | 26.9 | 0.05 | |||||
| Pyrenochaeta | 5.4 | ||||||
| Aspergillus sp. | Aspergillus | 3.5 | 7.5 | 2.1 | |||
| Acremonium ovobatum | |||||||
| 85% Serpula lacrymans 1 | 93% Serpula lacrymans 1 | ||||||
| M51 2 | 23 | Gypsum board | NA 2 | 56.6 | |||
| Acremonium | 40.7 | 0.2 | |||||
| Penicillium | 2.5 | 0.6 | 0.6 | ||||
| Cladosporium sp. 3 | <1% 3 | ||||||
| M52 2 | 23 | Gypsum board | NA 2 | 45 | |||
| Acremonium | 31.8 | 0.2 | |||||
| Pyrenochaeta | 14.7 | ||||||
| Talaromyces | 6.9 | 0.07 | |||||
| Cladosporium sp. | |||||||
| M53 | 24 | Wood | Stachybotrys | 44 | 0.4 | 20 | |
| Fusarium | 23 | ||||||
| Penicillium | 13.6 | 46.5 | 22.1 | ||||
| Acremonium | 13.2 | 7.6 | 5 | ||||
| Chaetomium sp. | Chaetomium | 5.8 | 15.9 | 18.4 | |||
| M61 | 29 | Gypsum board; black mold | Chaetomium globosum Chaetomium murorum | Chaetomium | 95.6 | 4.7 | 4.8 |
| Mucor | 1.5 | 5.4 | 7 | ||||
| Aspergillus sp. | <1% 3 | ||||||
| Pseudoallescheria sp. | |||||||
| M62 | 30 | Gypsum board; dark reddish mold | Chaetomium globosum | Chaetomium | 66.7 | 14.7 | 3.6 |
| Aspergillus versicolor | Aspergillus | 16.6 | 10.9 | 19.6 | |||
| Penicillium | 6.8 | 13 | 19.9 | ||||
| Mucor | 4.8 | 6.4 | 16.2 | ||||
| Monodictys | 2.6 | 12.2 | 0.3 | ||||
| Stachybotrys chartarum3 | <1% 3 | ||||||
| Tritirachium sp. | |||||||
| M63 | 30 | Gypsum board; black mold | Chaetomium globosum | Chaetomium | 85.3 | 14.7 | 3.6 |
| Aspergillus versicolor | Aspergillus | 5.7 | 10.9 | 19.6 | |||
| Stachybotrys chartarum | Stachybotrys | 5.6 | 0.09 | 0.05 | |||
| Mucor | 2 | 6.4 | 16.2 | ||||
| M64 | 30 | Chipboard; black mold | Chaetomium sp. | Chaetomium | 51.1 | 14.7 | 3.6 |
| Monodictys sp. | Monodictys | 28.7 | 12.2 | 0.3 | |||
| Aspergillus | 19.5 | 10.9 | 19.6 | ||||
| Penicillium sp. 3 | <1% 3 | ||||||
| M65 | 30 | Wood; black mold | Debaryomyces | 44.9 | 37.6 | 0.9 | |
| Acremonium | 29.8 | 0.1 | |||||
| Cladosporium sp. | Cladosporium | 25.6 | 1.3 | 7.5 | |||
| M67 | 31 | Gypsum board; Dark green mold | Chaetomium sp. | Chaetomium | 56.9 | 1.2 | 9.9 |
| Aspergillus versicolor | Aspergillus | 42.4 | 4.3 | 7.1 | |||
| M68 | 32 | Gypsum board; black mold | Mucor | 50.2 | 14.3 | 2.8 | |
| Chaetomium globosum | Chaetomium | 28.2 | 0.7 | 0.09 | |||
| Penicillium sp. | Penicillium | 12.6 | 10.7 | 1.5 | |||
| Rhodotorula | 3.2 | 43.6 | 0.6 | ||||
| Fusarium | 2.4 | 2.4 | 0.006 | ||||
| Cladosporium sp. | Cladosporium | 1.4 | 3.6 | 5.7 | |||
| Rhizopus sp. | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin-Sanchez, P.M.; Nunez, M.; Estensmo, E.L.F.; Skrede, I.; Kauserud, H. Comparison of Methods to Identify and Monitor Mold Damages in Buildings. Appl. Sci. 2022, 12, 9372. https://doi.org/10.3390/app12189372
Martin-Sanchez PM, Nunez M, Estensmo ELF, Skrede I, Kauserud H. Comparison of Methods to Identify and Monitor Mold Damages in Buildings. Applied Sciences. 2022; 12(18):9372. https://doi.org/10.3390/app12189372
Chicago/Turabian StyleMartin-Sanchez, Pedro Maria, Maria Nunez, Eva Lena Fjeld Estensmo, Inger Skrede, and Håvard Kauserud. 2022. "Comparison of Methods to Identify and Monitor Mold Damages in Buildings" Applied Sciences 12, no. 18: 9372. https://doi.org/10.3390/app12189372
APA StyleMartin-Sanchez, P. M., Nunez, M., Estensmo, E. L. F., Skrede, I., & Kauserud, H. (2022). Comparison of Methods to Identify and Monitor Mold Damages in Buildings. Applied Sciences, 12(18), 9372. https://doi.org/10.3390/app12189372