
Decoding Structure–Odor Relationship Based on Hypergraph Neural Network and Deep Attentional Factorization Machine

Abstract
:1. Introduction
- (1)
- We propose a neural network model based on hypergraph neural networks and a deep attentional factorization machine for olfactory perception prediction, solving the complex interactions problem of the large number of molecular feature descriptors which is difficult for the existing methods to deal with.
- (2)
- Imitating the olfactory lateral inhibition of animals, we add a lateral inhibition-inspired attention mechanism to the convolutional neural network of the feature-semantic interaction module of molecule-odor descriptor to improve the prediction accuracy of olfactory perception.
- (3)
- We conduct experiments on real-world datasets. Experimental results show that our proposed HGAFMN achieves good results in olfactory perception prediction.
2. Related Work
2.1. Quantitative Structure–Odor Relationship
2.2. Graph Neural Networks-Based Learning
2.3. Lateral Inhibition
3. Materials and Methods
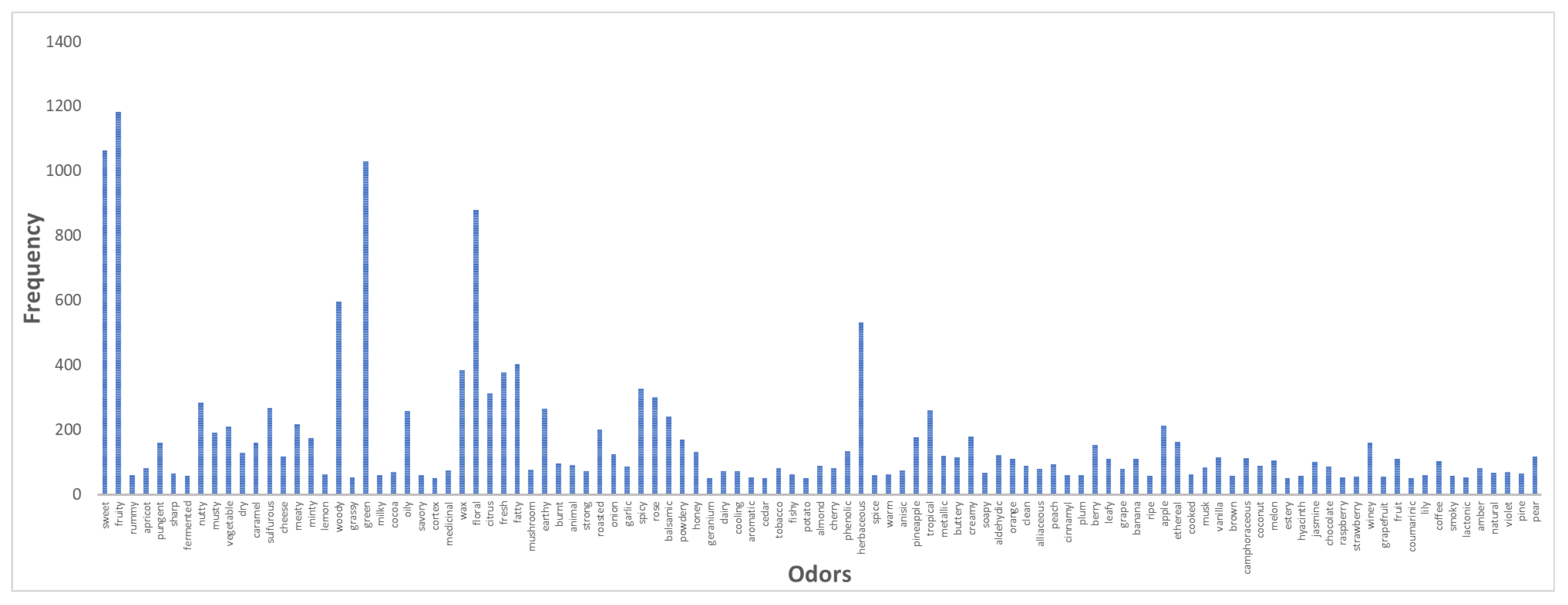
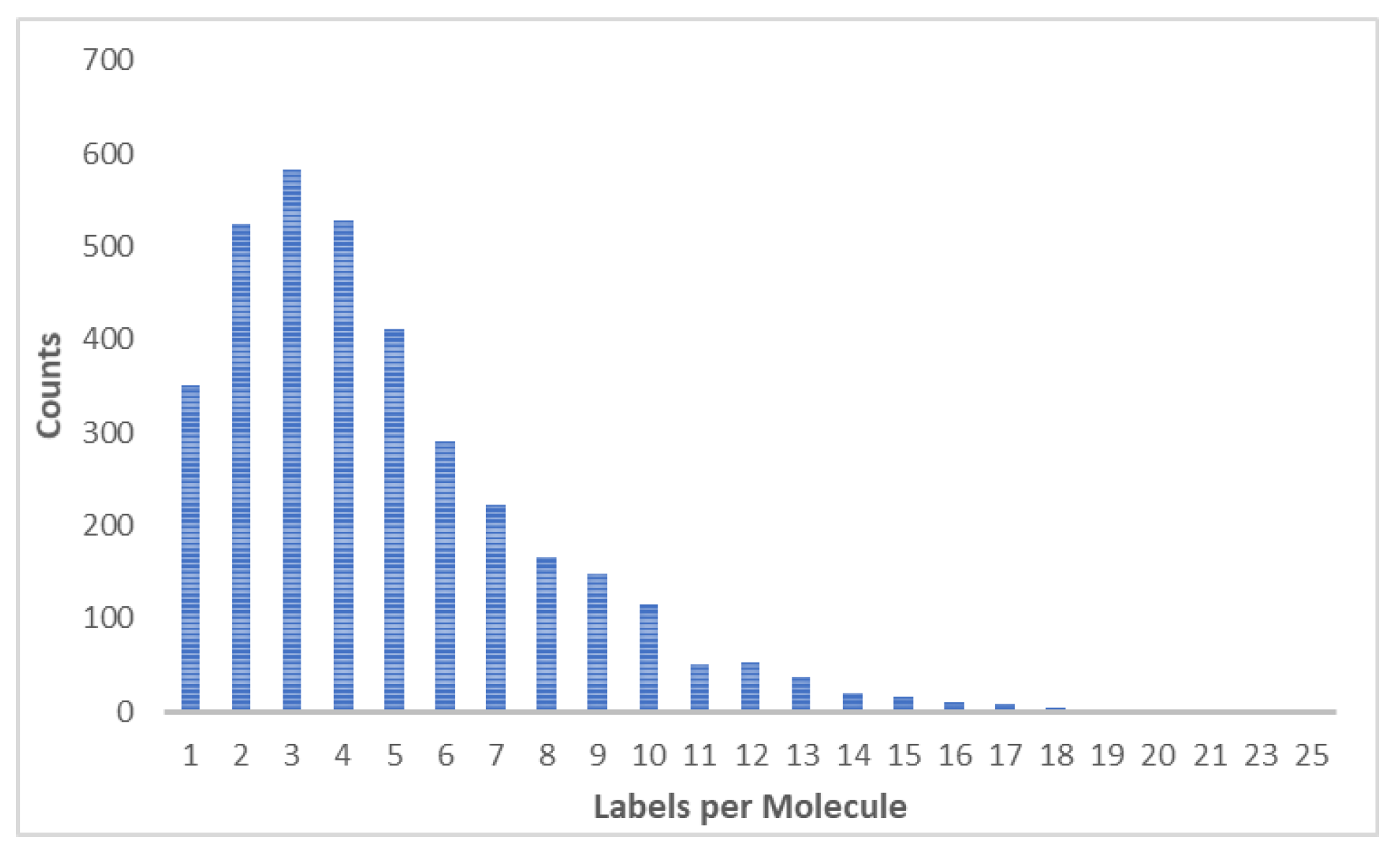
3.1. Datasets
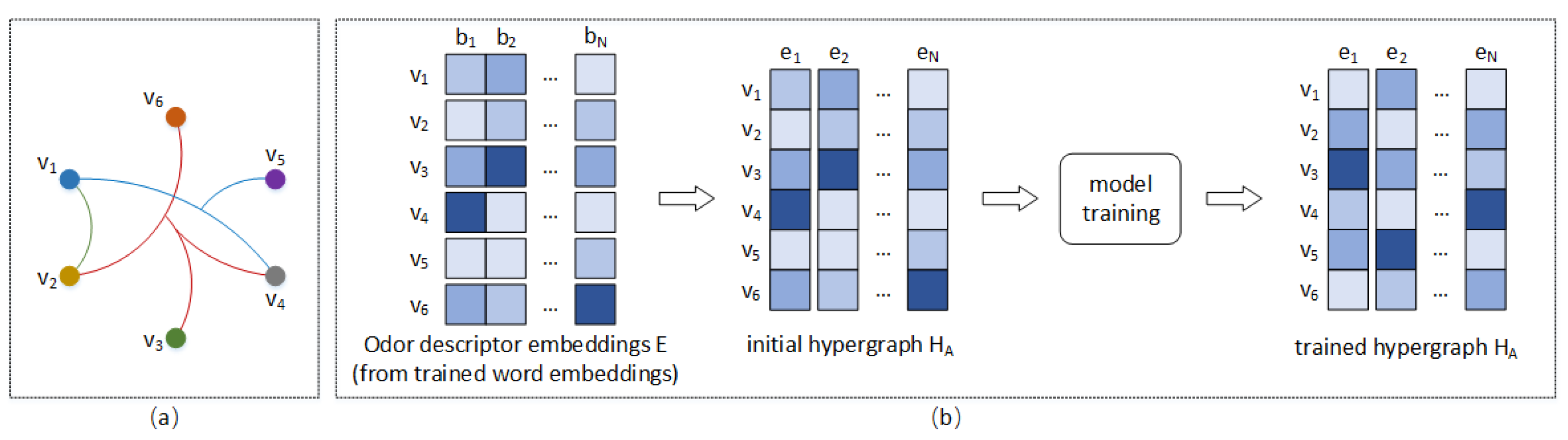
3.2. Feature-Semantic Interaction of Molecule-Odor Descriptor through HGNN
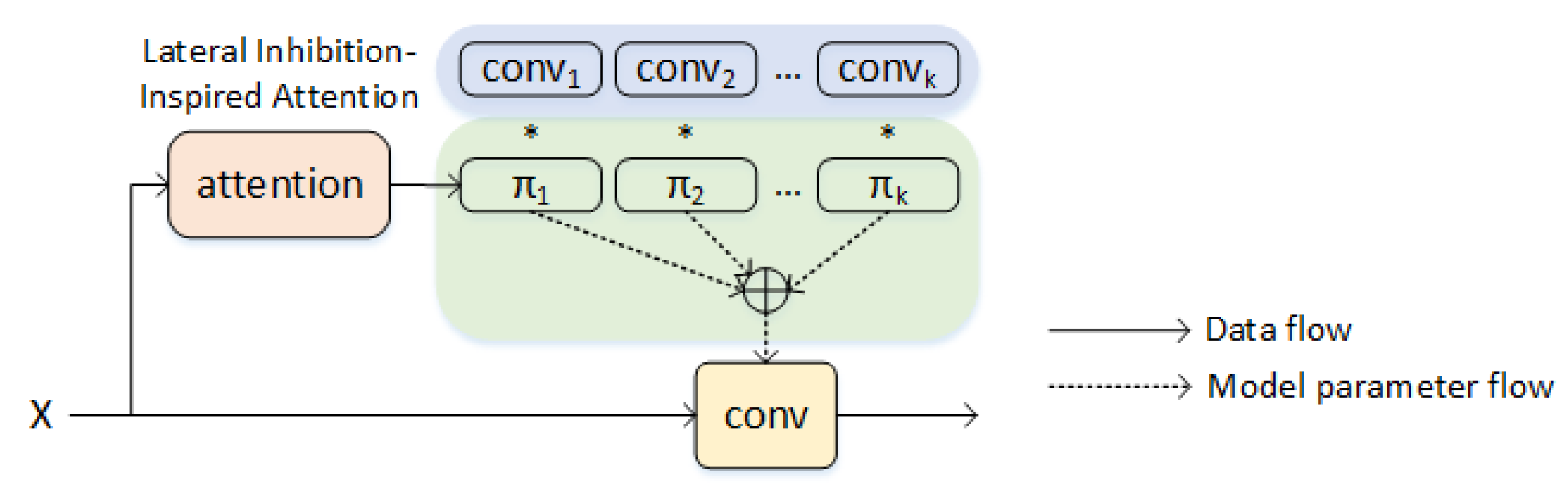
3.3. Weighted Deep Feature Combination Based on Attentional Factorization Machine
4. Experiments
4.1. Experimental Settings
4.1.1. Baselines and Evaluation Metrics
4.1.2. Hyper-Parameter Settings
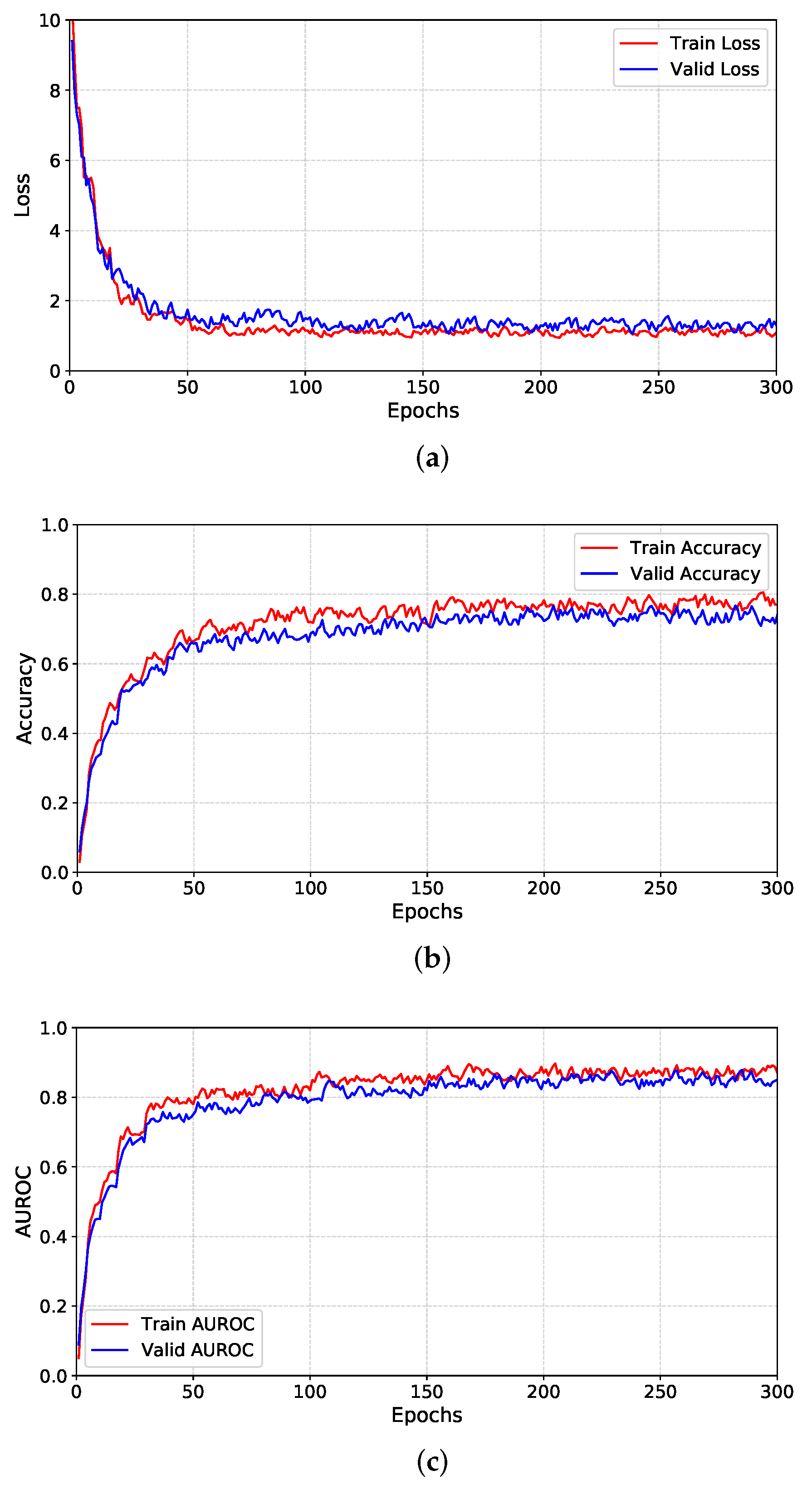
4.2. Performance Evaluation
4.3. Experiment Analysis
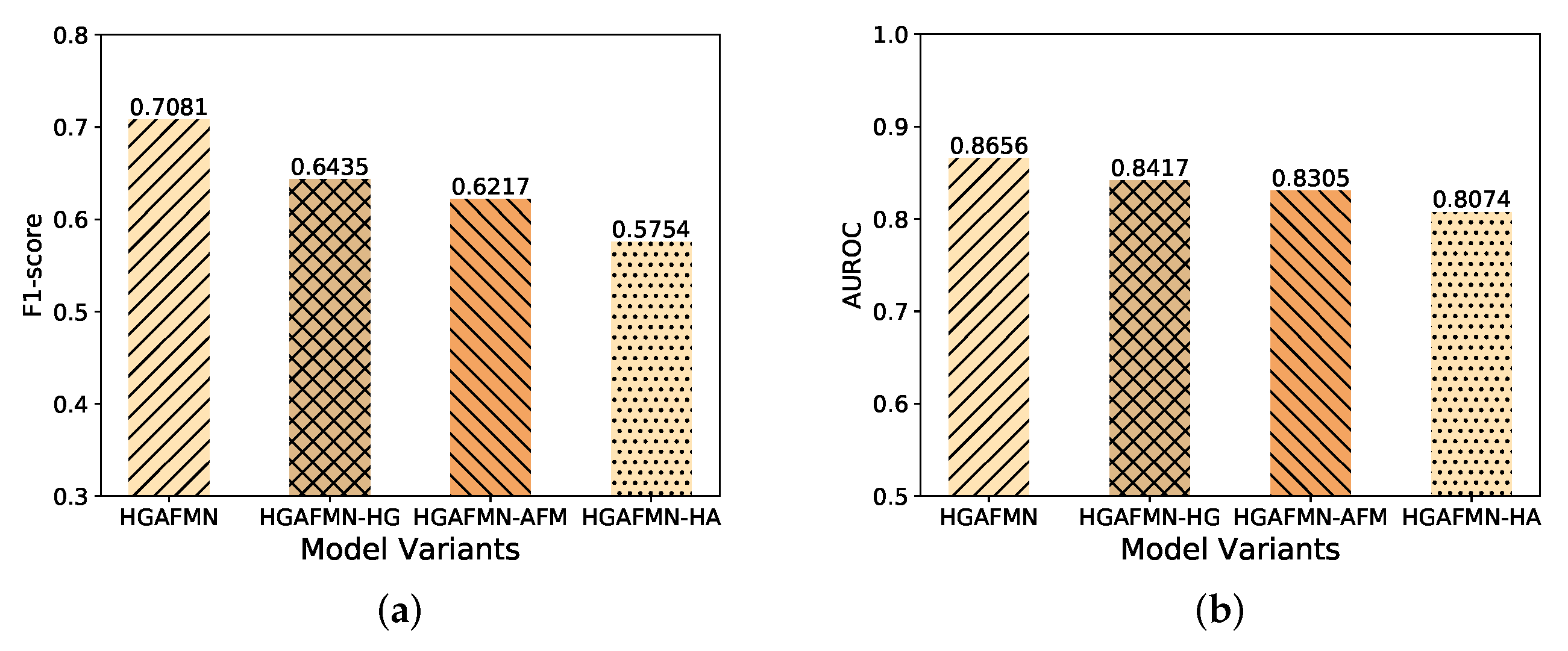
4.3.1. Ablation Study
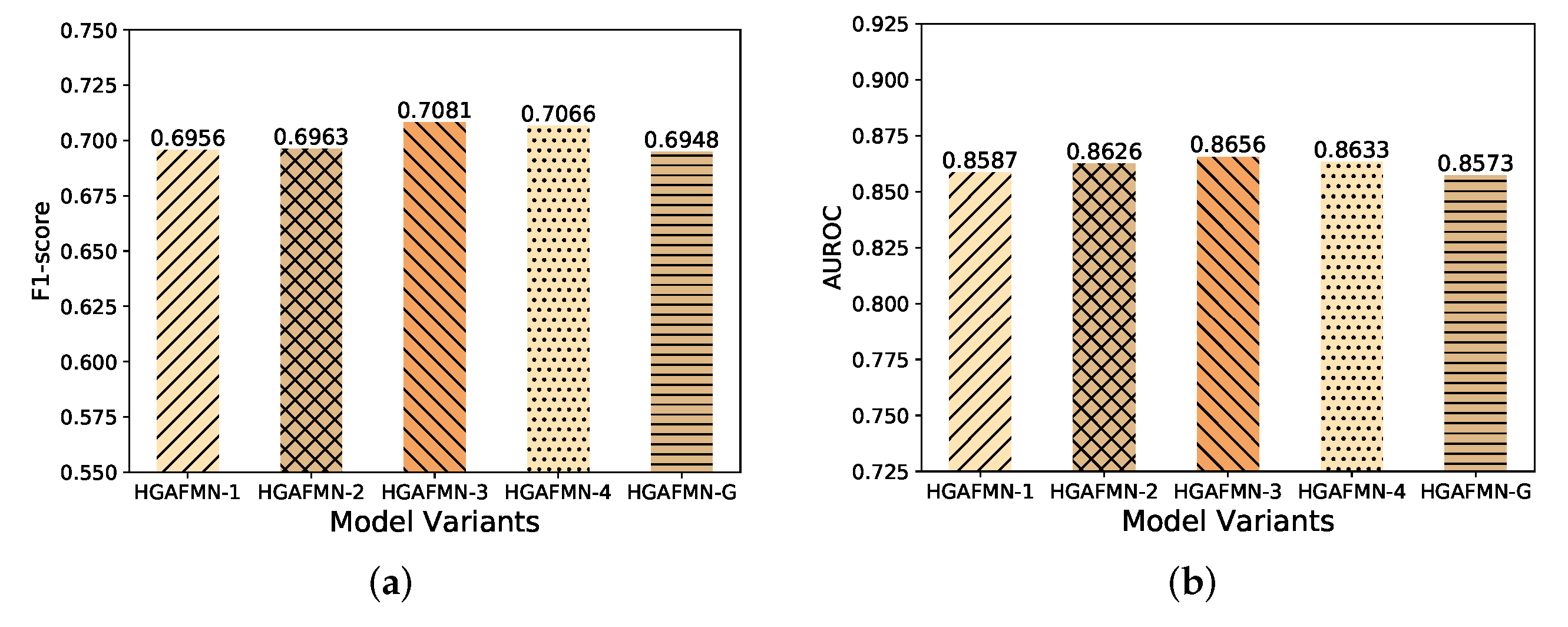
4.3.2. Performance Comparison w.r.t Layers of HGNNs
4.3.3. Performance Comparison with Respect to Hyper-Parameters
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, P.Y.; Sun, Y.; Axel, R.; Abbott, L.; Yang, G.R. Evolving the Olfactory System with Machine Learning. Neuron 2021, 109, 3879–3892.e5. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Luo, D.; Cheng, Y.; Wong, K.Y.; Hung, K. Identifying the Primary Odor Perception Descriptors by Multi-Output Linear Regression Models. Appl. Sci. 2021, 11, 3320. [Google Scholar] [CrossRef]
- Nozaki, Y.; Nakamoto, T. Predictive modeling for odor character of a chemical using machine learning combined with natural language processing. PLoS ONE 2018, 13, e0198475. [Google Scholar]
- Li, H.; Panwar, B.; Omenn, G.S.; Guan, Y. Accurate prediction of personalized olfactory perception from large-scale chemoinformatic features. Gigascience 2018, 7, gix127. [Google Scholar] [CrossRef]
- Sanchez-Lengeling, B.; Wei, J.N.; Lee, B.K.; Gerkin, R.C.; Aspuru-Guzik, A.; Wiltschko, A.B. Machine learning for scent: Learning generalizable perceptual representations of small molecules. arXiv 2019, arXiv:1910.10685. [Google Scholar]
- Sharma, A.; Kumar, R.; Ranjta, S.; Varadwaj, P.K. SMILES to smell: Decoding the structure–odor relationship of chemical compounds using the deep neural network approach. J. Chem. Inf. Model. 2021, 61, 676–688. [Google Scholar] [CrossRef]
- Feng, Y.; You, H.; Zhang, Z.; Ji, R.; Gao, Y. Hypergraph neural networks. In Proceedings of the AAAI Conference on Artificial Intelligence, Honolulu, HI, USA, 27 January–1 February 2019; Volume 33, pp. 3558–3565. [Google Scholar]
- Bandyopadhyay, S.; Das, K.; Murty, M.N. Line Hypergraph Convolution Network: Applying Graph Convolution for Hypergraphs. arXiv 2020, arXiv:2002.03392. [Google Scholar]
- Urban, N.N. Lateral inhibition in the olfactory bulb and in olfaction. Physiol. Behav. 2002, 77, 607–612. [Google Scholar] [CrossRef]
- Shmuel, R.; Secundo, L.; Haddad, R. Strong, weak and neuron type dependent lateral inhibition in the olfactory bulb. Sci. Rep. 2019, 9, 1602. [Google Scholar] [CrossRef]
- Whitesell, J.D.; Sorensen, K.A.; Jarvie, B.C.; Hentges, S.T.; Schoppa, N.E. Interglomerular lateral inhibition targeted on external tufted cells in the olfactory bulb. J. Neurosci. 2013, 33, 1552–1563. [Google Scholar] [CrossRef]
- Keller, A.; Gerkin, R.C.; Guan, Y.; Dhurandhar, A.; Turu, G.; Szalai, B.; Mainland, J.D.; Ihara, Y.; Yu, C.W.; Wolfinger, R.; et al. Predicting human olfactory perception from chemical features of odor molecules. Science 2017, 355, 820–826. [Google Scholar] [CrossRef]
- Keller, A.; Vosshall, L.B. Olfactory perception of chemically diverse molecules. BMC Neurosci. 2016, 17, 55. [Google Scholar] [CrossRef]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef]
- Snitz, K.; Yablonka, A.; Weiss, T.; Frumin, I.; Khan, R.M.; Sobel, N. Predicting odor perceptual similarity from odor structure. PLoS Comput. Biol. 2013, 9, e1003184. [Google Scholar] [CrossRef]
- Jiang, D.; Wu, L.; Wen, T.; Li, J.; Jia, L.; Gao, Z. A Cheminformatic Compression Method for Multiple Odor Label in Intelligent Perception. In Proceedings of the 2021 IEEE International Conference on Smart Internet of Things (SmartIoT), Jeju, Korea, 13–15 August 2021; pp. 317–321. [Google Scholar]
- Boesveldt, S.; Olsson, M.J.; Lundström, J.N. Carbon chain length and the stimulus problem in olfaction. Behav. Brain Res. 2010, 215, 110–113. [Google Scholar] [CrossRef]
- Kermen, F.; Chakirian, A.; Sezille, C.; Joussain, P.; Le Goff, G.; Ziessel, A.; Chastrette, M.; Mandairon, N.; Didier, A.; Rouby, C.; et al. Molecular complexity determines the number of olfactory notes and the pleasantness of smells. Sci. Rep. 2011, 1, 206. [Google Scholar] [CrossRef]
- Gutiérrez, E.D.; Dhurandhar, A.; Keller, A.; Meyer, P.; Cecchi, G.A. Predicting natural language descriptions of mono-molecular odorants. Nat. Commun. 2018, 9, 4979. [Google Scholar] [CrossRef]
- Qiu, X.; Cheng, Y.; Luo, D.; He, C.; Angus, K.; Liu, Q. Odor-GCN: Graph Convolutional Network for Predicting Odor Impressions Based on Molecular Structures. Res. Sq. 2022. [Google Scholar] [CrossRef]
- Shang, L.; Tang, F.; Liu, C.; Chen, B.; Liu, L.; Hayashi, K. Machine-Learning-Based Olfactometry: Odor Descriptor Clustering Analysis for Olfactory Perception Prediction of Odorant Molecules. bioRxiv 2022. [Google Scholar] [CrossRef]
- Scarselli, F.; Gori, M.; Tsoi, A.C.; Hagenbuchner, M.; Monfardini, G. The graph neural network model. IEEE Trans. Neural Netw. 2008, 20, 61–80. [Google Scholar] [CrossRef]
- Wu, Z.; Pan, S.; Chen, F.; Long, G.; Zhang, C.; Philip, S.Y. A comprehensive survey on graph neural networks. IEEE Trans. Neural Netw. Learn. Syst. 2020, 32, 4–24. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Feng, Y.; Ji, R.; Zhao, X.; Tang, W.; Gao, Y. Dual channel hypergraph collaborative filtering. In Proceedings of the 26th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, Virtual Event, 6–10 July 2020; pp. 2020–2029. [Google Scholar]
- Wu, X.; Chen, Q.; Li, W.; Xiao, Y.; Hu, B. AdaHGNN: Adaptive Hypergraph Neural Networks for Multi-Label Image Classification. In Proceedings of the 28th ACM International Conference on Multimedia, Seattle, WA, USA, 12–16 October 2020; pp. 284–293. [Google Scholar]
- Ma, Z.; Jiang, Z.; Zhang, H. Hyperspectral Image Classification using Feature Fusion Hypergraph Convolution Neural Network. IEEE Trans. Geosci. Remote Sens. 2021, 60, 1–14. [Google Scholar] [CrossRef]
- Wang, J.; Ding, K.; Hong, L.; Liu, H.; Caverlee, J. Next-item recommendation with sequential hypergraphs. In Proceedings of the 43rd International ACM SIGIR Conference on Research and Development in Information Retrieval, Xi’an, China, 25–30 July 2020; pp. 1101–1110. [Google Scholar]
- Xia, X.; Yin, H.; Yu, J.; Wang, Q.; Cui, L.; Zhang, X. Self-Supervised Hypergraph Convolutional Networks for Session-based Recommendation. In Proceedings of the AAAI Conference on Artificial Intelligence, Virtually, 2–9 February 2021; Volume 35, pp. 4503–4511. [Google Scholar]
- Peng, D.; Zhang, S. GC–HGNN: A global-context supported hypergraph neural network for enhancing session-based recommendation. Electron. Commer. Res. Appl. 2022, 52, 101129. [Google Scholar] [CrossRef]
- Madine, M.; Rekik, I.; Werghi, N. Diagnosing Autism Using T1-W MRI With Multi-Kernel Learning and Hypergraph Neural Network. In Proceedings of the 2020 IEEE International Conference on Image Processing (ICIP), Abu Dhabi, United Arab Emirates, 25–28 October 2020; pp. 438–442. [Google Scholar]
- Xu, C.; Li, M.; Ni, Z.; Zhang, Y.; Chen, S. GroupNet: Multiscale Hypergraph Neural Networks for Trajectory Prediction with Relational Reasoning. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, New Orleans, LA, USA, 19–24 June 2022; pp. 6498–6507. [Google Scholar]
- Luo, L. Principles of Neurobiology; Garland Science: New York, NY, USA, 2020. [Google Scholar]
- Pashkovski, S.L.; Iurilli, G.; Brann, D.; Chicharro, D.; Drummey, K.; Franks, K.M.; Panzeri, S.; Datta, S.R. Structure and flexibility in cortical representations of odour space. Nature 2020, 583, 253–258. [Google Scholar] [CrossRef]
- Aqrabawi, A.J.; Kim, J.C. Olfactory memory representations are stored in the anterior olfactory nucleus. Nat. Commun. 2020, 11, 1246. [Google Scholar] [CrossRef]
- Mao, Z.H.; Massaquoi, S.G. Dynamics of winner-take-all competition in recurrent neural networks with lateral inhibition. IEEE Trans. Neural Netw. 2007, 18, 55–69. [Google Scholar] [CrossRef]
- Cao, C.; Huang, Y.; Wang, Z.; Wang, L.; Xu, N.; Tan, T. Lateral inhibition-inspired convolutional neural network for visual attention and saliency detection. In Proceedings of the Thirty-Second AAAI Conference on Artificial Intelligence, New Orleans, LA, USA, 2–7 February 2018. [Google Scholar]
- Păiș, V. RACAI at SemEval-2022 Task 11: Complex named entity recognition using a lateral inhibition mechanism. In Proceedings of the 16th International Workshop on Semantic Evaluation (SemEval-2022), Seattle, WA, USA, 14–15 July 2022; pp. 1562–1569. [Google Scholar]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Landrum, G. RDKit: A Software Suite for Cheminformatics, Computational Chemistry, and Predictive Modeling. 2013. Available online: http://www.rdkit.org (accessed on 10 August 2021).
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep residual learning for image recognition. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Las Vegas, NV, USA, 27–30 June 2016; pp. 770–778. [Google Scholar]
- Nielsen, C.J. Effect of scenario and experience on interpretation of mach bands. J. Endod. 2001, 27, 687–691. [Google Scholar] [CrossRef]
- Müller, N.G.; Mollenhauer, M.; Rösler, A.; Kleinschmidt, A. The attentional field has a Mexican hat distribution. Vis. Res. 2005, 45, 1129–1137. [Google Scholar] [CrossRef]
- Casanova, M.F.; Buxhoeveden, D.; Gomez, J. Disruption in the inhibitory architecture of the cell minicolumn: Implications for autisim. Neuroscientist 2003, 9, 496–507. [Google Scholar] [CrossRef]
- Chen, T.; Xu, M.; Hui, X.; Wu, H.; Lin, L. Learning semantic-specific graph representation for multi-label image recognition. In Proceedings of the IEEE/CVF International Conference on Computer Vision, Seoul, Korea, 27–2 November 2019; pp. 522–531. [Google Scholar]
- Pennington, J.; Socher, R.; Manning, C.D. Glove: Global vectors for word representation. In Proceedings of the 2014 Conference on Empirical Methods in Natural Language Processing (EMNLP), Doha, Qatar, 25–29 October 2014; pp. 1532–1543. [Google Scholar]
- Chen, L.; Wu, L.; Hong, R.; Zhang, K.; Wang, M. Revisiting Graph Based Collaborative Filtering: A Linear Residual Graph Convolutional Network Approach. In Proceedings of the AAAI Conference on Artificial Intelligence, New York, NY, USA, 7–12 February 2020; Volume 34, pp. 27–34. [Google Scholar]
- Guo, H.; Tang, R.; Ye, Y.; Li, Z.; He, X. DeepFM: A factorization-machine based neural network for CTR prediction. arXiv 2017, arXiv:1703.04247. [Google Scholar]
- Xiao, J.; Ye, H.; He, X.; Zhang, H.; Wu, F.; Chua, T.S. Attentional factorization machines: Learning the weight of feature interactions via attention networks. arXiv 2017, arXiv:1708.04617. [Google Scholar]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention is all you need. Adv. Neural Inf. Process. Syst. 2017, 30, 5998–6008. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout: A simple way to prevent neural networks from overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]
- Simonyan, K.; Zisserman, A. Very Deep Convolutional Networks for Large-Scale Image Recognition. In Proceedings of the 3rd International Conference on Learning Representations, ICLR Conference Track Proceedings, San Diego, CA, USA, 7–9 May 2015. [Google Scholar]
- Chollet, F. Xception: Deep learning with depthwise separable convolutions. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Honolulu, HI, USA, 21–26 July 2017; pp. 1251–1258. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models | Precision | Recall | F1 | AUROC |
|---|---|---|---|---|
| DREAM Winner [4] | 0.3063 | 0.3110 | 0.3082 | 0.8304 |
| GNN-QSOR [5] | 0.3614 | 0.3701 | 0.3653 | 0.8618 † |
| DNN-CNN [6] | 0.5294 † | 0.5307 † | 0.5301 † | 0.7851 |
| Our Proposal | 0.6552 | 0.7701 | 0.7081 | 0.8656 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Zhao, Q.; Ma, M.; Xu, J. Decoding Structure–Odor Relationship Based on Hypergraph Neural Network and Deep Attentional Factorization Machine. Appl. Sci. 2022, 12, 8777. https://doi.org/10.3390/app12178777
Wang Y, Zhao Q, Ma M, Xu J. Decoding Structure–Odor Relationship Based on Hypergraph Neural Network and Deep Attentional Factorization Machine. Applied Sciences. 2022; 12(17):8777. https://doi.org/10.3390/app12178777
Chicago/Turabian StyleWang, Yu, Qilong Zhao, Mingyuan Ma, and Jin Xu. 2022. "Decoding Structure–Odor Relationship Based on Hypergraph Neural Network and Deep Attentional Factorization Machine" Applied Sciences 12, no. 17: 8777. https://doi.org/10.3390/app12178777
APA StyleWang, Y., Zhao, Q., Ma, M., & Xu, J. (2022). Decoding Structure–Odor Relationship Based on Hypergraph Neural Network and Deep Attentional Factorization Machine. Applied Sciences, 12(17), 8777. https://doi.org/10.3390/app12178777