Abstract
A molecule is the smallest particle in a chemical element or compound that possesses the element or compound’s chemical characteristics. There are numerous challenges associated with the development of molecular simulations of fluid characteristics for industrial purposes. Fluid characteristics for industrial purposes find applications in the development of various liquid household products, such as liquid detergents, drinks, beverages, and liquid health medications, amongst others. Predicting the molecular properties of liquid pharmaceuticals or therapies to address health concerns is one of the greatest difficulties in drug development. Computational tools for precise prediction can help speed up and lower the cost of identifying new medications. A one-dimensional deep convolutional gated recurrent neural network (1D-CNN-GRU) was used in this study to offer a novel forecasting model for molecular property prediction of liquids or fluids. The signal data from molecular properties were pre-processed and normalized. A 1D convolutional neural network (1D-CNN) was then built to extract the characteristics of the normalized molecular property of the sequence data. Furthermore, gated recurrent unit (GRU) layers processed the extracted features to extract temporal features. The output features were then passed through several fully-connected layers for final prediction. For both training and validation, we used molecular properties obtained from the Kaggle database. The proposed method achieved a better prediction accuracy, with values of 0.0230, 0.1517, and 0.0693, respectively, in terms of the mean squared error (MSE), root mean square error (RMSE), and mean absolute error (MAE).
1. Introduction
Predicting molecular properties such as atomization energy is one of the most significant challenges in quantum physical chemistry [1]. Indeed, it has sparked considerable interest in important fields of chemical science, physical science, and computer science, as it accelerates technological improvements in the process of exploring materials with desirable properties, which includes pharmaceutical research with a clear objective and new material manufacturing [2]. Density functional theory (DFT) has been extensively used in the literature to predict molecular properties. It is a common belief that subatomic interactions between components (for example, molecules) cause molecular coherence and synchronization, which are strongly connected to their fundamental features [3]. Numerous subatomic particle techniques based on DFT have been devised, to describe the quantum interactions of Kohn and Sham [4]. On the other hand, DFTs are computationally expensive, since they often require specialized functions to identify particle interactions, which takes a long time. For instance, experimental data have revealed that predicting the characteristics of just one molecule with 20 atoms takes approximately an hour [5]. Predictions that emanate from a huge number of molecules in a chemical compound space can be quite cumbersome and time-consuming. Efficient methods are needed to make the prediction process easy and fast. The majority of healthcare liquid medications have chemical ingredients. Efficient approaches can aid in the prediction of a large number of molecules in the chemical components of possible liquid medications.
Unlike the previously described approaches (density functional theory and subatomic particle techniques), machine learning (ML) allows for the exact and quick prediction of molecules, which can subsequently be used to filter millions of organic molecules of interest using descriptors generated from energies [6]. Many machine learning approaches, including artificial neural networks, Gaussian process regression, kernel ridge regression [6], and quantum machine learning (QML) [7], have been shown to achieve an acceptable balance of accuracy and computing cost. Other studies have demonstrated that integrating information from quantum chemistry techniques (also known as delta learning) [8] and training a model on multiple qualities (also known as transfer learning) [9] may anticipate the difference between low- and high-fidelity computations. There have also been several recent articles [10,11,12] on using machine learning to anticipate the energy of large organic molecules. However, the effectiveness of these approaches for estimating the energy of large organic molecules has not been well evaluated.
Scientists have previously demonstrated the potential of these data-driven methods for molecular property prediction [13,14], motivated by the impressive growth of deep learning in various fields such as pattern recognition, computational linguistics, and environmental and human science [15,16,17,18,19]. In principle, these methods depend on rule-based feature extraction (for example, a bag of atom bonds) or viewing molecules as lattice-shaped formations, such as two-dimensional images or text. Unfortunately, few of them actually consider the underlying quantum synergies of molecules, resulting in significant information leakage, and leaving the molecular property prediction puzzle largely unsolved.
Regrettably, there are numerous technological and domain obstacles in the way. To start with, quantum interactions in molecules, such as attention pull, exchange aversion, and electrically charged contact, are immensely complicated, particularly in large molecules [20]. Analytical approaches to model them are difficult. Additionally, the volume of labeled molecular data is relatively constrained compared to classic tasks, such as systems that use artificial intelligence to train computers to understand and interpret visual worlds, necessitating a generalizable approach for prediction. Finally, we are frequently presented with labeled data for small calculations that are costly in practice. As a result, it is vital to recognize this imbalance and provide a transferrable solution for predicting the properties of large molecules using a model that has been trained on smaller molecules.
To solve these problems, this research proposes a one-dimensional deep convolutional gated recurrent neural network (1D-CNN-GRU) for predicting the intramolecular coupling constants of molecules and for increasing the properties of molecules of possible liquid medications/drugs and other generic molecules, by directly adding their quantum interactions. In the feature extraction step, it also extracts molecular property features based on sequence data characteristics. Unlike previous feature extraction approaches, CNN can extract data features with minimum user effort or professional knowledge. Finally, a property may be predicted using the whole interaction description from all levels (for example, the additional energy required to fragment a molecule into separate atoms). Extensive testing was performed on both balanced and unbalanced molecular datasets, with the experimental results confirming the usefulness of our suggested technique. Furthermore, 1D-CNN-GRU demonstrated a greater capability for generalizability and portability. The suggested model can naturally transmit molecular interaction information at each level. The contributions of this work are outlined below:
- This study proposed a one-dimensional deep convolutional gated recurrent neural network (1D-CNN-GRU) for improving the properties of chemical compounds in possible liquid drugs/medications.
- A comparison of the performance of molecular property systems based on a recurrent neural network, long short-term memory (LSTM), gated recurrent unit (GRU), one-dimensional convolutional neural network (1DCNN), and the proposed one-dimensional convolutional gated recurrent unit neural network (1D-CNN-GRU) was conducted.
- A 1D-CNN-GRU optimization algorithm that can accurately predict the molecular properties of molecules is presented.
- The efficacy of the suggested model was assessed using various performance criteria.
2. Related Work
Several studies have been conducted on predicting molecular characteristics. Faber et al. [13] investigated the impact of regressors and chemical characterizations, while developing rapid machine learning (ML) models of thirteen electronic ground-state parameters of organic molecules. Learning curves that highlight out-of-sample faults as a function of training dataset size, encompassing up to 118k distinct molecules, were used to assess the performance of each regressor/representation/property combination. They observed that, with the exception of the largest vibrational frequency, where RF performed best, gated convolution networks (GC), gated graph networks (GG), and kernel ridge regression (KRR) had the best performance across all attributes. There was no optimal representation and regressor combination for all attributes (however, a subsequent work with improved GG-based models predicted the best results for all properties). The research had one drawback: ML models for predicting features of molecules in non-equilibrium or stressed configurations can require significantly more training data.
McDonagh et al. [21] described a new approach for estimating the dynamic electron correlation energy of an atom or a link in a molecule, using topological atoms. They used the machine learning technique Kriging to predict these dynamic quantum mechanical energy components (Gaussian process regression with a non-zero mean function). The actual energy values were calculated by combining the MP2 two-particle density matrix with the interacting quantum atoms (IQA) approach. They presented examples as the final step in realizing the full potential of IQA for molecular modeling. This key result enables us to consider situations in which displacement is the most significant intermolecular interaction. The results of these instances indicated a novel method for generating diffusion possibilities for molecular simulation.
Huang and von Lilienfeld [22] investigated works that used modern machine learning approaches based on synthetic data, which is frequently generated using quantum mechanics-based methodologies, and model architectures inspired by quantum mechanics to address this challenge. Quantum mechanics-based machine learning (QML) combines the computational efficacy of empirical proxy models with a structure-based interpretation of matter. To achieve universality and transferability across CCS, they strictly mimicked fundamental physics. While the latest estimations of quantum issues enforce drastic processing constraints, recent QML-based discoveries suggest the possibility of a significant speedup, without jeopardizing quantum mechanics’ prediction accuracy.
Montavon et al. [23] used a learning-from-scratch technique for the prediction of molecular energy transfer in psychoactive chemical space. The original molecular structures were used to determine subatomic molecule energies. According to the study, creating an elastic estimation method and ensuring normalization probabilistically instead of analytically, as per usual, has advantages. Their findings increased the cutting edge by almost a factor of three, bringing statistical methods closer to chemical precision. The input depiction became deeper as the total amount of information in the input distribution increased. The multilayer neural network performed better, implying that the neural network’s absence of a powerful added feature prior should be balanced by a huge volume of data. The neural network operates best with random Coulomb matrices, which are inherently the best depictions, because each molecule is connected with a full distribution of Coulomb matrices. The inefficiency of Gaussian kernel ridge regression reduces gradually as the training data increases, whereas the neural network can benefit more from the new data. The techniques’ downside is that the performance is still quite poor.
Goh et al. [24] presented the ChemNet for prediction, after converting chemical diagrams into 2D RGB images. However, in non-Euclidean space, where the intrinsic spatial and distance information of atoms is not completely acknowledged, this grid-like rearrangement frequently results in information loss for molecules. Their findings showed that, by using a pre-trained ChemNet that contains chemistry domain expertise, universally applicable neural networks can be used to more accurately predict new chemical compositions. ChemNet regularly beats the existing deep learning models trained on synthetic characteristics. These include molecular fingerprints, as well as the latest ConvGraph algorithm for various problems. Moreover, their parameter adjustment research indicated that ChemNet’s lower layers have acquired general chemical representations based on rule-based knowledge, enhancing its universal applicability to the prediction of unknown chemical attributes. The work’s drawback is that only AUC and RMSE were utilized to evaluate the system’s performance, which is not sufficient to validate the efficacy of the work.
Sun et al. [25] conducted a thorough examination of graph convolutional networks and their application in pharmaceutical research and molecular bioinformatics. They discussed current applications from four perspectives: molecular property and activity prediction, interaction prediction, synthesis prediction, and de novo drug creation. They gave a quick overview of the scientific concept of graph convolutional networks, before illustrating different components based on numerous interpretations. The representative used in narcotic problems was then summarized. The present limitations and future opportunities of using graph convolutional networks in drug development were also discussed.
Schütt et al. [14] introduced SchNet, a deep learning framework with continuous convolutional layers that was specifically built to model atomistic systems. They demonstrated SchNet’s capability by correctly predicting a number of properties for molecules and materials across chemical space, where the model learns chemically plausible embeddings of atom types from throughout the periodic table. Finally, they used SchNet to predict potential-energy areas and energy-conserving force fields for molecular dynamics simulation of small molecules, and they used this to perform an exemplary study of the quantum-mechanical properties of C20-fullerene, which would have been impossible to perform using regular ab initio molecular dynamics. SchNet provides accurate and rapid forecasts. It can also put the learnt representation to the test using local chemical potentials.
The performance of the General AMBER force field (GAFF) in exploring liquid dynamics was compared by Wang and Hou [26]. Diffusion coefficients were estimated for 17 solvents, five chemical compounds in a dilute environment, four proteins in organic solvents, and nine chemical molecules in non-aqueous solutions. To compute the diffusion coefficients of solutes in solutions, a realistic sampling technique was presented and verified. The major molecular diffusion settings, such as simulation box sizes and wrapping molecular diffusion snapshot coordinates into basic simulation boxes, were investigated. The authors’ quantitative approach of aggregating the mean square displacement (MSD) gathered in many short-MD simulations was effective in predicting diffusion coefficients of solutes at infinite dilution, according to the scientists. The work had some drawbacks. These included that the work had no performance measures other than average unsigned error, root-mean-square error, and correlation coefficient, which were used to assess the method’s effectiveness.
The following are some of the ways in which this work improves on past research: This paper employs a one-dimensional deep convolutional gated recurrent neural network (1D-CNN-GRU) to provide a unique forecasting model for the predicting chemical properties of potential liquid drugs or medication. The signal data for molecular properties is preprocessed and standardized. A 1D convolutional neural network (1D-CNN) is then created to extract the features of the normalized molecular property sequence data. Furthermore, the retrieved features are processed by the gated recurrent unit (GRU) layers to extract temporal characteristics. For final prediction, the output characteristics are routed through numerous completely connected layers.
3. Materials and Methods
3.1. Molecular Property Dataset
The molecular characteristics between atom pairs in molecules, the coupling type, and any other features to create the molecule structure were obtained from the Kaggle database [27]. As indicated in Table 1, the dataset contains both numerical and text data types. The id data type is an integer, whereas the molecule name data type is a combination of numbers and characters. The atom index 0 and atom index 1 columns are both integers, the coupling type columns are both numbers and characters, and the scalar coupling constant column is a float number. As indicated in Figure 1, the scalar coupling constant is the target variable.

Table 1.
Descriptions of Datasets.
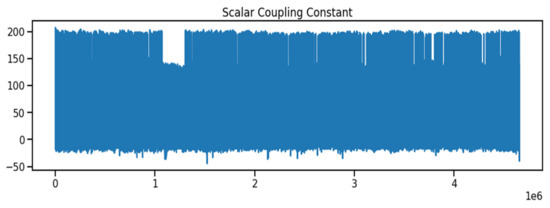
Figure 1.
The Scalar Coupling Constant in the Molecular Property.
3.2. Data Normalization
Data normalization, which lowers data to a range of 0–1, is one of the most used methods of data processing. Normalization is often employed in the processing of several assessment indexes procedures, to eliminate data unit restrictions and make the comparison and weighting of different indicators easier [28]. The following are some of the benefits of data normalization.
- Data normalization aids in the improvement of data speed, accuracy, and efficiency, as well as the removal of undesired outliers.
- It aids in the reduction of data alteration issues.
- Data normalization reduces duplication, by ensuring that only relevant data is stored in each table.
The mathematical expression of data normalization is:
where is the maximum value of the input data and is the minimum value of the input data.
3.3. The Proposed Model
The tremendous feature extraction capacity of a convolutional neural network (CNN) [29] allows it to mine data extensively. A CNN is a two-dimensional image processing and financial signal processing algorithm [30]. To extract pixel information and achieve image identification and classification, a convolution kernel flows smoothly over an image. As the input data for predicting the remaining useful life of chemical properties are mostly one-dimensional, these features cannot be retrieved using a two-dimensional convolution kernel. As a result, a 1D-CNN [31] model is utilized to collect sensitive information and achieve the feature extraction of molecular properties in the feature extraction stage, based on the properties of the sequence data. In comparison to classic feature extraction approaches, CNN can extract data characteristics without requiring a lot of user effort or expert knowledge. Figure 2 shows the overview of the proposed one-dimensional convolutional gated recurrent unit neural network, and Table 2 shows the proposed model (1D-CNN-GRU) parameters.
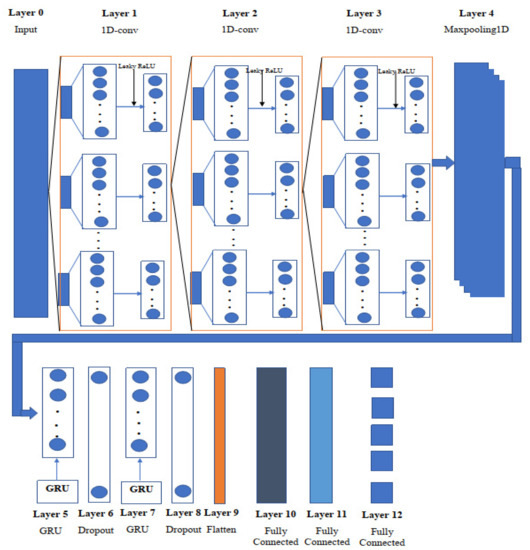
Figure 2.
Schematic diagram of the one-dimensional convolutional gated recurrent neural network.
Table 2.
Proposed Model (1D-CNN-GRU) Parameters.
In this study, the molecular property dataset is adjusted before being input into the proposed one-dimensional convolutional gated recurrent neural network. The ID-CNN GRU architecture, dataset, training, and optimization algorithms will be detailed in depth in the next sections. All experiments were carried out using the Python programming language, which includes libraries such as numpy, pandas, seaborn, matplotlib, joyplot, sklearn, keras, and math.
3.4. 1D-CNN-GRU Architecture
To construct the unified neural network, the network consists of 12 layers: three 1D convolutional neural network (1D-CNN) layers, two Gated Recurrent Unit (GRU) [32] levels, two dropout layers, one maxpooling, one flatten layer, and three fully connected layers. The signals are initially routed via the first convolutional layer. The kernel utilized in the first layer of our proposed 1D-Convolutional GRU network is set to 2, which indicates that all weights will be shared by every stride of the input layer of atom index 0 and atom index 1, as well as the scalar coupling constant outputs. In the kernel window, input values multiplied by weights are added together and utilized to build the feature map value. The first convolution layer’s output is 2 × 128, but it is also the input to the following convolutional layer. The filter size is lowered to 64 in the second 1D-convolutional layer, from 128 in the first, and the kernel size is adjusted to 3 for all 1D convolutional layers. In addition, the input size in the third 1D-convolutional layer is 2 × 64, with padding set to the same value in all three 1D-convolutional layers.
The mathematical expression of the 1D-convolutional layer is as follows:
where represents the Leaky ReLU (Rectified Linear Unit) activation function, represents the number of feature maps in the layer, represents the one-dimensional convolutional neural network operation with same padding, represents the trainable one-dimensional convolutional kernel, represent th feature map, and represents the bias of the the feature map.
In all 1D-convolutional neural networks, the feature maps are subsequently input into the activation leaky ReLU function. The following is the mathematical equation for Leaky ReLU:
β()={βx, for 0 < x ≤ 0}
Layer 4 uses maxpooling to assist downsampling the input representation, by taking the maximum value over a spatial window of size, with hyperparameters such as pool size set to 3, padding set to “same”, and strides set to 2.
The maxpooling can be expressed as:
where is the neuron before maxpooling operation, is the neuron after maxpooling operation, and is the size of the pooling.
A gated recurrent unit (GRU) was implemented in the fifth layer with 128 neurons to overcome the vanishing gradient problem of 1D-CNN, and the activity regularizer of layer 2 was adjusted to 0.001, to penalize the layer output. Update and reset gates are the two gates used by GRU. These two gates determine which data are sent to the output. The network unit plugs in the update gate with input and multiplies it by its own weight . The same is true for , which stores data from earlier (t − 1) units and multiplies it by its own weight U(z). Both results were combined together, and the result was squashed using a Leaky ReLU activation function with an alpha of 0.001. The update gate assists the model in determining how much past data should be passed on to the future. The mathematical expression of the update gates is:
where is the update gate at time step is the current sample input at time step is the weight of the update gate, is the previous hidden state at time step , and is the weight of the update gate.
In the GRU model, a reset gate is utilized to determine how much information from the past should be forgotten. This formula is identical to the one used for the update gate. The weights and gates are what make the difference. We plugged in and , multiply with their respective weights, added the results, and applied the Leaky ReLU activation function as before.
The mathematical expression of the reset gate is as follows:
where is the reset gate at time step is the current sample input at time step is the weight of the reset gate, is the previous hidden state at time step , and is the weight of the reset gate.
To keep important information from the past, new memory content is introduced that replaces the tanh activation function with a Leaky ReLU activation function . This involves multiply the input with a weight and with a weight Furthermore, the element-wise product of reset gate and previous hidden state are calculated. This decides which time steps should be removed from the preceding ones.
The mathematical expression is as follows:
where is the current memory at time step is the current sample input at time step is the weight of networks, is the previous hidden state at time step , and is the weight of the previous hidden state.
The update gate is necessary to retrieve the current final memory. It decides what to gather from the present memory content and past stages This is accomplished by multiplying the update gate and element-by-element and multiplying and element-by-element. After this, the two are added together.
The mathematical expression is as follows:
where is the current memory at time step , and is the previous hidden state at time step Layer 6 receives a matrix input from the GRU layer, which has 128 neurons, and a dropout layer is employed to reduce overfitting using a dropout of 0.2. At the end of each cycle, the dropout picks a few nodes at random and eliminates them, together with all of their incoming and outgoing connections.
The mathematical expression of the dropout layer can be expressed as:
where is the Bernouli random variable that deletes the weight with a probability between 0 and 1. After using the dropout layer, the GRU layer was considered in layer 7 by feeding the layer with 1 128 vector and using the step described in layer 5 with the addition of a kernel initializer set to random uniform with a seed of 42. Layer 8 also uses the dropout layer to prevent the network from overfitting. In layer 9, a flatten layer is used to reduce the input data to a single dimension. The single dimension from the flatten layer is input into the fully connected layer 10 with 512 nodes, and the leaky ReLU activation function is used in all the remaining activation functions. Layer 11 is made up of 256 nodes and is fully linked. Data is supplied into the final fully connected layers with a linear activation function, after passing through Layer 11 for the final prediction.
3.5. Optimization Algorithm
An optimizer is used to minimize the model loss function value and discover the best solution to acquire appropriate model parameters. The Adam (adaptive moment estimation) [33] optimizer is one of the most widely used in neural networks because it outperforms the previous optimizers by combining the best properties of the AdaGrad (adaptive gradient algorithm) [34] and RMSProp (root mean squared propagation) [35,36] algorithms to create an optimization algorithm that can handle sparse gradients in noisy problems.
where is the learning rate, , , are the exponential decay rates for the moment estimates, is the aggregate of gradients at t, is the aggregate of gradients at time t-1, is the derivation of loss function, derivation of weight at time is the weight at time weight at time and is the sum of the square of past gradients.
4. Results and Discussion
Pharmaceutical industries are witnessing a major transformation with the introduction of machine learning and deep learning techniques. While many see this new technology as a potential threat, it could be the solution to our persistent pharmaceutical deficits. Indeed, AI has already proven to be useful in several aspects of pharmaceutical research, from assisting researchers to find new possible treatments, to predicting which pharmaceuticals will fail clinical testing. There is really no doubt that these innovations will have a profound impact on the development of drugs, as more pharmaceutical firms embrace them. Drug development is a time-consuming and expensive venture. Moreover, it is a gradual procedure, that starts with the discovery of a promising molecule, and finishes with the creation of a new molecular entity. This research is important to pharmaceutical companies, particularly in the production of liquid medications and drugs for medicinal purposes, because it aids in the discovery of an effective particle, whose end purpose is to affect the human body and demonstrate its effectiveness, pureness, and relevance in medical therapy. The preceding conditions ensure that the new pharmaceuticals that are approved by the authorities improve patients’ wellbeing, not only through curing their disease but also through guaranteeing that the treatment does not cause serious side effects and lead to other problems, such as potential complications.
The simulation results achieved in this paper can be used to improve the quality and contents of liquid and solid drugs, by determining the molecular properties of the drugs. Both medication reactions in the body over time, and drug effects on the body or on microorganisms within or on the body, are influenced by physico-chemical and molecular characteristics. This could also address challenging issues around medication safety. In this regard, the current tendency in drug development is to take absorption, distribution, metabolism, excretion, and toxicity (ADMET) qualities into account alongside target affinity. To help medicinal chemists prioritize drug ideas, the idea of “drug-likeness” establishes acceptable limitations of fundamental features stated as a set of rules. Liquid drug makers investigating the treatments for illnesses such as Alzheimer’s, cancer, and other challenging conditions are interested in learning about new targets that could be the primary goal of a new drug development initiative.
As an algorithm-based strategy, deep learning incorporates numerous mathematical and computational ideas. It can analyze vast volumes of data and has unwavering coordination and matching powers. It can be used to enhance drug discovery tasks such as target recognition, drug design, and drug repurposing. Instead of rendering scientists’ jobs obsolete, it serves a crucial role in developing their talents. This is due to the need for subject matter experts in the data structuring for AI analysis, as well as benefitting the review process and end-to-end result confirmation. Conventional drug development can only analyze a small number of tests or data at a time, increasing the risk of prejudice.
The performance of the recurrent neural network (RNN), long short term memory (LSTM), gated recurrent unit (GRU), one-dimensional convolutional neural network (1D-CNN), and the proposed 1D convolutional gated recurrent unit neural network (1D-CNN-GRU) molecular property systems is explained in this section. Table 3 shows the summary statistics for each of the columns of molecular properties. The mean, standard deviation, minimum, 25%, 50%, 75%, and maximum are analyzed. The mean of atom_index_1 is small compared to other molecular properties. Atom_index_1 also has a small volatility of 3.27 compared with other molecular properties, which indicating that the atom index fluctuates slowly and tends to be more stable, while Id has a maximum value according to Table. The minimum value of the molecular property dataset is within the range of 0 to −44. The coupling type consists of 3JHC, 2JHC, 1JHC, 3JHH, 2JHH, 3JHN, 2JHN, and 1JHN, as shown in the bar graph of Figure 3. 3JHC came in first position with the highest value of 1511207, followed by 2JHC with the value of 1140867, while 1JHN came last with the lowest value of 43680. The top 10 molecule names for molecular properties is shown in Figure 4. The molecule name that tops the list is dsgdb9nsd_042139, having the highest, followed by dsgdb9nsd_096580, dsgdb9nsd_121391, dsgdb9nsd_122665, and dsgdb9nsd_123139.
Table 3.
Summary Statistics of the Molecular Properties Dataset.
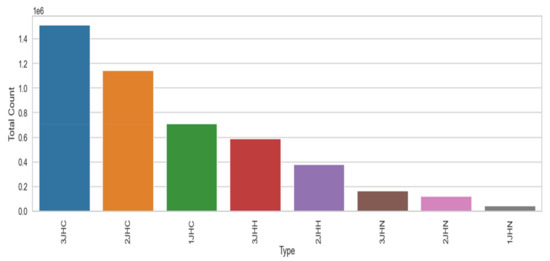
Figure 3.
The coupling type of molecular property.
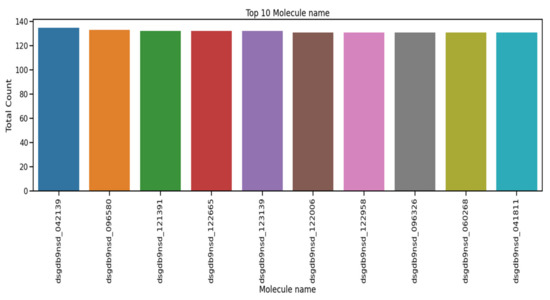
Figure 4.
Top 10 molecule names for molecular properties.
Figure 5 shows the difference in the density of atom_index 0 and 1, which are aligned to the same horizontal scale. The graph revealed the distribution of atom index_0 and atom_index_1.
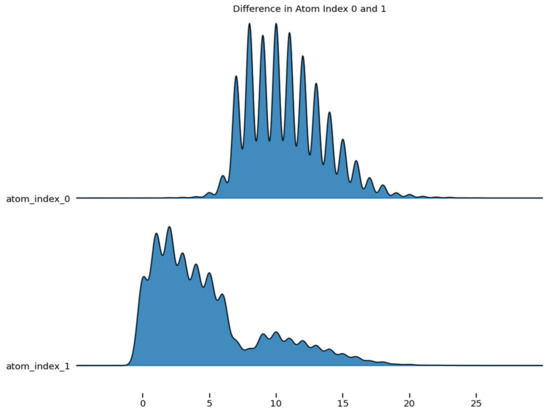
Figure 5.
Difference in the atom index of molecular properties.
The training loss and validation loss of deep learning algorithms considered in this study, such as RNN, LSTM, GRU, 1D-CNN, and the proposed method based on the 1D-CNN-GRU model when it was applied to the target variable of the scalar coupling constant are given in Figure 6, Figure 7, Figure 8, Figure 9 and Figure 10. It can be seen from Figure 10 that the proposed 1D-CNN-GRU training loss and validation loss decreased at the lowest speed. The training and validation loss values of the proposed model were obviously smaller than the values of the other deep learning models, thus obtaining better training and validation accuracies. However, although the training and testing performance of the standard 1DCNN was higher to that of the proposed model and combining them with GRU greatly improved the performance. The performance evaluation and errors of the different models, such as LSTM, RNN, GRU, 1D-CNN, and 1D-CNN-GRU, are presented in Table 4. It is noticeable from Table 4 that the metrics of MSE, RMSE, and MAE for the proposed 1D-CNN-GRU were relatively small. Due to the combination of 1D-CNN and GRU network parameter initialization, the output features of the proposed 1D-CNN-GRU model had a significant improvement over all other deep learning models. Figure 11, Figure 12, Figure 13, Figure 14 and Figure 15 is the prediction result of all the deep learning models considered in this study. Figure 15 shows that 1D-CNN-GRU can predict the scalar coupling constant better than the other deep learning models.
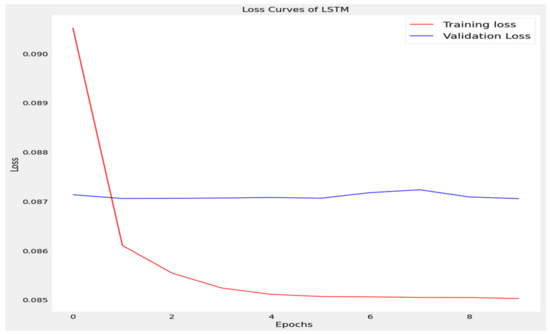
Figure 6.
Loss curve of long short term memory (LSTM).
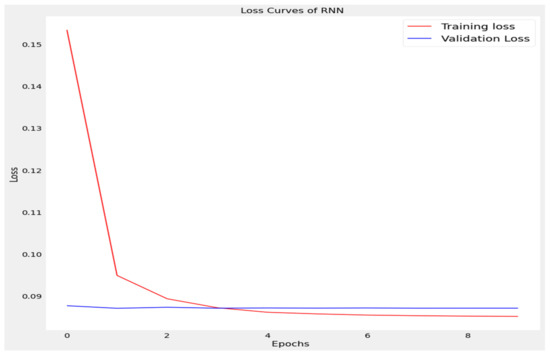
Figure 7.
Loss curve of recurrent neural network (RNN).
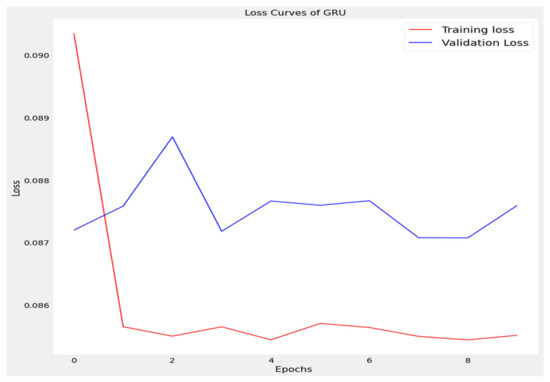
Figure 8.
Loss curve of gated recurrent unit (GRU).
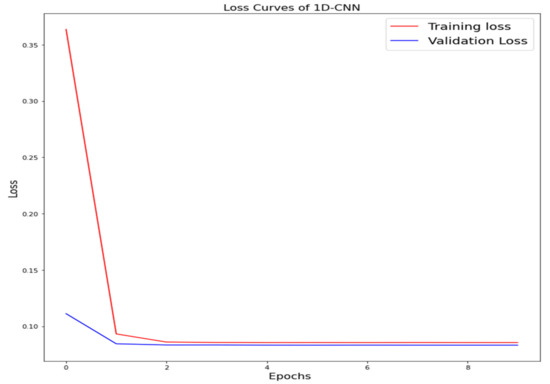
Figure 9.
Loss curve of one-dimensional convolutional neural network (1D-CNN).
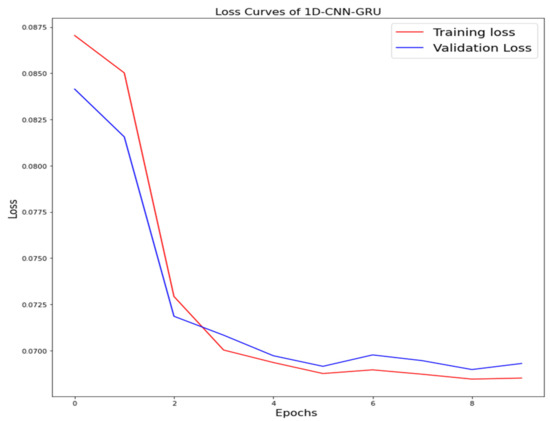
Figure 10.
Loss curve of one-dimensional convolutional gated recurrent unit neural network (1D-CNN-GRU).
Table 4.
Performance Evaluation of LSTM, RNN, GRU, 1D-CNN, and 1D-CNN-GRU.
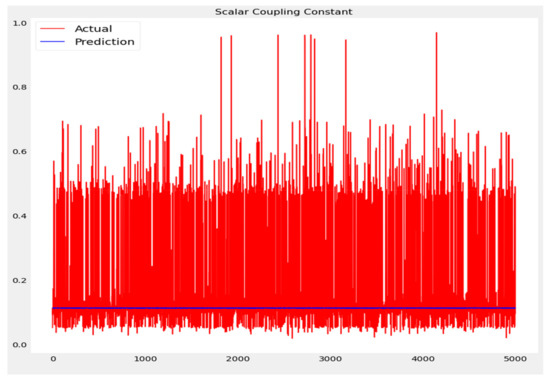
Figure 11.
Actual and prediction values of LSTM.
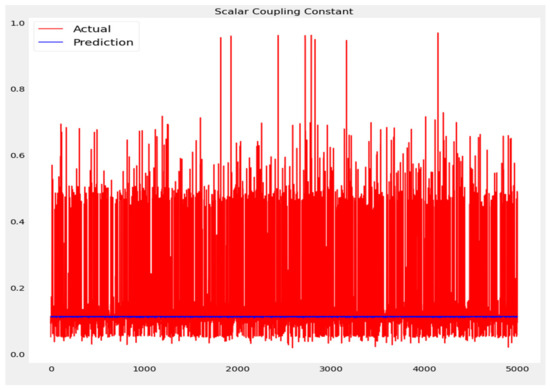
Figure 12.
Actual and prediction values of RNN.
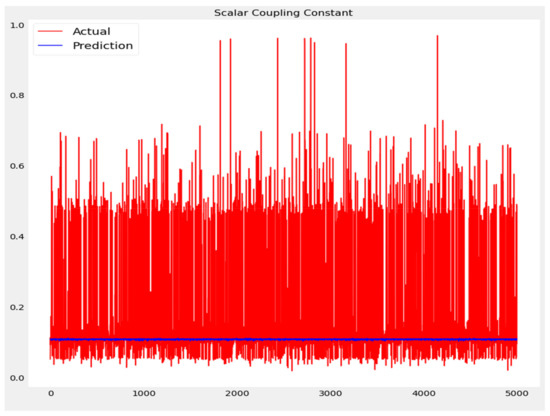
Figure 13.
Actual and prediction values of GRU.
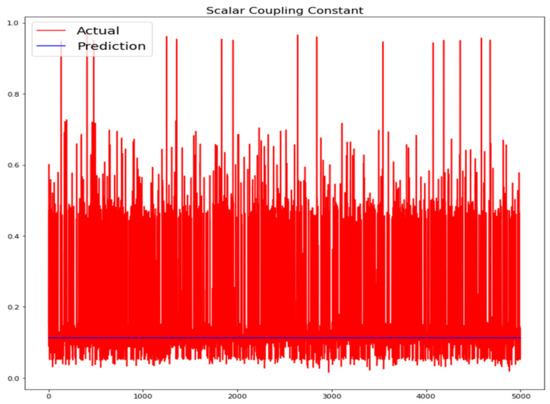
Figure 14.
Actual and prediction values of 1D-CNN.
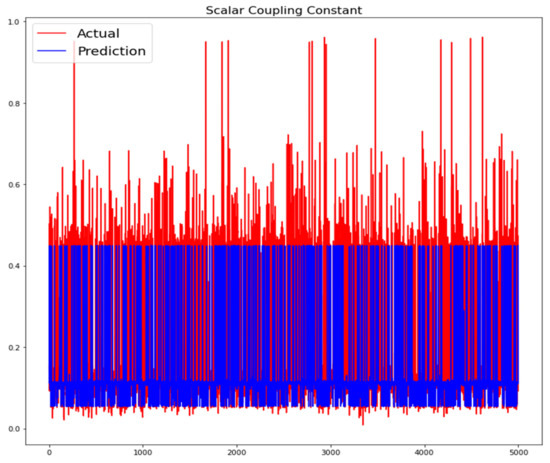
Figure 15.
Actual and prediction values of 1D-CNN-GRU.
5. Conclusions
A fundamental component of a successful drug development process is computational prediction of the molecular characteristics. In the chemical and pharmaceutical industries, simultaneous accurate and efficient prediction of molecular characteristics throughout the chemical compound space is a fundamental component of rational compound design.
For molecular property signal analysis, a 1D-CNN-GRU model is presented in this study. To build an end-to-end network that can predict scalar coupling constant signals, the proposed model combines a 1D-CNN with a GRU. The 1D-CNN-GRU offers a powerful signal feature extraction capability, as well as the capacity to forecast successive scalar coupling constant signals. Experiments on a molecular property data set gathered from Kaggle were used to validate the proposed model performance. When compared to other models, such as LSTM, RNN, GRU, and 1D-CNN, the suggested model obtained the lowest prediction error: 0.0230, 0.1517, and 0.0693 for MSE, RMSE, and MAE, respectively.
Despite the fact that the presented model has made significant progress in the field of molecular property prediction, it still has limitations that must be addressed in the future. There should be a means to increase the value of the input variables, such as atom index 0 and atom index 1. Other hyperparameters may be incorporated in both 1D- CNN and GRU as well. Based on these restrictions, future studies will concentrate on two areas: first, the proposed model may be improved by modifying it and using alternative optimizers and Bayesian optimization for the more complicated molecular property dataset. The proposed model can also benefit from dimension reduction.
Author Contributions
Conceptualization, D.O.O. and E.G.D.; methodology, D.O.O.; software, D.O.O.; validation, O.E., O.O.O., and O.O.O.; formal analysis, D.O.O.; investigation, D.O.O.; resources, O.O.O.; data curation, D.O.O.; writing—original draft preparation, O.O.O.; writing—review and editing, O.O.O.; visualization, D.O.O.; supervision, O.E., O.O.O.; project administration, D.O.O.; funding acquisition, E.G.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are available from the Tech, United States of America.
Acknowledgments
O.E. was supported by institutional research funding from Virginia Tech, United States of America.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Becke, A. The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design; John Wiley & Sons: Hoboken, NJ, USA, 2007. [Google Scholar]
- Oglic, D.; Garnett, R.; Gärtner, T. Active search in intensionally specified structured spaces. In Proceedings of the AAAI Conference on Artificial Intelligence, San Francisco, CA, USA, 4–9 February 2017; Volume 31. [Google Scholar]
- Thouless, D.J. The Quantum Mechanics of Many-Body Systems; Courier Corporation: Washington, DC, USA, 2014. [Google Scholar]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Gilmer, J.; Schoenholz, S.S.; Riley, P.F.; Vinyals, O.; Dahl, G.E. Neural message passing for quantum chemistry. In International Conference on Machine Learning; PMLR: New York City, NY, USA, 2017; pp. 1263–1272. [Google Scholar]
- Rupp, M. Machine learning for quantum mechanics in a nutshell. Int. J. Quantum Chem. 2015, 115, 1058–1073. [Google Scholar] [CrossRef]
- Zaspel, P.; Huang, B.; Harbrecht, H.; von Lilienfeld, O.A. Boosting quantum machine learning models with a multilevel combination technique: Pople diagrams revisited. J. Chem. Theory Comput. 2018, 15, 1546–1559. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, R.; Dral, P.O.; Rupp, M.; von Lilienfeld, O.A. Big data meets quantum chemistry approximations: The Δ-machine learning approach. J. Chem. Theory Comput. 2015, 11, 2087–2096. [Google Scholar] [CrossRef]
- Smith, J.S.; Nebgen, B.T.; Zubatyuk, R.; Lubbers, N.; Devereux, C.; Barros, K.; Tretiak, S.; Isayev, O.; Roitberg, A. Outsmarting Quantum Chemistry through Transfer Learning. 2019, pp. 1–32. Available online: www.chemrxiv.org (accessed on 10 March 2022).
- Smith, J.S.; Isayev, O.; Roitberg, A.E. ANI-1: An extensible neural network potential with DFT accuracy at force field computational cost. Chem. Sci. 2017, 8, 3192–3203. [Google Scholar] [CrossRef]
- Unke, O.T.; Meuwly, M. A reactive, scalable, and transferable model for molecular energies from a neural network approach based on local information. J. Chem. Phys. 2018, 148, 241708. [Google Scholar] [CrossRef]
- Zubatyuk, R.; Smith, J.S.; Leszczynski, J.; Isayev, O. Accurate and transferable multitask prediction of chemical properties with an atoms-in-molecules neural network. Sci. Adv. 2019, 5, eaav6490. [Google Scholar] [CrossRef]
- Faber, F.A.; Hutchison, L.; Huang, B.; Gilmer, J.; Schoenholz, S.S.; Dahl, G.E.; Vinyals, O.; Kearnes, S.; Riley, P.F.; von Lilienfeld, O.A. Prediction errors of molecular machine learning models lower than hybrid DFT error. J. Chem. Theory Comput. 2017, 13, 5255–5264. [Google Scholar] [CrossRef]
- Schütt, K.T.; Sauceda, H.E.; Kindermans, P.J.; Tkatchenko, A.; Müller, K.R. Schnet–A deep learning architecture for molecules and materials. J. Chem. Phys. 2018, 148, 241722. [Google Scholar] [CrossRef]
- Karpathy, A.; Toderici, G.; Shetty, S.; Leung, T.; Sukthankar, R.; Fei-Fei, L. Large-scale video classification with convolutional neural networks. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Columbus, OH, USA, 23–28 June 2014; pp. 1725–1732. [Google Scholar]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep residual learning for image recognition. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Las Vegas, NV, USA, 27–30 June 2016; pp. 770–778. [Google Scholar]
- Huang, B.; Von Lilienfeld, O.A. Communication: Understanding molecular representations in machine learning: The role of uniqueness and target similarity. J. Chem. Phys. 2016, 145, 161102. [Google Scholar] [CrossRef]
- Zhu, H.; Liu, Q.; Yuan, N.J.; Qin, C.; Li, J.; Zhang, K.; Zhou, G.; Wei, F.; Xu, Y.; Chen, E. Xiaoice band: A melody and arrangement generation framework for pop music. In Proceedings of the 24th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, London, UK, 19–23 August 2018; pp. 2837–2846. [Google Scholar]
- Liu, Q.; Huang, Z.; Huang, Z.; Liu, C.; Chen, E.; Su, Y.; Hu, G. Finding similar exercises in online education systems. In Proceedings of the 24th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, London, UK, 19–23 August 2018; pp. 1821–1830. [Google Scholar]
- Kollman, P. Theory of complex molecular interactions: Computer graphics, distance geometry, molecular mechanics, and quantum mechanics. Acc. Chem. Res. 1985, 18, 105–111. [Google Scholar] [CrossRef]
- McDonagh, J.L.; Silva, A.; Vincent, M.A.; Popelier, P.L.A. Machine learning of dynamic electron correlation energies from topological atoms. J. Chem. Theory Comput. 2017, 14, 216–224. [Google Scholar] [CrossRef]
- Huang, B.; von Lilienfeld, O.A. Ab initio machine learning in chemical compound space. Chem. Rev. 2021, 121, 10001–10036. [Google Scholar] [CrossRef]
- Montavon, G.; Hansen, K.; Fazli, S.; Rupp, M.; Biegler, F.; Ziehe, A.; Tkatchenko, A.; Lilienfeld, A.; Müller, K.R. Learning invariant representations of molecules for atomization energy prediction. Adv. Neural Inf. Process. Syst. 2012, 25, 440–448. [Google Scholar]
- Goh, G.B.; Siegel, C.; Vishnu, A.; Hodas, N. Using rule-based labels for weak supervised learning: A ChemNet for transferable chemical property prediction. In Proceedings of the 24th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, London, UK, 19–23 August 2018; pp. 302–310. [Google Scholar]
- Sun, M.; Zhao, S.; Gilvary, C.; Elemento, O.; Zhou, J.; Wang, F. Graph convolutional networks for computational drug development and discovery. Brief. Bioinform. 2019, 21, 919–935. [Google Scholar] [CrossRef]
- Wang, J.; Hou, T. Application of molecular dynamics simulations in molecular property prediction II: Diffusion coefficient. J. Comput. Chem. 2011, 32, 3505–3519. [Google Scholar] [CrossRef]
- Predicting Molecular Properties. Can You Measure the Magnetic Interactions between a Pair of Atoms? Kaggle. 2019. Available online: https://www.kaggle.com/c/champs-scalar-coupling (accessed on 20 March 2022).
- Singh, D.; Singh, B. Investigating the impact of data normalization on classification performance. Appl. Soft Comput. 2020, 97, 105524. [Google Scholar] [CrossRef]
- Oyewola, D.O.; Dada, E.G.; Misra, S.; Damaševičius, R. A novel data augmentation convolutional neural network for detecting malaria parasite in blood smear images. Appl. Artif. Intell. 2022, 36, 1–22. [Google Scholar] [CrossRef]
- Oyewola, D.O.; Dada, E.G.G.; Olaoluwa, O.E.; Al-Mustapha, K. Predicting Nigerian stock returns using technical analysis and machine learning. Eur. J. Electr. Eng. Comput. Sci. 2019, 3. [Google Scholar] [CrossRef]
- Yao, D.; Li, B.; Liu, H.; Yang, J.; Jia, L. Remaining useful life prediction of roller bearings based on improved 1D-CNN and simple recurrent unit. Measurement 2021, 175, 109166. [Google Scholar] [CrossRef]
- Wei, M.; Gu, H.; Ye, M.; Wang, Q.; Xu, X.; Wu, C. Remaining useful life prediction of lithium-ion batteries based on Monte Carlo Dropout and gated recurrent unit. Energy Rep. 2021, 7, 2862–2871. [Google Scholar] [CrossRef]
- Arouri, Y.; Sayyafzadeh, M. An adaptive moment estimation framework for well placement optimization. Comput. Geosci. 2022, 1–17. [Google Scholar] [CrossRef]
- Xue, Y.; Wang, Y.; Liang, J. A self-adaptive gradient descent search algorithm for fully-connected neural networks. Neurocomputing 2022, 478, 70–80. [Google Scholar] [CrossRef]
- Nguyen, L.C.; Nguyen-Xuan, H. Deep learning for computational structural optimization. ISA Trans. 2020, 103, 177–191. [Google Scholar] [CrossRef]
- Oyewola, D.O.; Ibrahim, A.; Kwanamu, J.; Dada, E.G. A new auditory algorithm in stock market prediction on oil and gas sector in Nigerian stock exchange. Soft Comput. Lett. 2021, 3, 100013. [Google Scholar] [CrossRef]















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).