
Structural and Functional Shift in Soil Bacterial Community in Response to Long-Term Compost Amendment in Paddy Field



 ,
, 
Abstract
1. Introduction
2. Materials and Methods
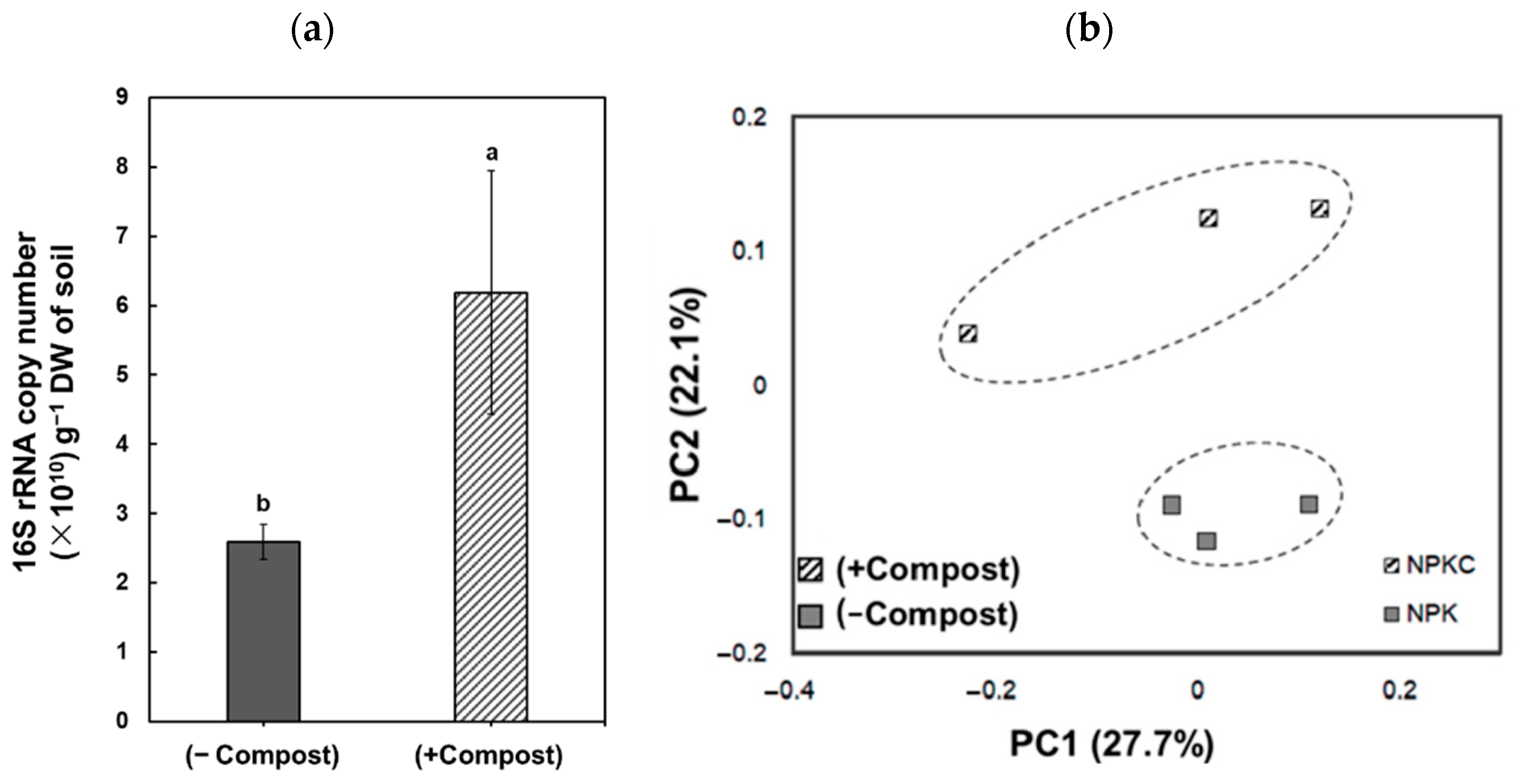
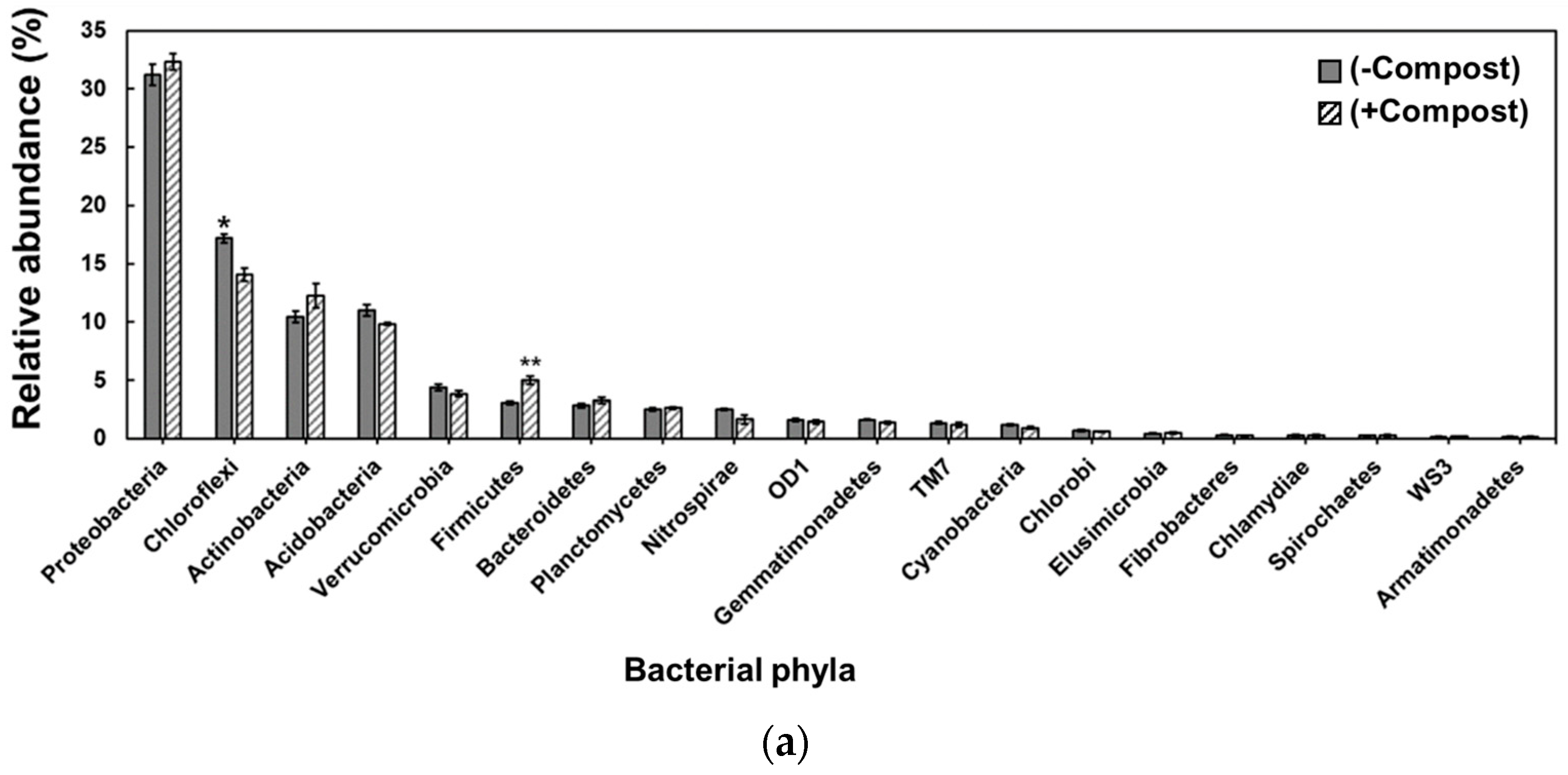
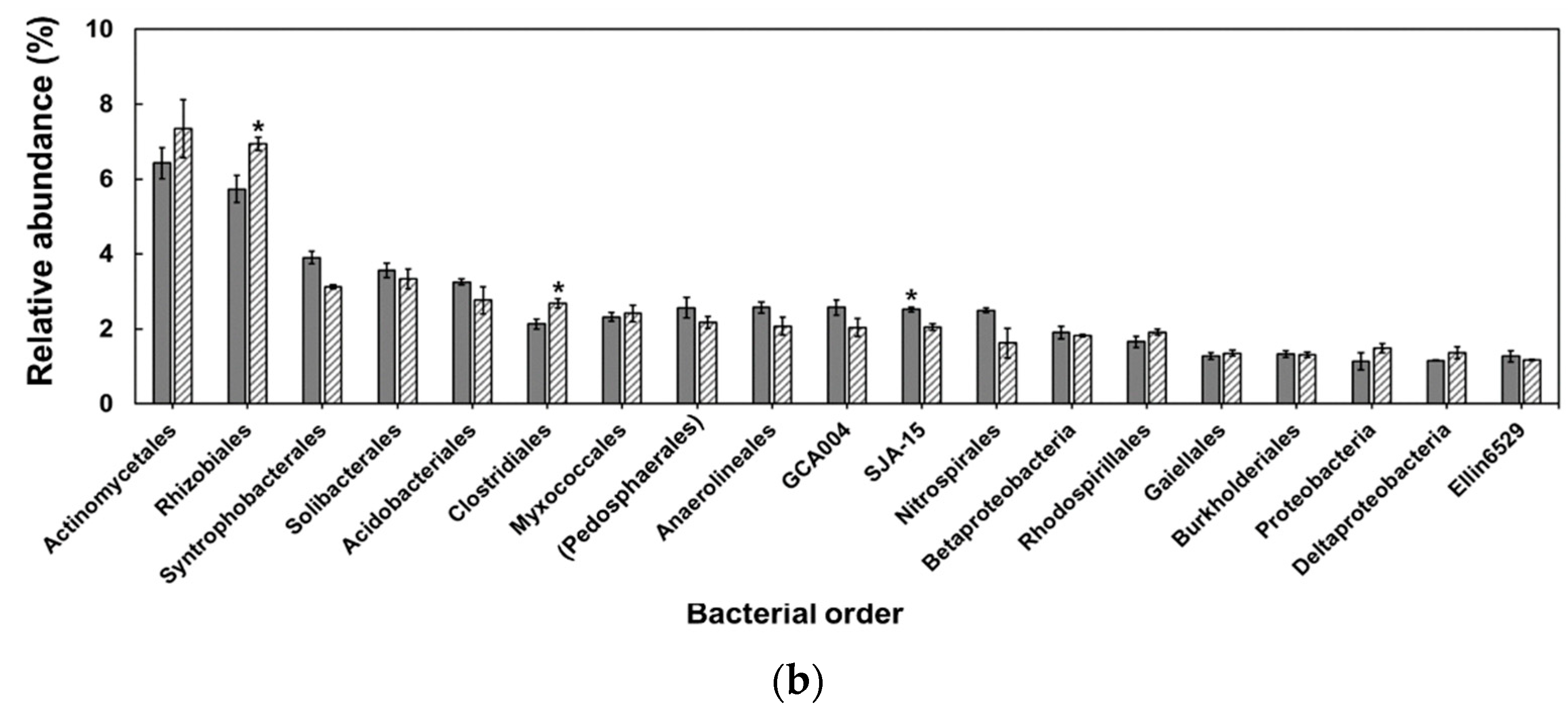
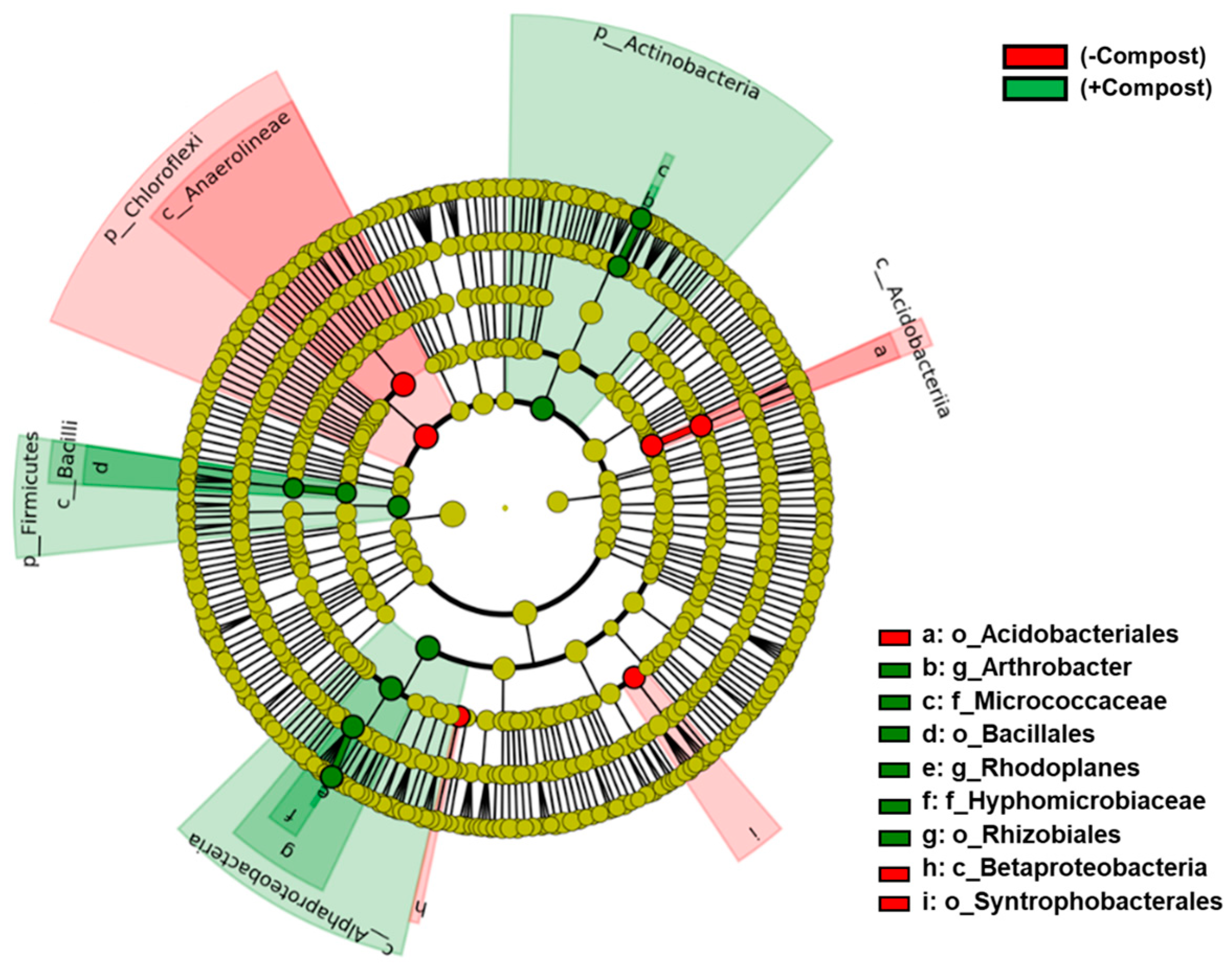
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fierer, N.; Lauber, C.L.; Ramirez, K.S.; Zaneveld, J.; Bradford, M.A.; Knight, R. Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. ISME J. 2012, 6, 1007–1017. [Google Scholar] [CrossRef]
- Zhang, Y.; Dong, S.; Gao, Q.; Liu, S.; Zhou, H.; Ganjurjav, H.; Wang, X. Climate change and human activities altered the diversity and composition of soil microbial community in alpine grasslands of the Qinghai-Tibetan Plateau. Sci. Total Environ. 2016, 562, 353–363. [Google Scholar] [CrossRef]
- Janssens, I.A.; Dieleman, W.; Luyssaert, S.; Subk, J.A.; Reichstein, M.; Ceulemans, R.; Ciais, P.; Dolman, A.J.; Grace, J.; Matteucci, G.; et al. Reduction of forest soil respiration in response to nitrogen deposition. Nat. Geosci. 2010, 3, 315–322. [Google Scholar] [CrossRef]
- Yadav, R.L.; Yadav, D.S.; Singh, R.M.; Kumar, A. Long term effects of inorganic fertilizer inputs on crop productivity in a rice-wheat cropping system. Nutr. Cycl. Agroecosyst. 1998, 51, 193–200. [Google Scholar] [CrossRef]
- Zhou, J.; Guan, D.; Zhou, B.; Zhao, B.; Ma, M.; Qin, J.; Jiang, X.; Chen, S.; Cao, F.; Shen, D.; et al. Influence of 34-years of fertilization on bacterial communities in an intensively cultivated black soil in northeast China. Soil Biol. Biochem. 2015, 90, 42–51. [Google Scholar] [CrossRef]
- Zhou, J.; Jiang, X.; Zhou, B.; Zhao, B.; Ma, M.; Guan, D.; Li, J.; Chen, S.; Cao, F.; Shen, D.; et al. Four years of nitrogen fertilization decreases fungal diversity and alters fungal community composition in black soil in northeast China. Soil Biol. Biochem. 2016, 95, 135–143. [Google Scholar] [CrossRef]
- Liu, M.; Sui, X.; Hu, Y.; Feng, F. Microbial community structure and the relationship with soil carbon and nitrogen in an original Korean pine forest of Changbai Mountain, China. BMC Microbiol. 2019, 19, 218. [Google Scholar] [CrossRef]
- Mäder, P.; Fliessbach, A.; Dubois, D.; Gunst, L.; Fried, P.; Niggli, U. Soil fertility and biodiversity in organic farming. Science 2002, 296, 1694–1697. [Google Scholar] [CrossRef] [PubMed]
- Barthod, J.; Rumpel, C.; Dignac, M.F. Composting with additives to improve organic amendments. A review. Agron. Sustain. Dev. 2018, 38, 17. [Google Scholar] [CrossRef]
- Van Zwieten, L. The long-term role of organic amendments in addressing soil constraints to production. Nutr. Cycl. Agroecosyst. 2018, 111, 99–102. [Google Scholar] [CrossRef]
- Tilman, D.; Reich, P.B.; Knops, J.M. Biodiversity and ecosystem stability in a decadelong grassland experiment. Nature 2006, 441, 629–632. [Google Scholar] [CrossRef]
- Ryan, L.F.; Daniel, B.R.; Dharni, V.; Mark, A.W.; Larry, T. Rhizogenic Fe-C redox cycling: A hypothetical biogeochemical mechanism that drives crustal weathering in upland soils. Biogeochemistry 2008, 87, 127–141. [Google Scholar]
- Poulsen, P.H.; Al-Soud, W.A.; Bergmark, L.; Magid, J.; Hansen, L.H.; Sørensen, S.J. Effects of fertilization with urban and agricultural organic wastes in a field trial–Prokaryotic diversity investigated by pyrosequencing. Soil Biol. Biochem. 2013, 57, 784–793. [Google Scholar] [CrossRef]
- Yuan, H.; Ge, T.; Zhou, P.; Liu, S.; Roberts, P.; Zhu, H.; Zou, Z.; Tong, C.; Wu, J. Soil microbial biomass and bacterial and fungal community structures responses to long-term fertilization in paddy soils. J. Soils Sediments 2013, 13, 877–886. [Google Scholar] [CrossRef]
- Biswas, B.; Pandey, N.; Bisht, Y.; Singh, R.; Kumar, J.; Bhaskar, T. Pyrolysis of agricultural biomass residues: Comparative study of corn cob, wheat straw, rice straw and rice husk. Bioresour. Technol. 2017, 237, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.F.; Meng, J.; Wang, Q.X.; Zhang, W.M.; Cheng, X.Y.; Chen, W.F. Effects of straw and biochar addition on soil nitrogen, carbon, and super rice yield in cold waterlogged paddy soils of North China. J. Integr. Agric. 2017, 16, 1064–1074. [Google Scholar] [CrossRef]
- Xia, L.; Lam, S.K.; Yan, X.; Chen, D. How does recycling of livestock manure in agroecosystems affect crop productivity, reactive nitrogen losses, and soil carbon balance? Environ. Sci. Technol. 2017, 51, 7450–7457. [Google Scholar] [CrossRef] [PubMed]
- Samaddar, S.; Truu, J.; Chatterjee, P.; Truu, M.; Kim, K.; Kim, S.; Seshadri, S.; Sa, T. Long-term silicate fertilization increases the abundance of Actinobacterial population in paddy soils. Biol. Fertil. Soils 2019, 55, 109–120. [Google Scholar] [CrossRef]
- Liu, N.; Gumpertz, M.L.; Hu, S.; Ristaino, J.B. Long-term effects of organic and synthetic soil fertility amendments on soil microbial communities and the development of southern blight. Soil Biol. Biochem. 2007, 39, 2302–2316. [Google Scholar] [CrossRef]
- Huse, S.M.; Dethlefsen, L.; Huber, J.A.; Welch, D.M.; Relman, D.A.; Sogin, M.L. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 2008, 4, 1000255. [Google Scholar] [CrossRef]
- Ruijter, J.M.; Ramakers, C.; Hoogaars, W.M.H.; Karlen, Y.; Bakker, O.; Van den Hoff, M.J.B.; Moorman, A.F.M. Amplification efficiency: Linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009, 37, e45. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Opensource, platform-independent, community supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Samaddar, S.; Chatterje, P.; Truu, J.; Anandham, R.; Kim, S.; Sa, T. Long-term phosphorus limitation changes the bacterial community structure and functioning in paddy soils. Appl. Soil Ecol. 2019, 134, 111–115. [Google Scholar] [CrossRef]
- SAS Institute Inc. Base Sas 9.4 Procedures Guide: Statistical Procedures, 2nd ed.; SAS Institute Inc.: Cary, NC, USA, 2013. [Google Scholar]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, 1–18. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Li, W.X.; Wang, C.; Zheng, M.M.; Cai, Z.J.; Wang, B.R.; Shen, R.F. Fertilization strategies affect soil properties and abundance of N-cycling functional genes in an acidic agricultural soil. Appl. Soil Ecol. 2020, 156, 103704. [Google Scholar] [CrossRef]
- Calleja-Cervantes, M.E.; Fernández-González, A.J.; Irigoyen, I.; Fernández-López, M.; Aparicio-Tejo, P.M.; Menéndez, S. Thirteen years of continued application of composted organic wastes in a vineyard modify soil quality characteristics. Soil Biol. Biochem. 2015, 90, 241–254. [Google Scholar] [CrossRef]
- Tian, W.; Wang, L.; Li, Y.; Zhuang, K.; Li, G.; Zhang, J.; Xiao, X.; Xi, Y. Responses of microbial activity, abundance, and community in wheat soil after three years of heavy fertilization with manure-based compost and inorganic nitrogen. Agric. Ecosyst. Environ. 2015, 213, 219–227. [Google Scholar] [CrossRef]
- Hartmann, M.; Widmer, F. Community structure analyses are more sensitive to differences in soil bacterial communities than anonymous diversity indices. Appl. Environ. Microbiol. 2006, 72, 7804–7812. [Google Scholar] [CrossRef] [PubMed]
- Shade, A. Diversity is the question, not the answer. ISME J. 2017, 11, 1–6. [Google Scholar] [CrossRef]
- Janssen, P.H. Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl. Environ. Microbiol. 2006, 72, 1719–1728. [Google Scholar] [CrossRef]
- Spain, A.M.; Krumholz, L.R.; Elshahed, M.S. Abundance, composition, diversity and novelty of soil Proteobacteria. ISME J. 2009, 3, 992–1000. [Google Scholar] [CrossRef]
- Zhalnina, K.; Dias, R.; de Quadros, P.D.; Davis-Richardson, A.; Camargo, F.A.; Clark, I.M.; McGrath, S.P.; Hirsch, P.R.; Triplett, E.W. Soil pH determines microbial diversity and composition in the park grass experiment. Microb. Ecol. 2015, 69, 395–406. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, P.; Li, J.; Chen, Y.; Ying, X.; Liu, S. The effects of two organic manures on soil properties and crop yields on a temperate calcareous soil under a wheat-maize cropping system. Eur. J. Agron. 2009, 31, 36–42. [Google Scholar] [CrossRef]
- Tanahashi, T.; Murase, J.; Matsuya, K.; Hayashi, M.; Kimura, M.; Asakawa, S. Bacterial communities responsible for the decomposition of rice straw compost in a Japanese rice paddy field estimated by DGGE analysis of amplified 16S rDNA and 16S rRNA fragments. Soil Sci. Plant Nutr. 2005, 51, 351–360. [Google Scholar] [CrossRef]
- Sharmin, F.; Wakelin, S.; Huygens, F.; Hargreaves, M. Firmicutes dominate the bacterial taxa within sugar-cane processing plants. Sci. Rep. 2013, 3, 3107. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Su, W.; Chen, H.; Barberán, A.; Zhao, H.; Yu, M.; Yu, L.; Brookes, P.C.; Schadt, C.W.; Chang, S.X.; et al. Long-term nitrogen fertilization decreases bacterial diversity and favors the growth of Actinobacteria and Proteobacteria in agro-ecosystems across the globe. Glob. Change Biol. 2018, 24, 3452–3461. [Google Scholar] [CrossRef]
- Tsoy, O.V.; Ravcheev, D.A.; Čuklina, J.; Gelfand, M.S. Nitrogen fixation and molecular oxygen: Comparative genomic reconstruction of transcription regulation in Alphaproteobacteria. Front. Microbiol. 2016, 7, 1343. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Beiko, R.G.; Langille, M.G. Predicting the functional potential of the microbiome from marker genes using PICURSt. In Microbiome Analysis; Beiko, R., Hsiao, W., Parkinson, J., Eds.; Humana Press: New York, NY, USA, 2018; Volume 1849, pp. 169–177. [Google Scholar]
- Pii, Y.; Borruse, L.; Brusetti, L.; Crecchio, C.; Cesco, S.; Mimmo, T. The interaction between iron nutrition, plant species and soil type shapes the rhizosphere microbiome. Plant Physiol. Biochem. 2016, 99, 39–48. [Google Scholar] [CrossRef]
- Zarraonaindia, I.; Owens, S.M.; Weisenhorn, P.; West, K.; Hampton-Marcell, J.; Lax, S.; Bokulich, N.A.; Mills, D.A.; Martin, G.; Taghavi, S.; et al. The soil microbiome influences grapevine-associated microbiota. mBio 2015, 6, e02527-14. [Google Scholar] [CrossRef] [PubMed]
- Dube, J.P.; Valverde, A.; Steyn, J.M.; Cowan, D.A.; Van der Walls, J.E. Differences in bacterial diversity, composition and function due to long-term agriculture in soils in the eastern free State of South Africa. Diversity 2019, 11, 61. [Google Scholar] [CrossRef]
- Zhong, Y.; Yang, Y.; Liu, P.; Xu, R.; Rensing, C.; Fu, X.; Liao, H. Genotype and rhizobium inoculation modulate the assembly of soybean rhizobacterial communities. Plant Cell Environ. 2019, 42, 2028–2044. [Google Scholar] [CrossRef]
- Glick, B.R. Bacteria with ACC deaminase can promote plant growth and help to feed the world. Microbiol. Res. 2014, 169, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Pollmann, S.; Neu, D.; Weiler, E.W. Molecular cloning and characterization of an amidase from Arabidopsis thaliana capable of converting indole-3-acetamide into the plant growth hormone, indole-3-acetic acid. Phytochemistry 2003, 62, 293–300. [Google Scholar] [CrossRef]
- Koyama, H.; Kawamura, A.; Kihara, T.; Hara, T.; Takita, E.; Shibata, D. Overexpression of mitochondrial citrate synthase in Arabidopsis thaliana improved growth on a phosphorus-limited soil. Plant Cell Physiol. 2000, 41, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Barone, P.; Rosellini, D.; LaFayette, P.; Bouton, J.; Veronesi, F.; Parrott, W. Bacterial citrate synthase expression and soil aluminum tolerance in transgenic alfalfa. Plant Cell Rep. 2008, 27, 893–901. [Google Scholar] [CrossRef] [PubMed]
- López-Mondéjar, R.; Zühlke, D.; Becher, D.; Riedel, K.; Baldrian, P. Cellulose and hemicellulose decomposition by forest soil bacteria proceeds by the action of structurally variable enzymatic systems. Sci. Rep. 2016, 6, 25279. [Google Scholar] [CrossRef]
- Han, W.; He, M. The application of exogenous cellulase to improve soil fertility and plant growth due to acceleration of straw decomposition. Bioresour. Technol. 2010, 101, 3724–3731. [Google Scholar] [CrossRef] [PubMed]
- Tiquia, S.M.; Tam, N.F.Y.; Hodgkiss, I.J. Microbial activities during composting of spent pig-manure sawdust litter at different moisture contents. Bioresour. Technol. 1996, 55, 201–206. [Google Scholar] [CrossRef]
- Sunkar, R.; Bartels, D.; Kirch, H.H. Overexpression of a stress-inducible aldehyde dehydrogenase gene from Arabidopsis thaliana in transgenic plants improves stress tolerance. Plant J. 2003, 35, 452–464. [Google Scholar] [CrossRef]
- Schnurr, J.; Shockey, J. The acyl-CoA synthetase encoded by LACS2 is essential for normal cuticle development in Arabidopsis. Plant Cell 2004, 16, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Li, X.B. The GhACS1 gene encodes an acyl-CoA synthetase which is essential for normal microsporogenesis in early anther development of cotton. Plant J. 2009, 57, 473–486. [Google Scholar] [CrossRef] [PubMed]
- De Azevedo Souza, C.; Kim, S.S.; Koch, S.; Kienow, L.; Schneider, K.; McKim, S.M.; Haughn, G.W.; Kombrink, E.; Douglas, C.J. A novel fatty acyl-CoA synthetase is required for pollen development and sporopollenin biosynthesis in Arabidopsis. Plant Cell 2009, 21, 507–525. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, P.; Slabas, A.R.; Fawcett, T. Antisense expression of 3-oxoacyl-ACP reductase affects whole plant productivity and causes collateral changes in activity of fatty acid synthase components. Plant Cell Physiol. 2007, 48, 736–744. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
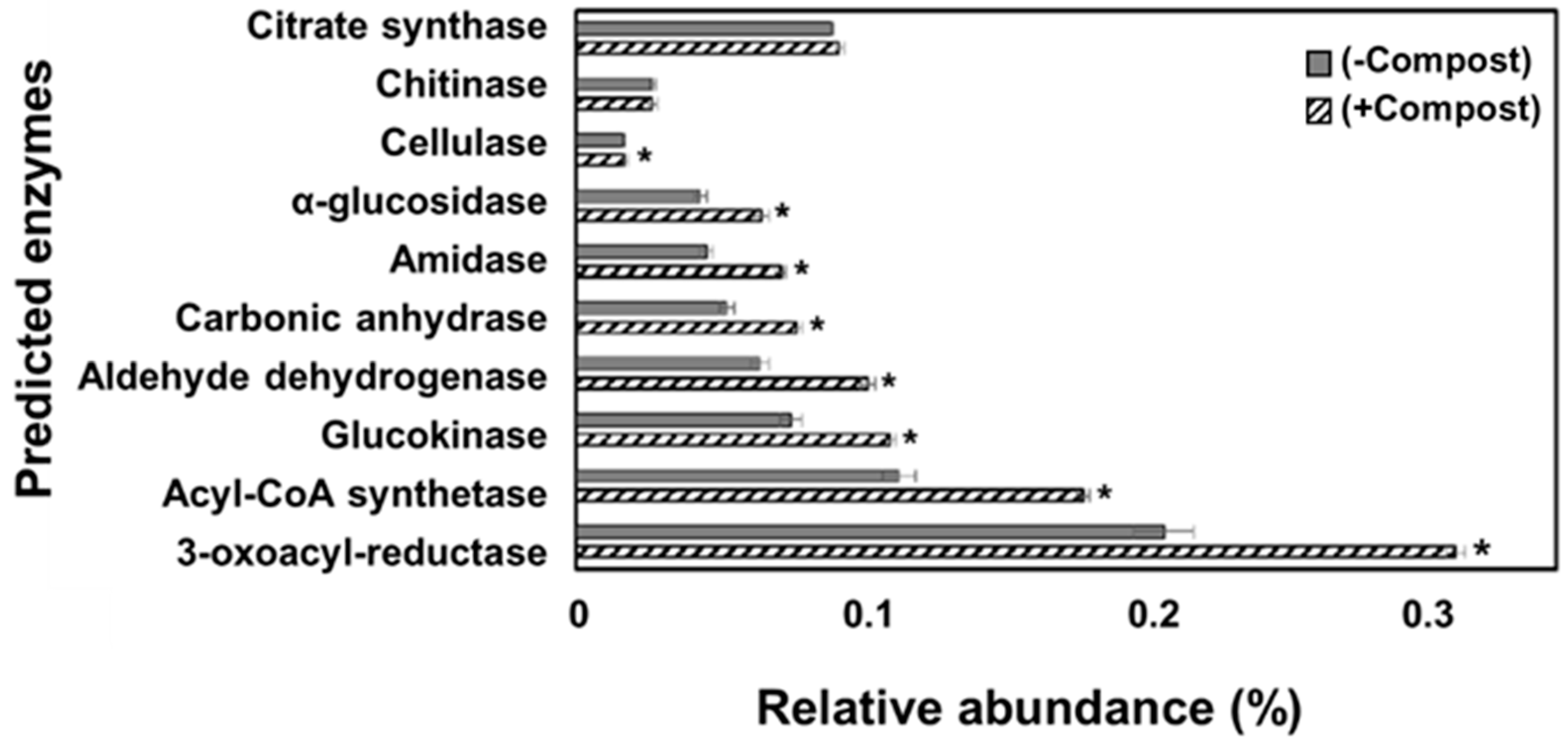{kind=link}
| Treatment | No. of Sequences | Goods Coverage (%) | No. of OTUs | Chao | Shannon’s Index |
|---|---|---|---|---|---|
| −Compost | 6567 | 98.6 | 782 ± 6.71 a | 816 ± 8.2 a | 6.02 ± 0.02 a |
| +Compost | 6567 | 98.8 | 750 ± 13.5 a | 775 ± 15 a | 5.98 ± 0.01 a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.; Samaddar, S.; Chatterjee, P.; Roy Choudhury, A.; Choi, J.; Choi, J.; Sa, T. Structural and Functional Shift in Soil Bacterial Community in Response to Long-Term Compost Amendment in Paddy Field. Appl. Sci. 2021, 11, 2183. https://doi.org/10.3390/app11052183
Kim S, Samaddar S, Chatterjee P, Roy Choudhury A, Choi J, Choi J, Sa T. Structural and Functional Shift in Soil Bacterial Community in Response to Long-Term Compost Amendment in Paddy Field. Applied Sciences. 2021; 11(5):2183. https://doi.org/10.3390/app11052183
Chicago/Turabian StyleKim, Sookjin, Sandipan Samaddar, Poulami Chatterjee, Aritra Roy Choudhury, Jeongyun Choi, Jongseo Choi, and Tongmin Sa. 2021. "Structural and Functional Shift in Soil Bacterial Community in Response to Long-Term Compost Amendment in Paddy Field" Applied Sciences 11, no. 5: 2183. https://doi.org/10.3390/app11052183
APA StyleKim, S., Samaddar, S., Chatterjee, P., Roy Choudhury, A., Choi, J., Choi, J., & Sa, T. (2021). Structural and Functional Shift in Soil Bacterial Community in Response to Long-Term Compost Amendment in Paddy Field. Applied Sciences, 11(5), 2183. https://doi.org/10.3390/app11052183