
‘SRS’ R Package and ‘q2-srs’ QIIME 2 Plugin: Normalization of Microbiome Data Using Scaling with Ranked Subsampling (SRS)




{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Theory
3. Method
3.1. Principle of SRS
- Scaling: feature counts (such as OTUs (operational taxonomic units), ASVs (amplicon sequence variants), or clades) are scaled sample-wise so that the sum of the scaled counts (Cscaled) for each sample is equal to the desired number of counts (Cmin).
- Ranked subsampling: the scaling step produces fractional values that must be converted into counts (integers). To do this, the Cscaled for each feature is split into the floor (Cint) and fractional part (Cfrac) of Cscaled. Because Cmin = ΣCscaled = ΣCint + ΣCfrac, it follows that Cmin ≥ ΣCint. Therefore, ΔC Cfrac values (where ΔC = Cmin − ΣCint) must be converted into additional counts (integers) so that Cmin can be reached. To do so, Cfrac values are ranked. Next, from the highest to the lowest rank, a count for each feature is added until ΔC counts have been added. After this step, all samples will have been normalized to Cmin counts.
- Special cases: (i) when Cfrac values involved in picking ΔC counts share the same rank across features, the counts are added for features based on the respective Cint ranks; (ii) when both Cfrac and its respective Cint values involved in picking ΔC counts share the same ranks across features, the counts are assigned randomly (without replacement). The specification of the seed that initializes the random process enables reproducible results.
3.2. ‘SRS’ R Package
3.2.1. SRS-Function
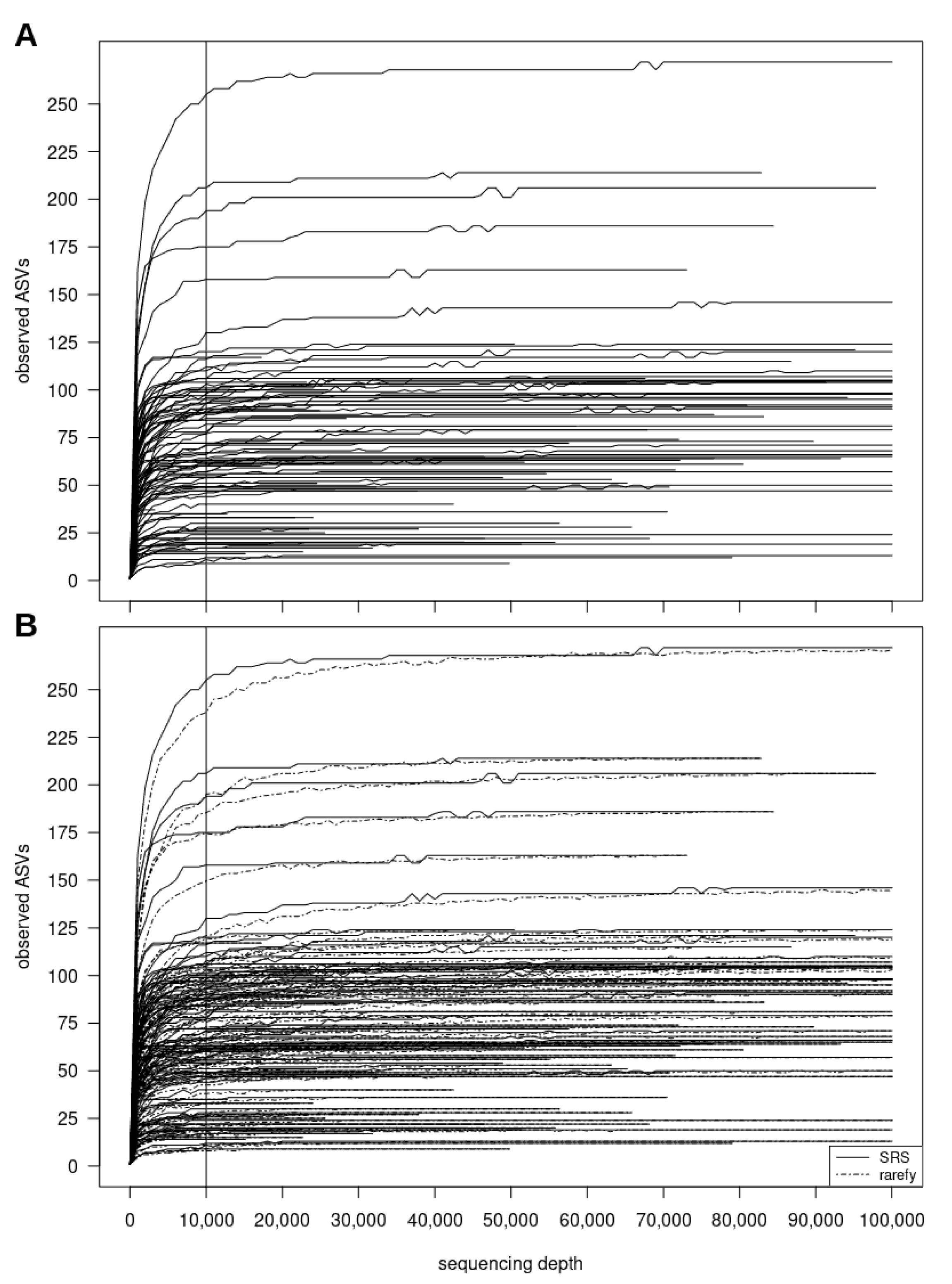
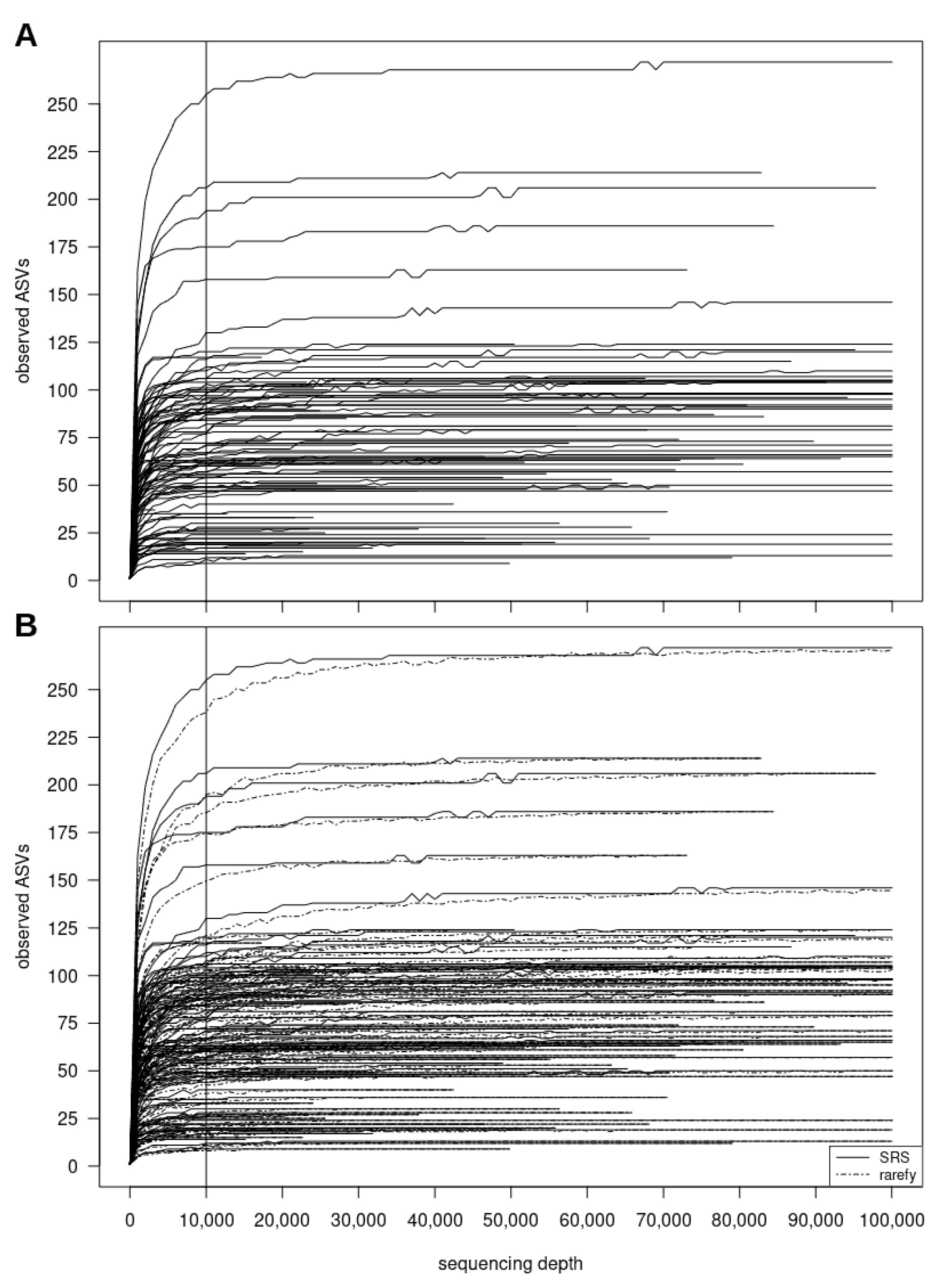
3.2.2. SRScurve-Function
max.sample.size = 0, rarefy.comparison = FALSE,
rarefy.repeats = 10, rarefy.comparison.legend = FALSE,
xlab = “sample size”, ylab = “richness”, label = FALSE,
col, lty,…)
3.2.3. SRS.shiny.app-Function
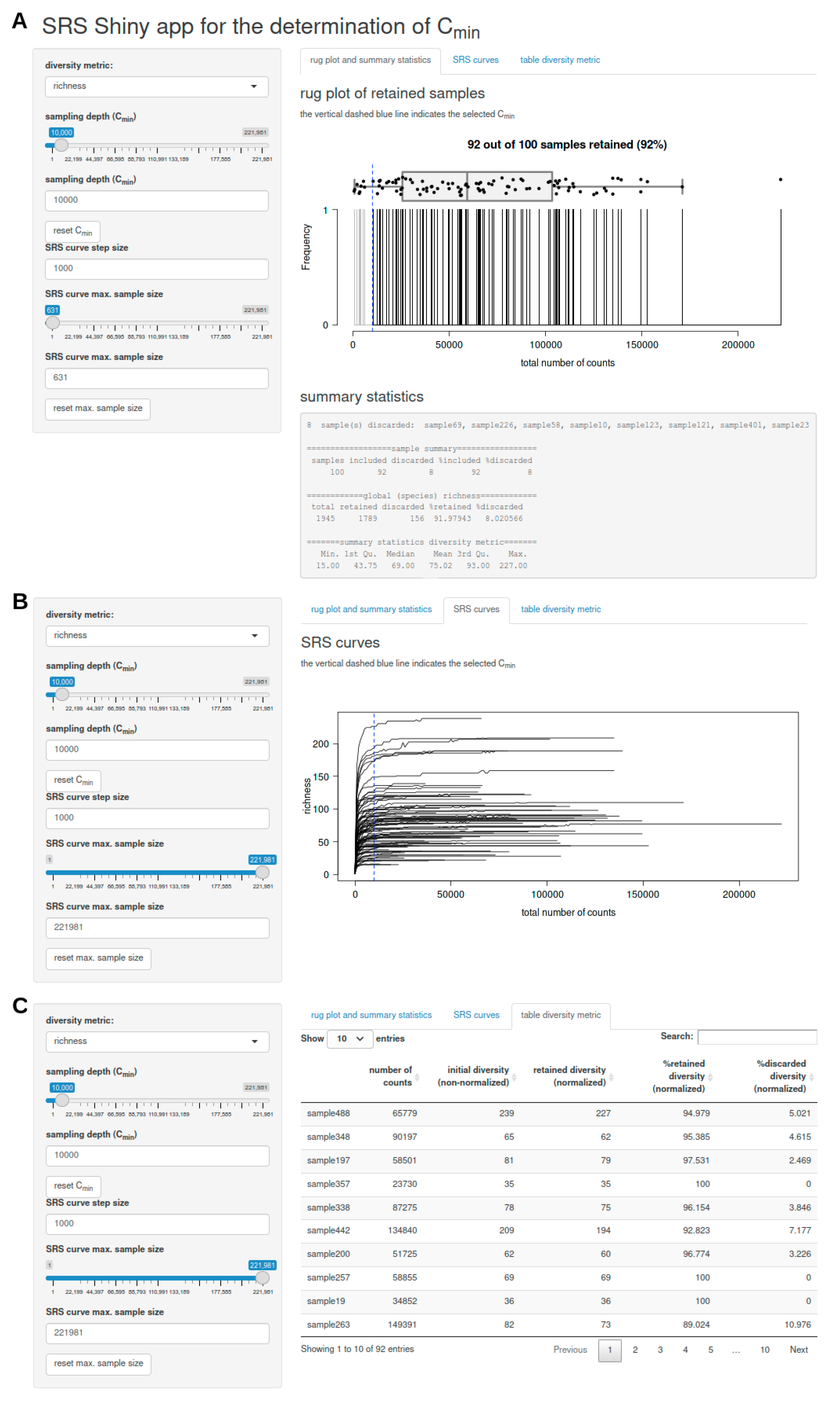
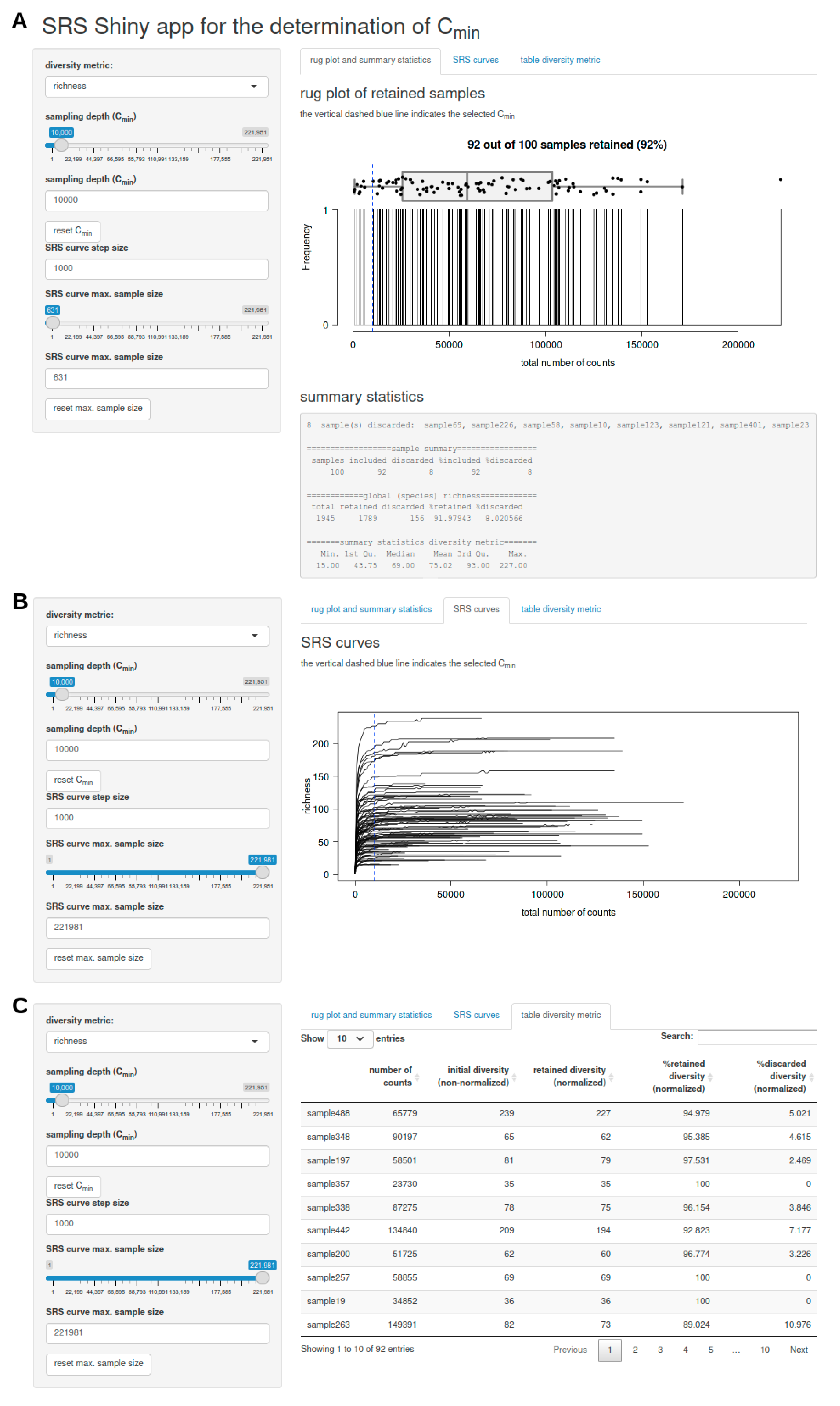
- a rug plot that shows the distribution of the number of counts per sample and displays discarded samples as well as summary statistics (including a list of discarded samples and descriptive statistics of the global feature richness and selected alpha diversity metric of the input dataset) in response to the selected Cmin (Figure 1A),
- a plot of SRS curves (SRScurve-function) that respond to the selected step size (step) and maximum sample size (max.sample.size) (Figure 1B), and
- an interactive table with sample names and the number of counts per sample as well as the initial diversity (non-normalized), retained diversity (normalized), %retained diversity (normalized), and %discarded diversity (normalized) of the selected alpha diversity metric in response to the selected Cmin (Figure 1C).
3.3. ‘q2-srs’ QIIME 2 Plugin
4. Results and Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human Gut Microbiome Viewed across Age and Geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N. Embracing the Unknown: Disentangling the Complexities of the Soil Microbiome. Nat. Rev. Microbiol. 2017, 15, 579–590. [Google Scholar] [CrossRef]
- Orsi, W.D. Ecology and Evolution of Seafloor and Subseafloor Microbial Communities. Nat. Rev. Microbiol. 2018, 16, 671–683. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Waste Not, Want Not: Why Rarefying Microbiome Data Is Inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef] [Green Version]
- Beule, L.; Karlovsky, P. Improved Normalization of Species Count Data in Ecology by Scaling with Ranked Subsampling (SRS): Application to Microbial Communities. PeerJ 2020, 8, e9593. [Google Scholar] [CrossRef] [PubMed]
- Cont, R.; Heidari, M. Optimal Rounding under Integer Constraints. arXiv 2014, arXiv:1501.00014. [Google Scholar]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. R Package Version 2.5-7. 2020. Available online: https://CRAN.R-project.org/package=vegan (accessed on 1 November 2021).
- Schloss, P.D. Identifying and Overcoming Threats to Reproducibility, Replicability, Robustness, and Generalizability in Microbiome Research. mBio 2018, 9, e00525-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Yang, J.; Park, J.; Jung, Y.; Chun, J. AMDB: A Database of Animal Gut Microbial Communities with Manually Curated Metadata. Nucleic Acids Res. 2021, gkab1009. [Google Scholar] [CrossRef] [PubMed]
- Beule, L.; Arndt, M.; Karlovsky, P. Relative Abundances of Species or Sequence Variants Can Be Misleading: Soil Fungal Communities as an Example. Microorganisms 2021, 9, 589. [Google Scholar] [CrossRef]
- Pontiller, B.; Pérez-Martínez, C.; Bunse, C.; Osbeck, C.M.G.; González, J.M.; Lundin, D.; Pinhassi, J. Taxon-Specific Shifts in Bacterial and Archaeal Transcription of Dissolved Organic Matter Cycling Genes in a Stratified Fjord. bioRxiv 2021. [Google Scholar] [CrossRef]
- Barreto Filho, M.M.; Walker, M.; Ashworth, M.P.; Morris, J.J. Structure and Long-Term Stability of the Microbiome in Diverse Diatom Cultures. Microbiol. Spectr. 2021, 9, e00269-21. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heidrich, V.; Karlovsky, P.; Beule, L. ‘SRS’ R Package and ‘q2-srs’ QIIME 2 Plugin: Normalization of Microbiome Data Using Scaling with Ranked Subsampling (SRS). Appl. Sci. 2021, 11, 11473. https://doi.org/10.3390/app112311473
Heidrich V, Karlovsky P, Beule L. ‘SRS’ R Package and ‘q2-srs’ QIIME 2 Plugin: Normalization of Microbiome Data Using Scaling with Ranked Subsampling (SRS). Applied Sciences. 2021; 11(23):11473. https://doi.org/10.3390/app112311473
Chicago/Turabian StyleHeidrich, Vitor, Petr Karlovsky, and Lukas Beule. 2021. "‘SRS’ R Package and ‘q2-srs’ QIIME 2 Plugin: Normalization of Microbiome Data Using Scaling with Ranked Subsampling (SRS)" Applied Sciences 11, no. 23: 11473. https://doi.org/10.3390/app112311473
APA StyleHeidrich, V., Karlovsky, P., & Beule, L. (2021). ‘SRS’ R Package and ‘q2-srs’ QIIME 2 Plugin: Normalization of Microbiome Data Using Scaling with Ranked Subsampling (SRS). Applied Sciences, 11(23), 11473. https://doi.org/10.3390/app112311473