
CO2 Capture by Reduced Graphene Oxide Monoliths with Incorporated CeO2 Grafted with Functionalized Polymer Brushes

 ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
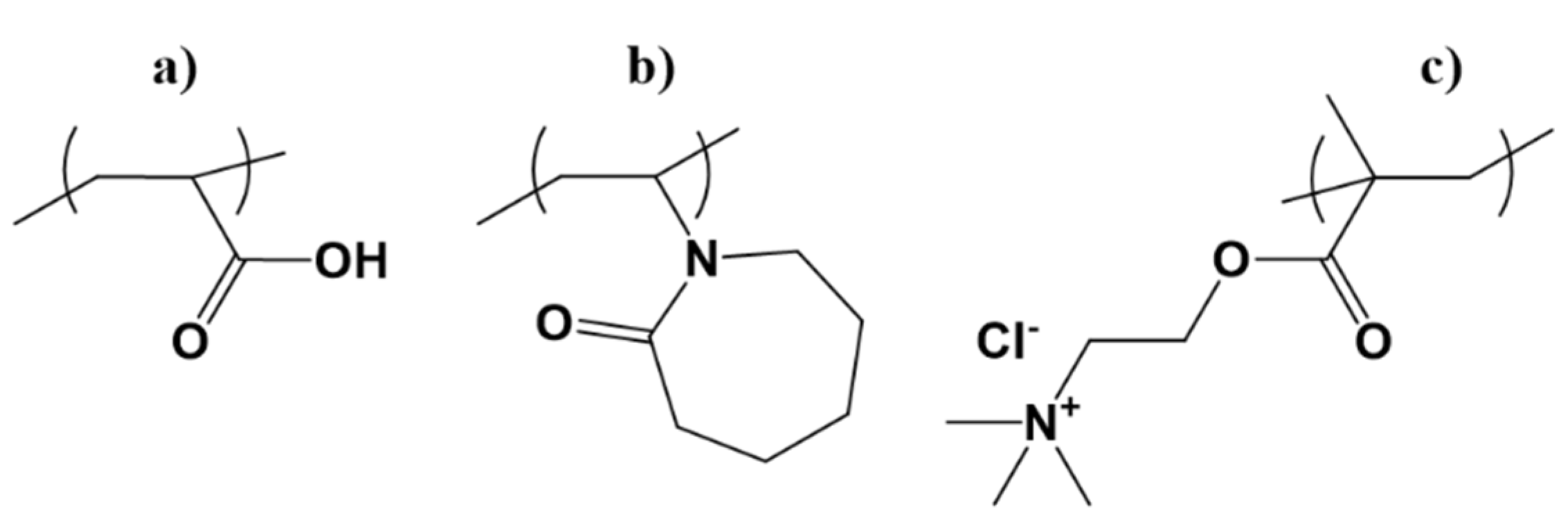
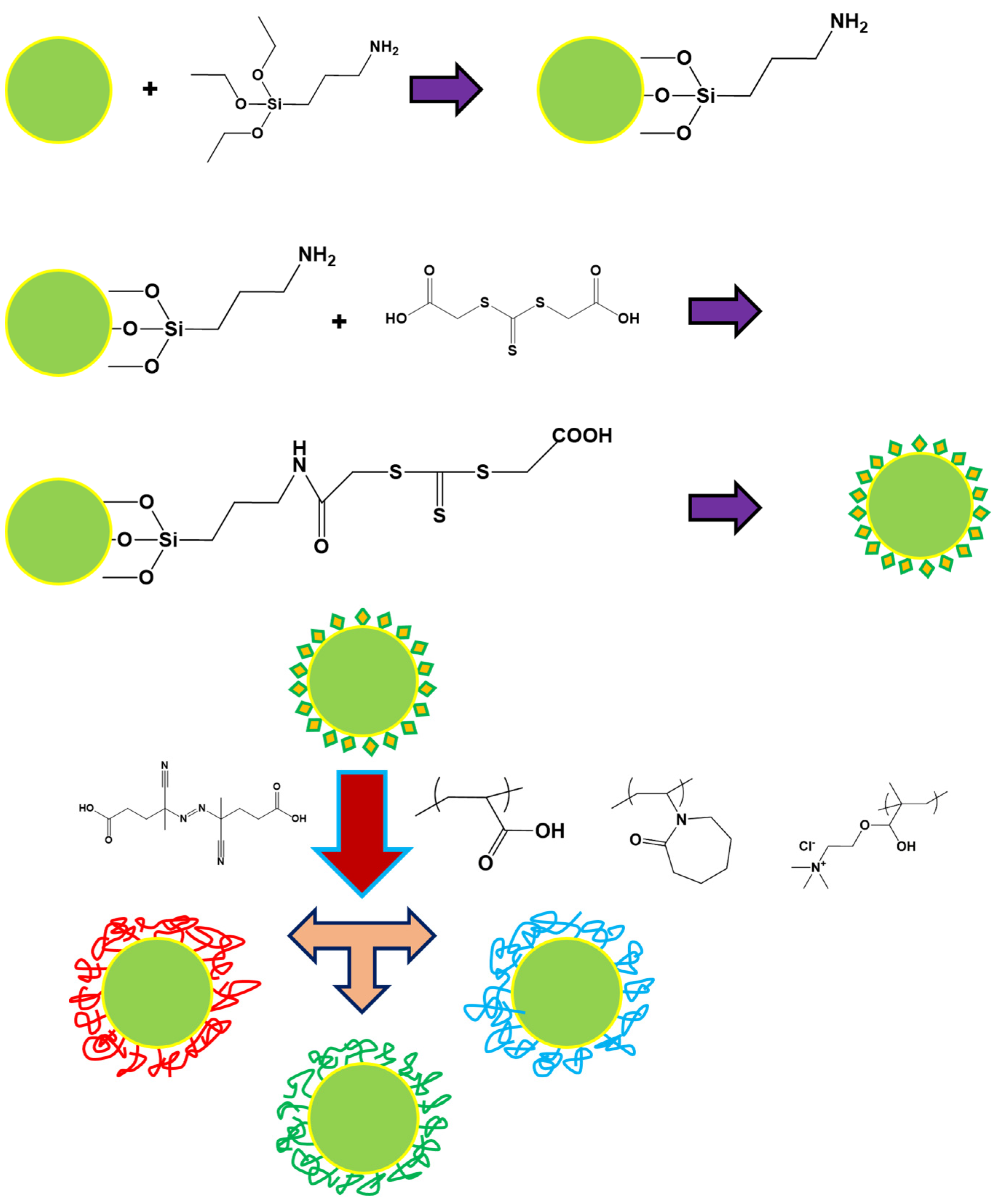
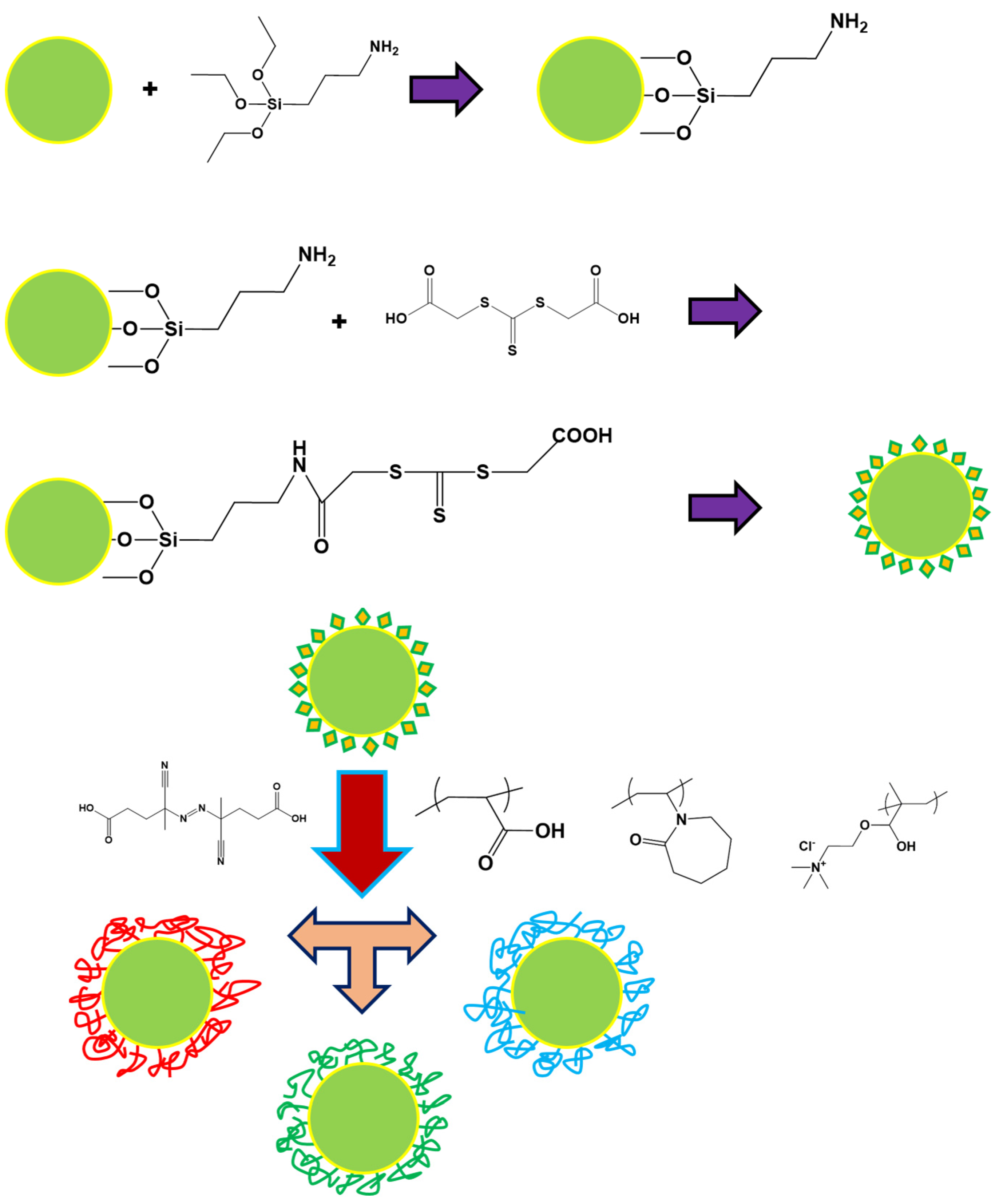
2.2. Synthesis of the Polymer Brushes from CeO2
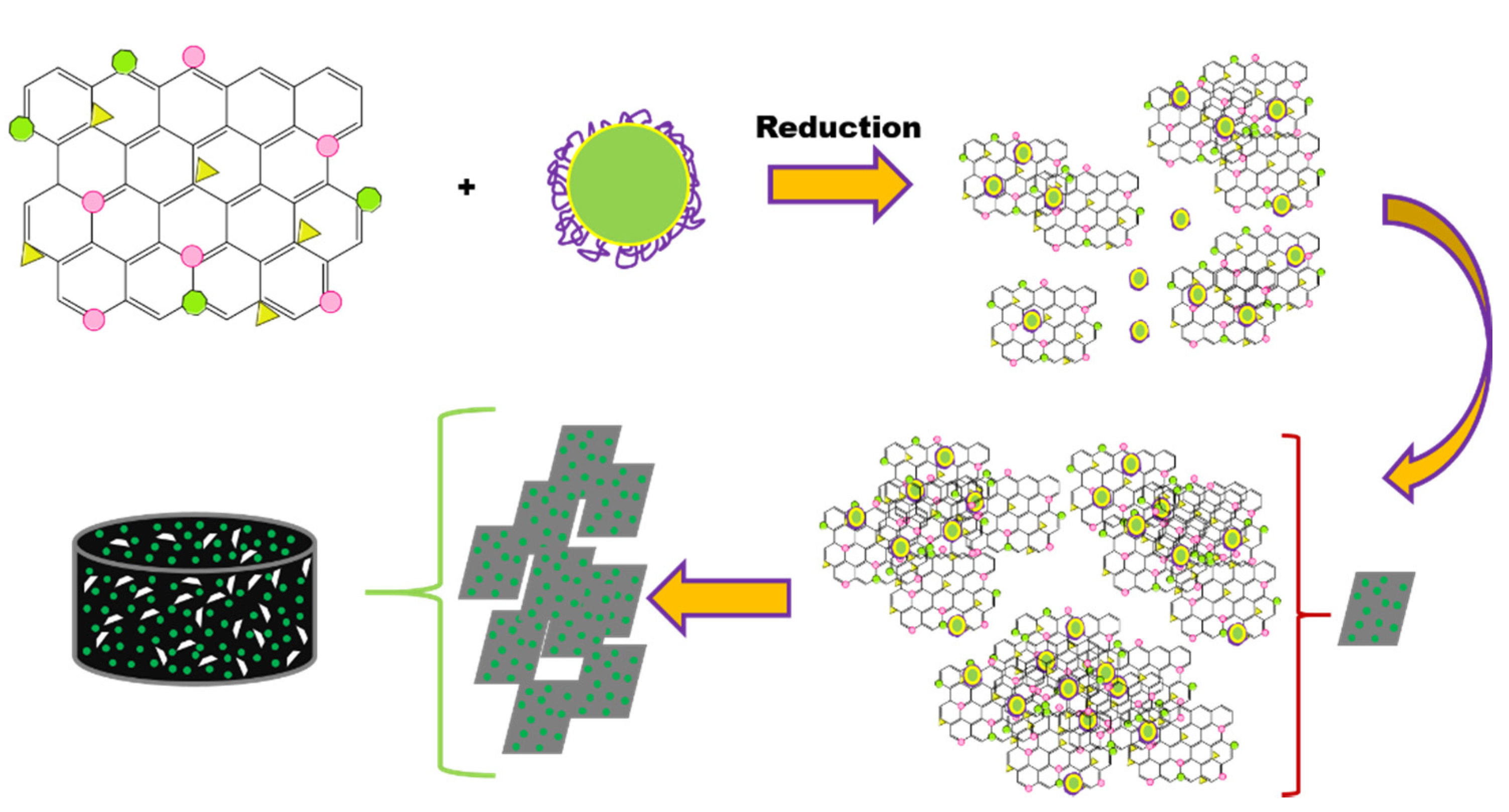
2.3. Synthesis of the Composite Monolith
2.4. Characterization
3. Results
3.1. Characterization of Bare and Grafted CeO2 Particles
3.1.1. Morphological Characterization
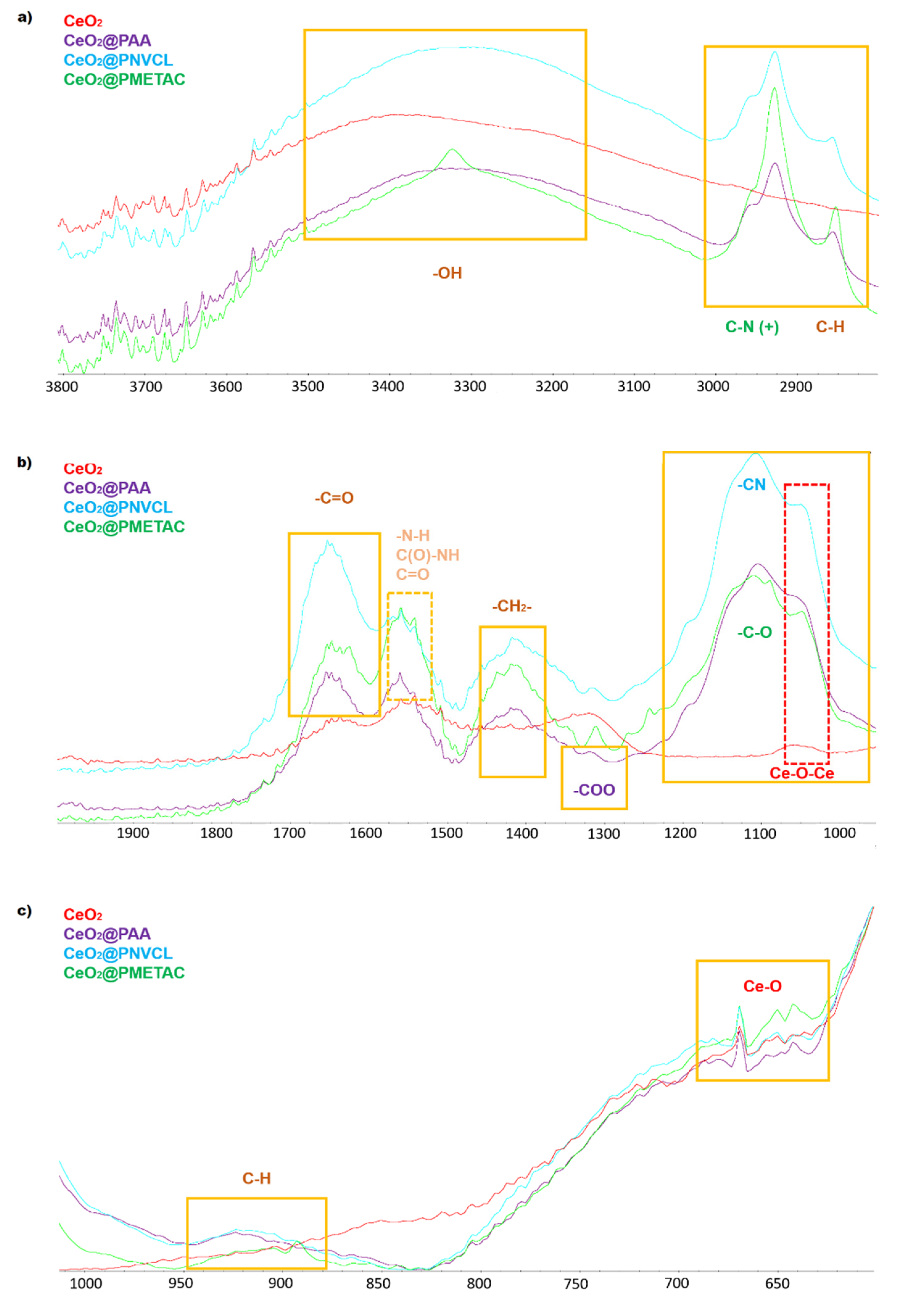
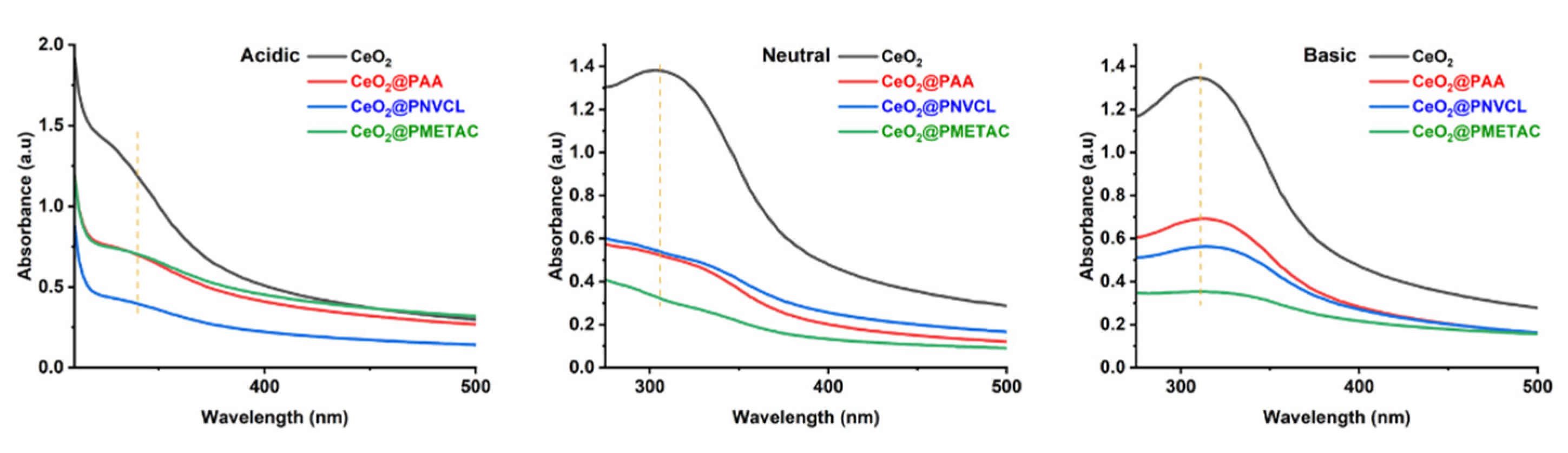
3.1.2. Spectroscopic Characterization
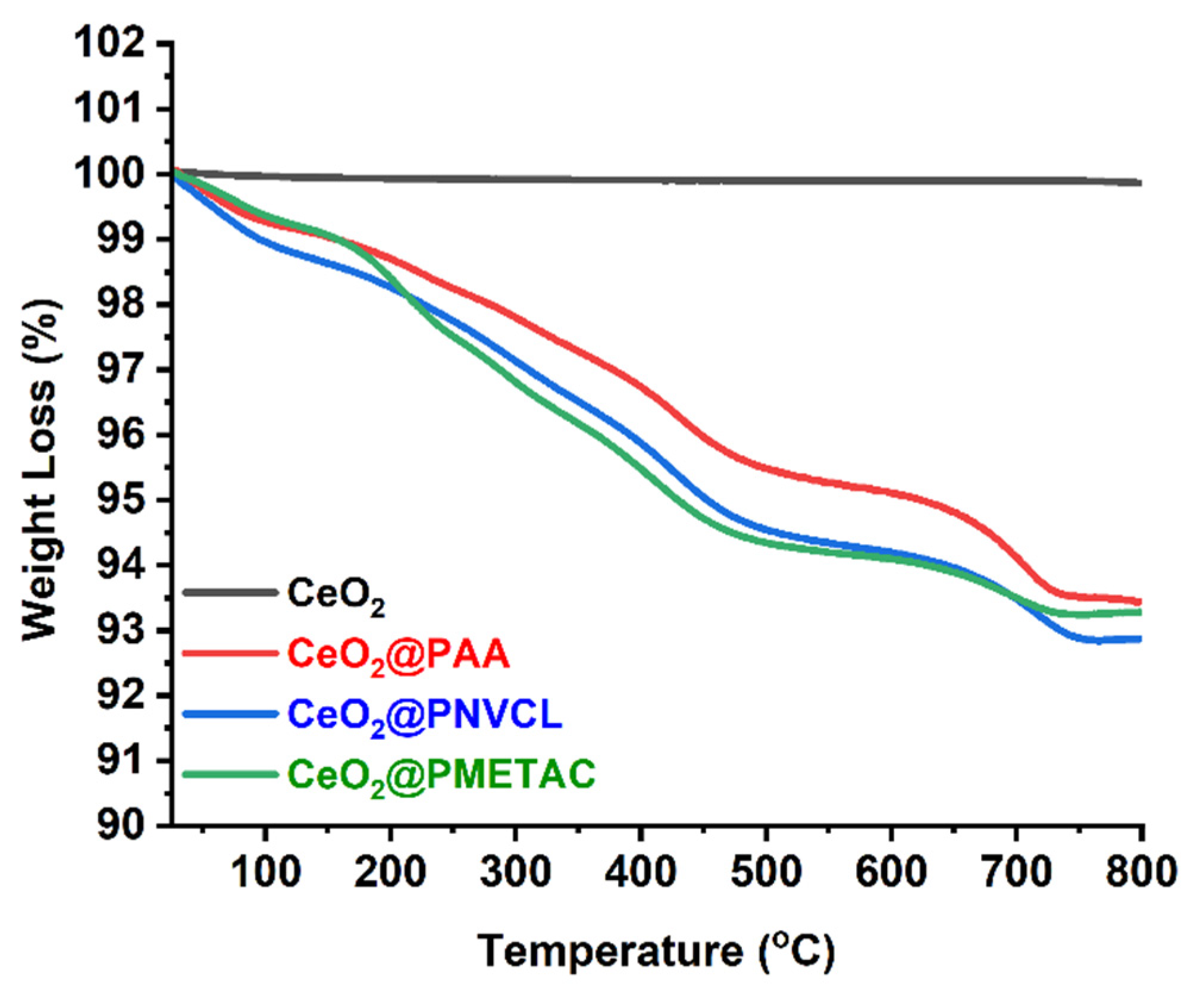
3.1.3. Molecular-Thermal Characterization
3.2. Characterization of rGO/CeO2/Polymer Monoliths
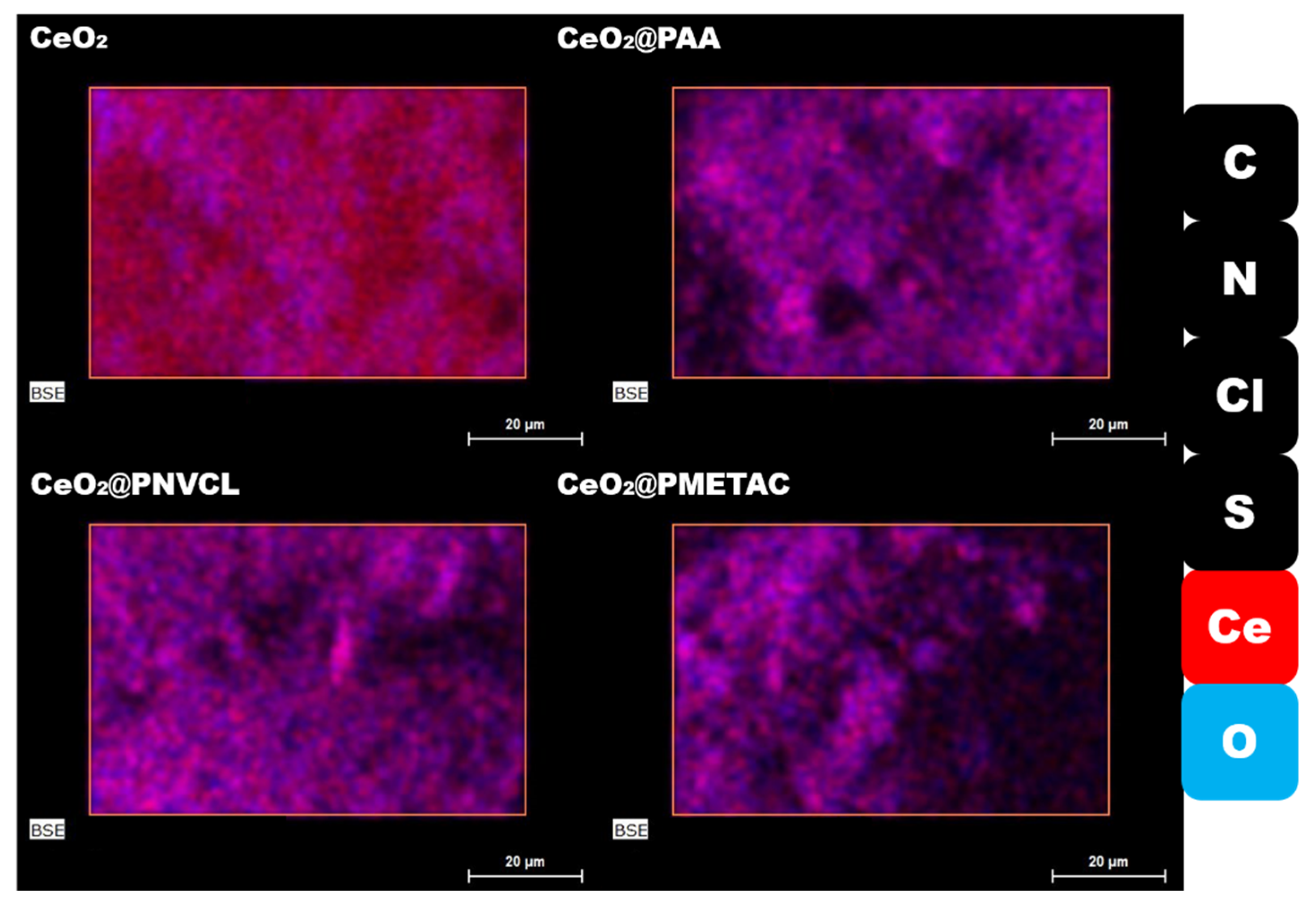
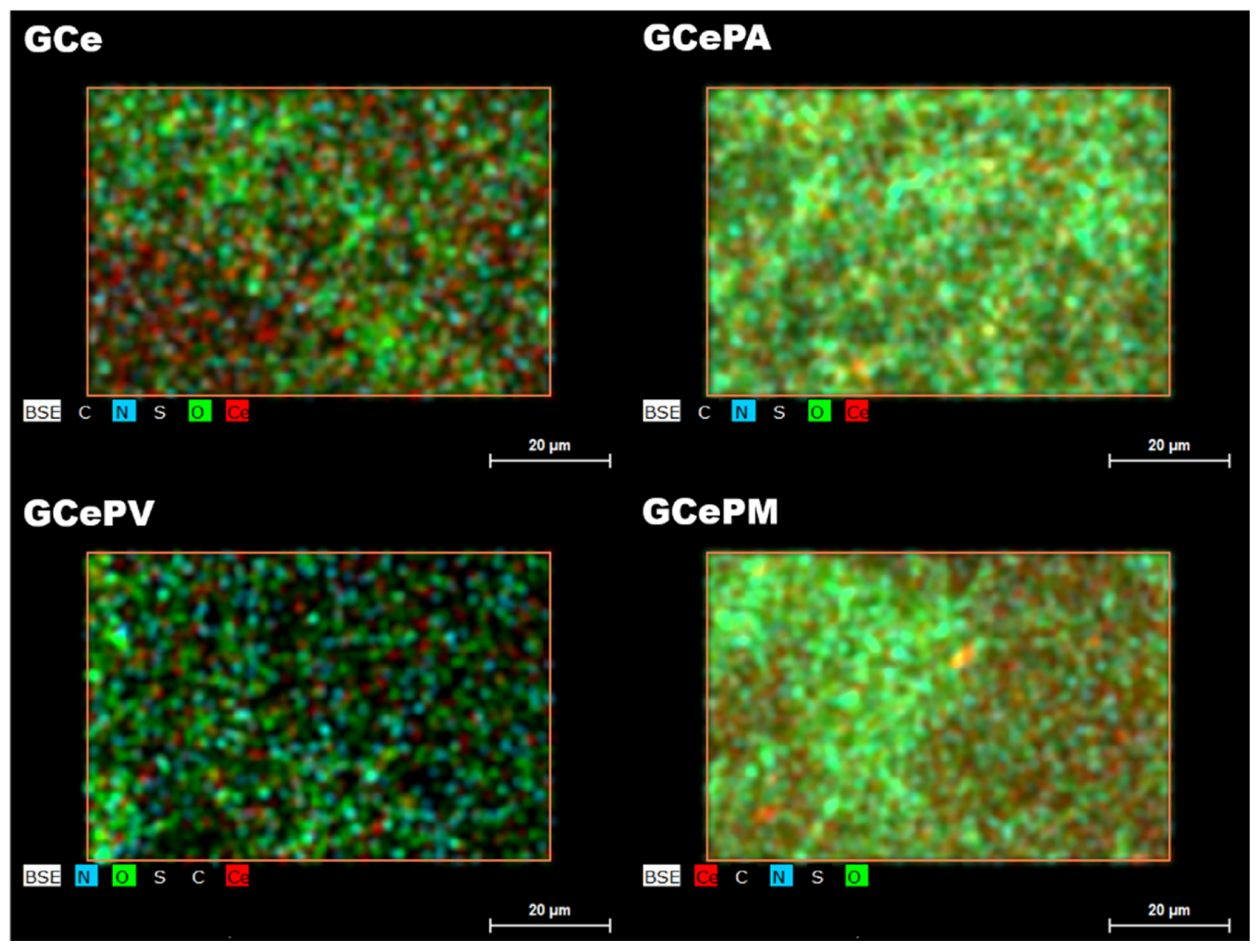
3.2.1. SEM/EDX
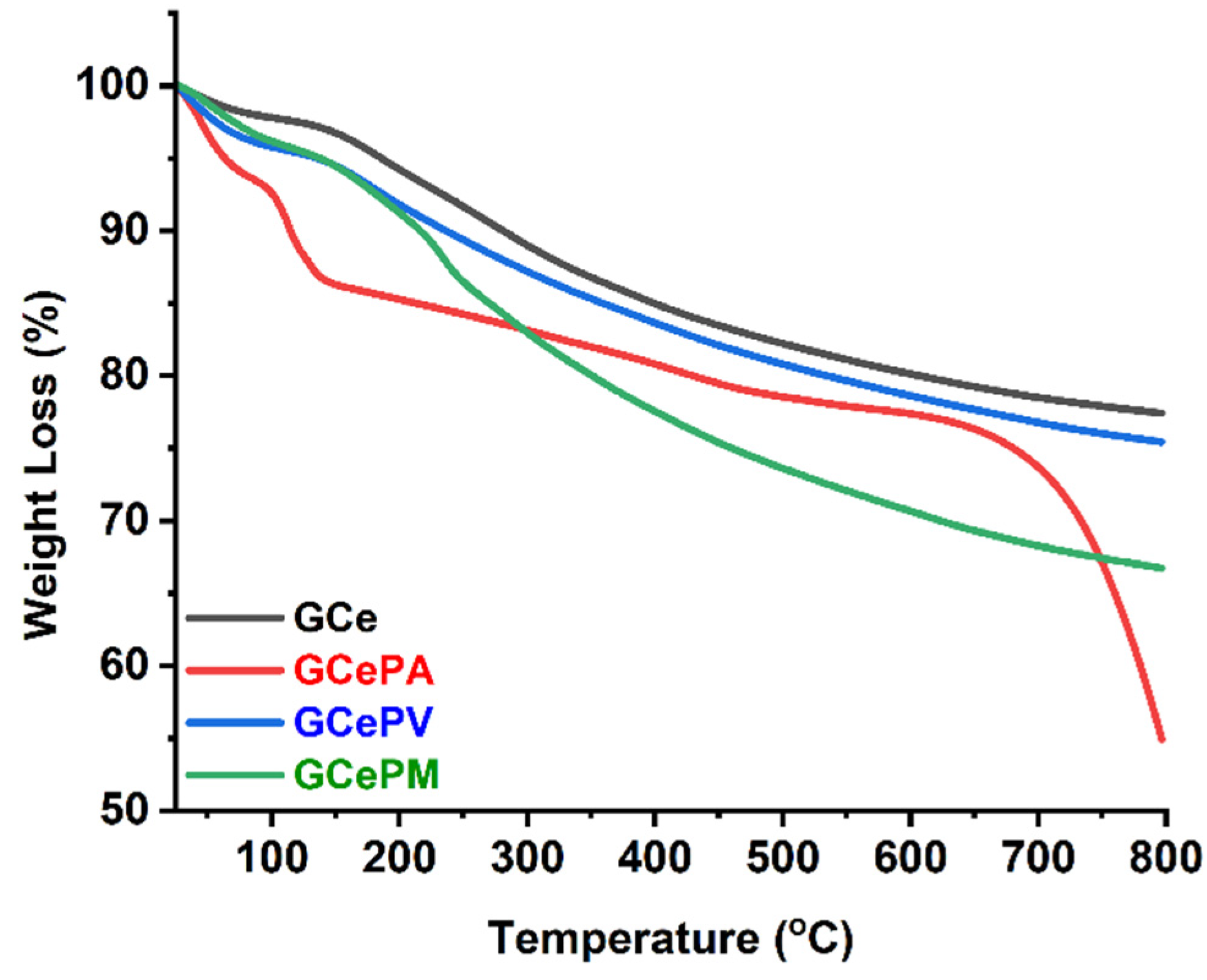
3.2.2. TGA
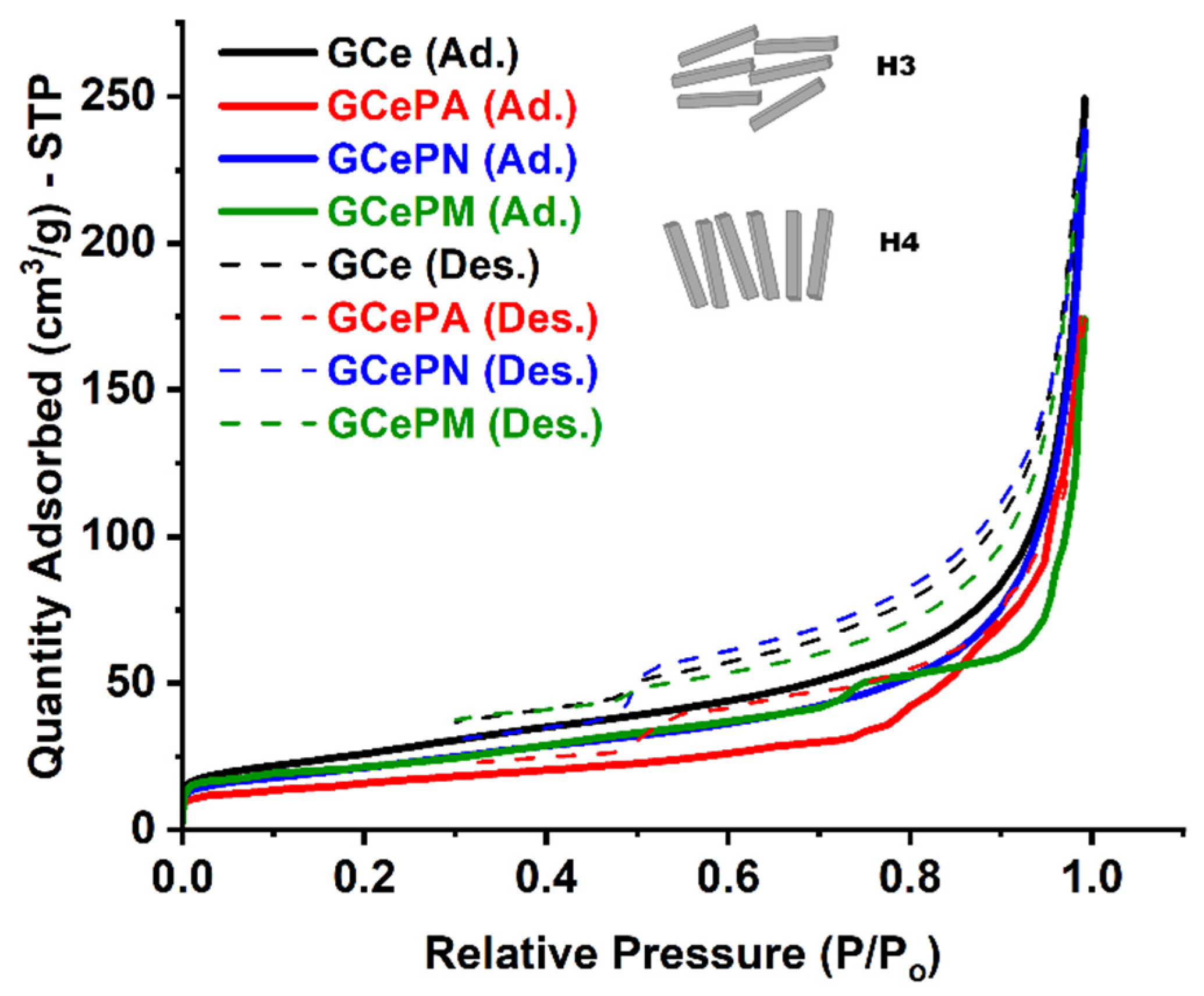
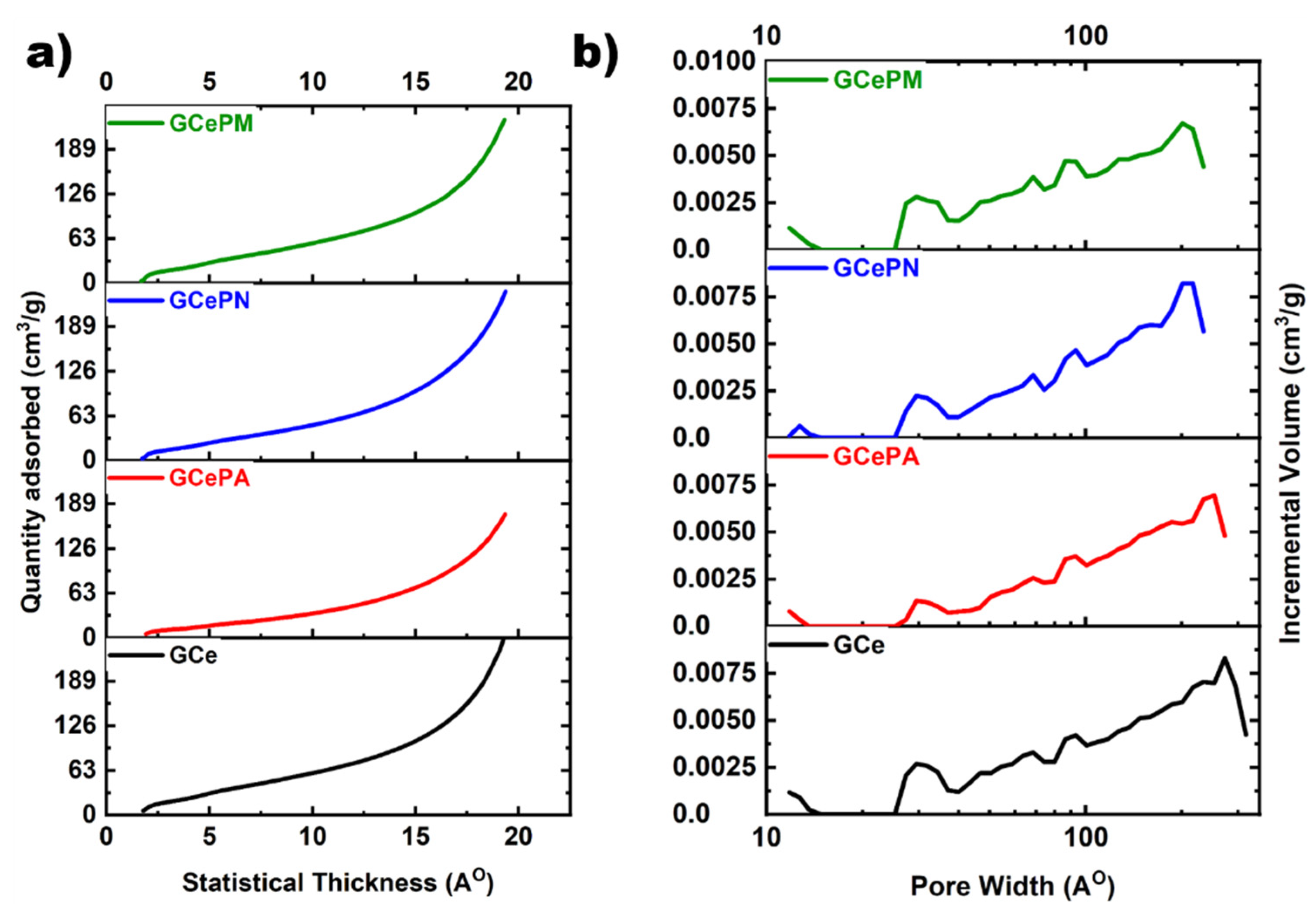
3.2.3. N2 Adsorption-Desorption
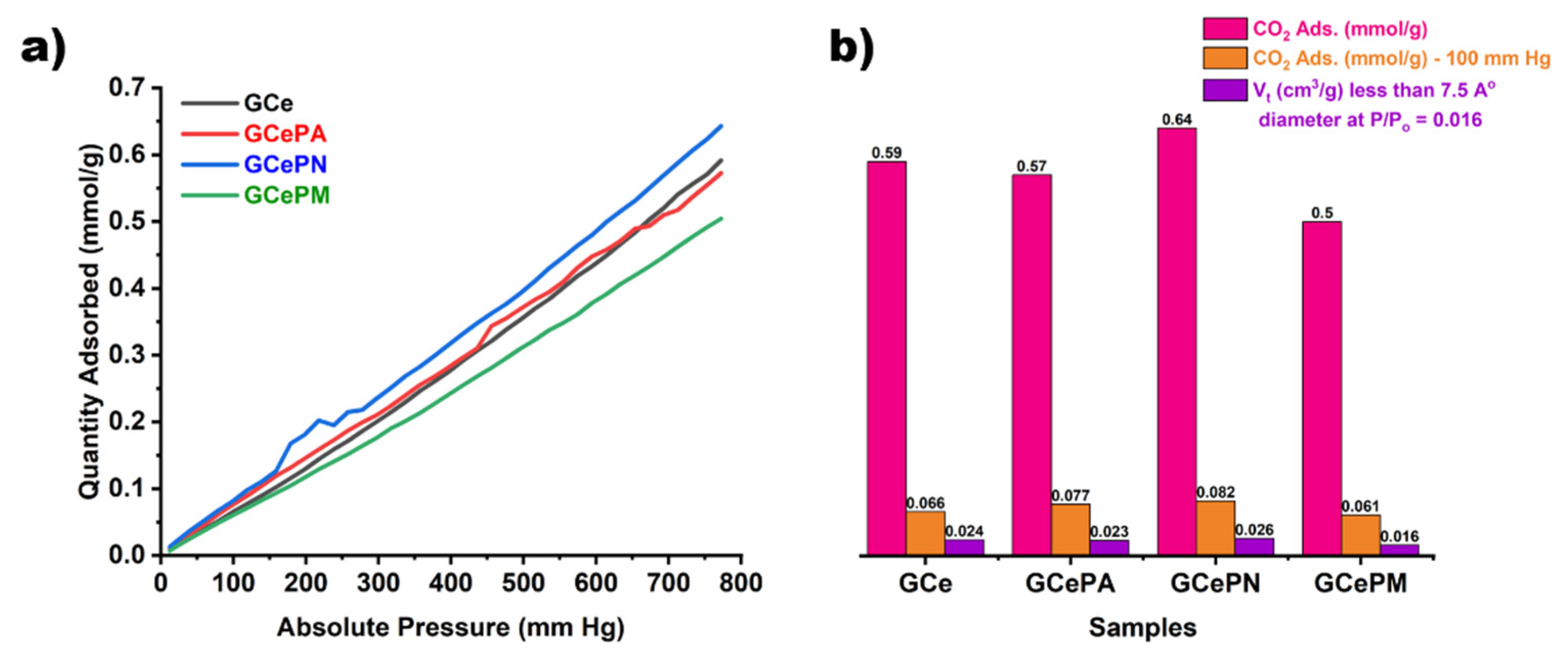
3.2.4. CO2 Adsorption
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ijaz, S.; Fahad Ehsan, M.; Naeem Ashiq, M.; Karamat, N.; He, T. Preparation of CdS@CeO2 core/shell composite for photocatalytic reduction of CO2 under visible-light irradiation. Appl. Surf. Sci. 2016, 390, 550–559. [Google Scholar] [CrossRef]
- Slostowski, C.; Marre, S.; Dagault, P.; Babot, O.; Toupance, T.; Aymonier, C. CeO2 nanopowders as solid sorbents for efficient CO2 capture/release processes. J. CO2 Util. 2017, 20, 52–58. [Google Scholar] [CrossRef] [Green Version]
- Liang, M.; Borjigin, T.; Zhang, Y.; Liu, B.; Liu, H.; Guo, H. Controlled assemble of hollow heterostructured g-C3N4@CeO2 with rich oxygen vacancies for enhanced photocatalytic CO2 reduction. Appl. Catal. B Environ. 2019, 243, 566–575. [Google Scholar] [CrossRef]
- Wang, H.; Guan, J.; Li, J.; Li, X.; Ma, C.; Huo, P.; Yan, Y. Fabricated g-C3N4/Ag/m-CeO2 composite photocatalyst for enhanced photoconversion of CO2. Appl. Surf. Sci. 2020, 506, 144931. [Google Scholar] [CrossRef]
- Yang, C.; Bebensee, F.; Chen, J.; Yu, X.; Nefedov, A.; Wöll, C. Carbon Dioxide Adsorption on CeO2(110): An XPS and NEXAFS Study. ChemPhysChem 2017, 18, 1874–1880. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Arai, Y.; Kado, S.; Kunimori, K.; Tomishige, K. Direct synthesis of organic carbonates from the reaction of CO2 with methanol and ethanol over CeO2 catalysts. Catal. Today 2006, 115, 95–101. [Google Scholar] [CrossRef]
- Tomishige, K.; Tamura, M.; Nakagawa, Y. CO2 Conversion with Alcohols and Amines into Carbonates, Ureas, and Carbamates over CeO2 Catalyst in the Presence and Absence of 2-Cyanopyridine. Chem. Rec. 2019, 19, 1354–1379. [Google Scholar] [CrossRef]
- Yoshikawa, K.; Kaneeda, M.; Nakamura, H. Development of Novel CeO2-based CO2 adsorbent and analysis on its CO2 adsorption and desorption mechanism. Energy Procedia 2017, 114, 2481–2487. [Google Scholar] [CrossRef]
- Liu, G.; Tatsuda, K.; Yoneyama, Y.; Tsubaki, N. Synthesis of mesoporous cerium compound for CO2 capture. E3S Web Conf. 2017, 22, 00106. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Wang, X.; Zhang, L.; Persson, K.M.; Chen, B.-Y.; Hsu, Y.; Chang, C.T. Near-visible-light-driven noble metal-free of reduced graphene oxide nanosheets over CeO2 nanowires for hydrogen production. J. Taiwan Inst. Chem. Eng. 2020, 107, 139–151. [Google Scholar] [CrossRef]
- Yang, S.-C.; Su, W.-N.; Rick, J.; Lin, S.D.; Liu, J.-Y.; Pan, C.-J.; Lee, J.-F.; Hwang, B.-J. Oxygen Vacancy Engineering of Cerium Oxides for Carbon Dioxide Capture and Reduction. ChemSusChem 2013, 6, 1326–1329. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, H.; Qin, Z.; Tian, S.; Ye, Z.; Ye, L.; Abroshan, H.; Li, G. TixCe1−xO2 nanocomposites: A monolithic catalyst for the direct conversion of carbon dioxide and methanol to dimethyl carbonate. Green Chem. 2019, 21, 4642–4649. [Google Scholar] [CrossRef]
- Kumari, N.; Sinha, N.; Haider, M.A.; Basu, S. CO2 Reduction to Methanol on CeO2 (110) Surface: A Density Functional Theory Study. Electrochim. Acta 2015, 177, 21–29. [Google Scholar] [CrossRef]
- Yoshikawa, K.; Sato, H.; Kaneeda, M.; Kondo, J.N. Synthesis and analysis of CO2 adsorbents based on cerium oxide. J. CO2 Util. 2014, 8, 34–38. [Google Scholar] [CrossRef]
- Politakos, N.; Barbarin, I.; Serrano Cantador, L.; Cecilia, J.A.; Mehravar, E.; Tomovska, R. Graphene-Based Monolithic Nanostructures for CO2 Capture. Ind. Eng. Chem. Res. 2020, 59, 8612–8621. [Google Scholar] [CrossRef]
- Politakos, N.; Barbarin, I.; Cordero-Lanzac, T.; Gonzalez, A.; Zangi, R.; Tomovska, R. Reduced Graphene Oxide/Polymer Monolithic Materials for Selective CO2 Capture. Polymers 2020, 12, 936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franchini, C.A.; Llorca, J.; Kuznetsov, A.; Silva, A.M. Outstanding dispersion of CeO2 on reduced graphene oxide. Implications for highly dispersed Pd catalysts. Diam. Relat. Mater. 2020, 109, 108061. [Google Scholar] [CrossRef]
- Radhakrishnan, S.; Kim, S.J. An enzymatic biosensor for hydrogen peroxide based on one-pot preparation of CeO2-reduced graphene oxide nanocomposite. RSC Adv. 2015, 5, 12937–12943. [Google Scholar] [CrossRef]
- Deng, D.; Chen, N.; Xiao, X.; Du, S.; Wang, Y. Electrochemical performance of CeO2 nanoparticle-decorated graphene oxide as an electrode material for supercapacitor. Ionics 2017, 23, 121–129. [Google Scholar] [CrossRef]
- Wang, S.; Gao, R.; Zhou, K. The influence of cerium dioxide functionalized reduced graphene oxide on reducing fire hazards of thermoplastic polyurethane nanocomposites. J. Colloid Interface Sci. 2019, 536, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Zhao, L.; Zhang, C.; Yan, Y.; Xian, Y. Controlled growth cerium oxide nanoparticles on reduced graphene oxide for oxygen catalytic reduction. Electrochim. Acta 2016, 191, 669–676. [Google Scholar] [CrossRef]
- Zhang, M.; Yuan, R.; Chai, Y.; Wang, C.; Wu, X. Cerium oxide–graphene as the matrix for cholesterol sensor. Anal. Biochem. 2013, 436, 69–74. [Google Scholar] [CrossRef]
- Catalán, J.; Fascineli, M.L.; Politakos, N.; Hartikainen, M.; Pereira Garcia, M.; Cáceres-Vélez, P.R.; Moreno, C.; Willian da Silva, S.; Morais, P.C.; Norppa, H.; et al. In vivo toxicological evaluation of polymer brush engineered nanoceria: Impact of brush charge. Nanotoxicology 2019, 13, 305–325. [Google Scholar] [CrossRef] [PubMed]
- Politakos, N.; Veloso, A.; González de San Román, E.; Cordero-Lanzac, T.; Qin, Z.; Leal, G.P.; Tomovska, R. Visible Light Photocatalysts Based on Manganese Doped TiO2 Integrated Within Monolithic Reduced Graphene Oxide/Polymer Porous Monolith. ChemistrySelect 2020, 5, 5873–5882. [Google Scholar]
- Portillo Moreno, O.; Gutiérrez Pérez, R.; Palomino Merino, R.; Chávez Portillo, M.; Hernandez Tellez, G.; Rubio Rosas, E.; Zamora Tototzintle, M. CeO2 nanoparticles growth by chemical bath and its thermal annealing treatment in air atmosphere. Optik 2017, 148, 142–150. [Google Scholar] [CrossRef]
- Latha, P.; Prakash, K.; Karuthapandian, S. Effective Photodegradation of CR & MO dyes by morphologically controlled Cerium oxide nanocubes under visible light Illumination. Optik 2018, 154, 242–250. [Google Scholar]
- Culica, M.E.; Chibac-Scutaru, A.L.; Melinte, V.; Coseri, S. Cellulose Acetate Incorporating Organically Functionalized CeO2 NPs: Efficient Materials for UV Filtering Applications. Materials 2020, 13, 2955. [Google Scholar] [CrossRef] [PubMed]
- Jing Yang, W.; Pranantyo, D.; Neoh, K.-G.; Kang, E.-T.; Lay-Ming Teo, S.; Rittschof, D. Layer-by-Layer Click Deposition of Functional Polymer Coatings for Combating Marine Biofouling. Biomacromolecules 2012, 13, 2769–2780. [Google Scholar] [CrossRef] [PubMed]
- Supeno, S.; Daik, R.; El-Sheikh, S.M. Cationic quaternization of cellulose with methacryloyloxy ethyl trimethyl ammonium chloride via ATRP method. AIP Conf. Proc. 2014, 1614, 178. [Google Scholar]
- Kavitha, T.; Kang, I.-K.; Park, S.-Y. Poly(N-vinyl caprolactam) grown on nanographene oxide as an effective nanocargo for drug delivery. Colloids Surf. B Biointerfaces 2014, 115, 37–45. [Google Scholar] [CrossRef]
- Hsiao, M.-H.; Lin, K.-H.; Liu, D.-M. Improved pH-responsive amphiphilic carboxymethyl-hexanoyl chitosan–poly(acrylic acid) macromolecules for biomedical applications. Soft Matter 2013, 9, 2458–2466. [Google Scholar] [CrossRef]
- Mueen, R.; Morlando, A.; Qutaish, H.; Lerch, M.; Cheng, Z.; Konstantinov, K. ZnO/CeO2 nanocomposite with low photocatalytic activity as efficient UV filters. J. Mater. Sci. 2020, 55, 6834–6847. [Google Scholar] [CrossRef]
- Ren, Z.; Peng, F.; Li, J.; Liang, X.; Chen, B. Morphology-Dependent Properties of Cu/CeO2 Catalysts for the Water-Gas Shift Reaction. Catalysts 2017, 7, 48. [Google Scholar] [CrossRef] [Green Version]
- Sagadevan, S.; Johan, M.R.; Lett, J.A. Fabrication of reduced graphene oxide/CeO2 nanocomposite for enhanced electrochemical performance. Appl. Phys. A 2019, 125, 315. [Google Scholar] [CrossRef]
- Nurhasanah, I.; Safitri, W.; Arifin, Z.; Subagio, A.; Windarti, T. Antioxidant activity and dose enhancement factor of CeO2 nanoparticles synthesized by precipitation method. IOP Conf. Ser. Mater. Sci. Eng. 2018, 432, 012031. [Google Scholar] [CrossRef]
- Pop, O.L.; Mesaros, A.; Vodnar, D.C.; Suharoschi, R.; Tăbăran, F.; Magerușan, L.; Tódor, I.S.; Diaconeasa, Z.; Balint, A.; Ciontea, L.; et al. Cerium Oxide Nanoparticles and Their Efficient Antibacterial Application In Vitro against Gram-Positive and Gram-Negative Pathogens. Nanomaterials 2020, 10, 1614. [Google Scholar] [CrossRef]
- Hobza, P.; Havlas, Z. Blue-Shifting Hydrogen Bonds. Chem. Rev. 2000, 100, 4253–4264. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.K.; Mohapatra, M.; Singh, A.K.; Anand, S. Hydrothermal Synthesis of Single Crystalline Nano CeO2 and Its Structural, Optical, and Electronic Characterization. Mater. Manuf. Process. 2010, 25, 982–989. [Google Scholar] [CrossRef]
- Pettinger, N.W.; Empey, J.M.; Fröbel, S.; Kohler, B. Photoreductive dissolution of cerium oxide nanoparticles and their size-dependent absorption properties. Phys. Chem. Chem. Phys. 2020, 22, 5756–5764. [Google Scholar] [CrossRef]
- Seixas de Melo, J.; Costa, T.; Francisco, A.; Maçanita, A.L.; Gago, S.; Gonçalves, I.S. Dynamics of short as compared with long poly(acrylic acid) chains hydrophobically modified with pyrene, as followed by fluorescence techniques. Phys. Chem. Chem. Phys. 2007, 9, 1370–1385. [Google Scholar] [CrossRef]
- Kesal, D.; Christau, S.; Krause, P.; Möller, T.; Von Klitzing, R. Uptake of pH-Sensitive Gold Nanoparticles in Strong Polyelectrolyte Brushes. Polymers 2016, 8, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yurdakal, S.; Garlisi, C.; Özcan, L.; Bellardita, M.; Palmisano, G. Chapter 4—(Photo) catalyst Characterization Techniques: Adsorption Isotherms and BET, SEM, FTIR, UV–Vis, Photoluminescence, and Electrochemical Characterizations. In Heterogeneous Photocatalysis Relationships with Heterogeneous Catalysis and Perspectives; Elsevier Science: Amsterdam, The Netherlands, 2019; pp. 87–152. [Google Scholar]
- Wang, P.; Jiang, Z.; Chen, L.; Yin, L.; Li, Z.; Zhang, C.; Tang, X.; Wang, G. Pore structure characterization for the Longmaxi and Niutitang shales in the Upper Yangtze Platform, South China: Evidence from focused ion beam–He ion microscopy, nano-computerized tomography and gas adsorption analysis. Mar. Pet. Geol. 2016, 77, 1323–1337. [Google Scholar] [CrossRef]
- Storck, S.; Bretinger, H.; Maier, W.F. Characterization of micro- and mesoporous solids by physisorption methods and pore-size analysis. Appl. Catal. A Gen. 1998, 174, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Teixeira Gouveia, L.G.; Borges Agustini, C.; Perez-Lopez, O.W.; Gutterres, M. CO2 adsorption using solids with different surface and acid-base properties. J. Environ. Chem. Eng. 2020, 8, 103823. [Google Scholar] [CrossRef]
- Kovacevic, M.; Mojet, B.L.; van Ommen, G.J.; Lefferts, L. Effects of Morphology of Cerium Oxide Catalysts for Reverse Water Gas Shift Reaction. Catal. Lett. 2016, 146, 770–777. [Google Scholar] [CrossRef] [Green Version]
- Ju, T.-J.; Wang, C.-H.; Lin, S.D. Insights into the CO2 deoxygenation to CO over oxygen vacancies of CeO2. Catal. Sci. Technol. 2019, 9, 2118–2124. [Google Scholar] [CrossRef]
- Lee, S.M.; Lee, Y.H.; Moon, D.H.; Ahn, J.Y.; Nguyen, D.D.; Chang, S.W.; Kim, S.S. Reaction Mechanism and Catalytic Impact of Ni/CeO2–x Catalyst for Low-Temperature CO2 Methanation. Ind. Eng. Chem. Res. 2019, 58, 8656–8662. [Google Scholar] [CrossRef]
- Xing, W.; Liu, C.; Zhou, Z.; Zhou, J.; Wang, G.; Zhuo, S.; Xue, Q.; Song, L.; Yan, Z. Oxygen-containing functional group-facilitated CO2 capture by carbide-derived carbons. Nanoscale Res. Lett. 2014, 9, 189. [Google Scholar] [CrossRef] [Green Version]
- Singh, G.; Lee, J.; Karakoti, A.; Bahadur, R.; Yi, J.; Zhao, D.; AlBahily, K.; Vinu, A. Emerging trends in porous materials for CO2 capture and conversion. Chem. Soc. Rev. 2020, 49, 4360–4404. [Google Scholar] [CrossRef]
- Ko, Y.G.; Shin, S.S.; Choi, U.S. Primary, secondary, and tertiary amines for CO2 capture: Designing for mesoporous CO2 adsorbents. J. Colloid Interface Sci. 2011, 361, 594–602. [Google Scholar] [CrossRef]
- Molavi, H.; Eskandari, A.; Shojaei, A.; Mousavi, S.A. Enhancing CO2/N2 adsorption selectivity via post-synthetic modification of NH2-UiO-66(Zr). Microporous Mesoporous Mater. 2018, 257, 193–201. [Google Scholar] [CrossRef]
- Saka, C. Overview on the Surface Functionalization Mechanism and Determination of Surface Functional Groups of Plasma Treated Carbon Nanotubes. Crit. Rev. Anal. Chem. 2018, 48, 1–14. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Grafting Density (%) a | Mw b | Z−Potential c (mV) |
|---|---|---|---|
| CeO2 | − | − | −25.4 |
| CeO2@PAA | 5.83 | 36,500 44,500 | −21.3 |
| CeO2@PNVCL | 6.09 | 27,000 46,500 | −6.99 |
| CeO2@PMETAC | 6.08 | 26,000 43,500 | −0.14 |
| Scheme | C | O | N | Ce | S |
|---|---|---|---|---|---|
| rGO | 71.2 | 22.3 | 6.4 | - | 0.1 |
| GCe | 84.5 | 11.2 | 0.5 | 3.7 | 0.1 |
| GCePA | 60.1 | 31.8 | 0.7 | 6.7 | 0.7 |
| GCePN | 78.2 | 17.0 | 0.9 | 3.8 | 0.1 |
| GCePM | 71.7 | 21.7 | 0.6 | 5.8 | 0.2 |
| Samples | SSA a (m2/g) | Total Pore Volume b (cm3/g) | Average Pore Size c (nm) | Percentage of Micropores d (Volume) | Percentage of Micropores e (Area) |
|---|---|---|---|---|---|
| GCe | 95.1 | 0.175 | 7.4 | 5.1 | 11.1 |
| GCePA | 52.6 | 0.120 | 9.1 | 5.6 | 13.2 |
| GCePN | 77.5 | 0.166 | 8.6 | 5.5 | 7.4 |
| GCePM | 89.7 | 0.165 | 7.4 | 7.0 | 9.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Politakos, N.; Serrano Cantador, L.; Cecilia, J.A.; Barbarin, I.; Tomovska, R. CO2 Capture by Reduced Graphene Oxide Monoliths with Incorporated CeO2 Grafted with Functionalized Polymer Brushes. Appl. Sci. 2021, 11, 11154. https://doi.org/10.3390/app112311154
Politakos N, Serrano Cantador L, Cecilia JA, Barbarin I, Tomovska R. CO2 Capture by Reduced Graphene Oxide Monoliths with Incorporated CeO2 Grafted with Functionalized Polymer Brushes. Applied Sciences. 2021; 11(23):11154. https://doi.org/10.3390/app112311154
Chicago/Turabian StylePolitakos, Nikolaos, Luis Serrano Cantador, Juan Antonio Cecilia, Iranzu Barbarin, and Radmila Tomovska. 2021. "CO2 Capture by Reduced Graphene Oxide Monoliths with Incorporated CeO2 Grafted with Functionalized Polymer Brushes" Applied Sciences 11, no. 23: 11154. https://doi.org/10.3390/app112311154
APA StylePolitakos, N., Serrano Cantador, L., Cecilia, J. A., Barbarin, I., & Tomovska, R. (2021). CO2 Capture by Reduced Graphene Oxide Monoliths with Incorporated CeO2 Grafted with Functionalized Polymer Brushes. Applied Sciences, 11(23), 11154. https://doi.org/10.3390/app112311154