
Converting Biomedical Text Annotated Resources into FAIR Research Objects with an Open Science Platform

 ,
,  , ,
, ,  and
and 
Abstract
1. Introduction
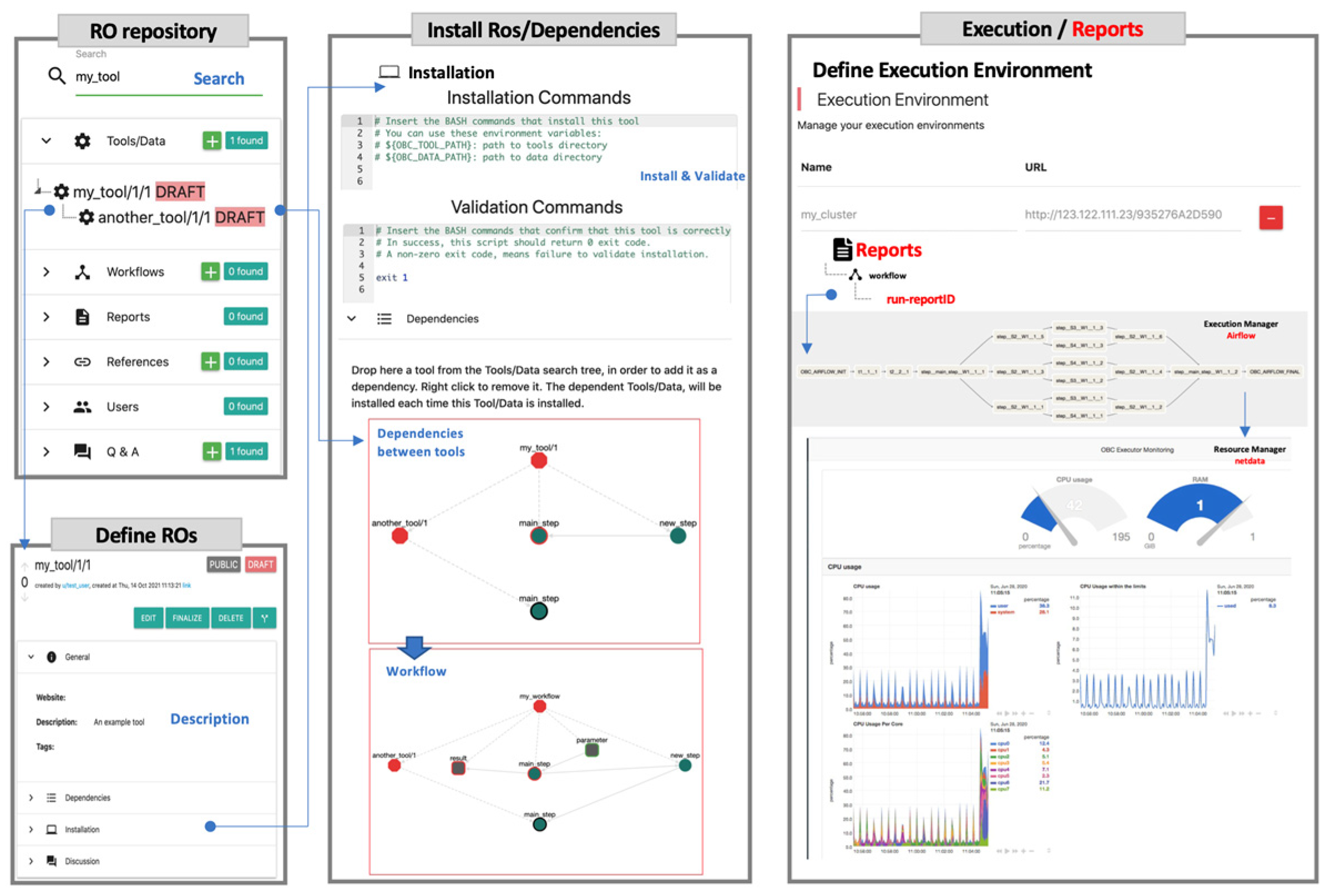
2. FAIRification of ROs with OpenBio.eu
3. Imported Resources for Biomedical Text Annotation
3.1. Imported Corpora
3.2. Imported Ontologies
3.3. Imported Tools for Biomedical Text Annotation
4. Working with OpenBio.eu
4.1. Importing ROs in OpenBio.eu
4.2. Synthesizing Workflows in OpenBio.eu
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bornmann, L.; Mutz, R. Growth rates of modern science: A bibliometric analysis based on the number of publications and cited references. J. Assoc. Inf. Sci. Technol. 2015, 66, 2215–2222. [Google Scholar] [CrossRef]
- Huang, C.-C.; Lu, Z. Community challenges in biomedical text mining over 10 years: Success, failure and the future. Brief. Bioinform. 2016, 17, 132–144. [Google Scholar] [CrossRef]
- Munafò, M.R.; Nosek, B.A.; Bishop, D.V.M.; Button, K.S.; Chambers, C.D.; Percie du Sert, N.; Simonsohn, U.; Wagenmakers, E.-J.; Ware, J.J.; Ioannidis, J.P.A. A manifesto for reproducible science. Nat. Hum. Behav. 2017, 1, 21. [Google Scholar] [CrossRef]
- Freedman, L.P.; Cockburn, I.M.; Simcoe, T.S. The economics of reproducibility in preclinical research. PLOS Biol. 2015, 13, 1–9. [Google Scholar] [CrossRef]
- Luque, C.; Luna, J.M.; Luque, M.; Ventura, S. An advanced review on text mining in medicine. WIREs Data Min. Knowl. Discov. 2019, 9, e1302. [Google Scholar] [CrossRef]
- Jovanović, J.; Bagheri, E. Semantic annotation in biomedicine: The current landscape. J. Biomed. Semantics 2017, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Neves, M.; Leser, U. A survey on annotation tools for the biomedical literature. Brief. Bioinform. 2014, 15, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.G.; Howsmon, D.; Zhang, B.; Hahn, J.; McGuinness, D.; Hendler, J.; Ji, H. Entity linking for biomedical literature. BMC Med. Inform. Decis. Mak. 2015, 15 (Suppl 1), S4. [Google Scholar] [CrossRef] [PubMed]
- Comeau, D.C.; Islamaj Doğan, R.; Ciccarese, P.; Cohen, K.B.; Krallinger, M.; Leitner, F.; Lu, Z.; Peng, Y.; Rinaldi, F.; Torii, M.; et al. BioC: A minimalist approach to interoperability for biomedical text processing. Database 2013, 2013, bat064. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, J.M.; Bader, G.D. Transfer learning for biomedical named entity recognition with neural networks. Bioinformatics 2018, 34, 4087–4094. [Google Scholar] [CrossRef] [PubMed]
- Tomori, S.; Ninomiya, T.; Mori, S. Domain Specific Named Entity Recognition Referring to the Real World by Deep Neural Networks. In Proceedings of the 54th Annual Meeting of the Association for Computational Linguistics (Volume 2: Short Papers), Berlin, Germany, 7–12 August 2016; Association for Computational Linguistics: Berlin, Germany, 2016; pp. 236–242. [Google Scholar]
- Maldonado, R.; Goodwin, T.R.; Skinner, M.A.; Harabagiu, S.M. Deep Learning Meets Biomedical Ontologies: Knowledge Embeddings for Epilepsy. AMIA Annu. Symp. Proc. AMIA Symp. 2018, 2017, 1233–1242. [Google Scholar] [PubMed]
- Sousa, D.; Couto, F.M. BiOnt: Deep Learning Using Multiple Biomedical Ontologies for Relation Extraction. Adv. Inf. Retr. 2020, 367–374. [Google Scholar]
- Tseytlin, E.; Mitchell, K.; Legowski, E.; Corrigan, J.; Chavan, G.; Jacobson, R.S. NOBLE—Flexible concept recognition for large-scale biomedical natural language processing. BMC Bioinform. 2016, 17, 32. [Google Scholar] [CrossRef]
- Almeida, J.S.; Hajagos, J.; Saltz, J.; Saltz, M. Serverless OpenHealth at data commons scale-traversing the 20 million patient records of New York’s SPARCS dataset in real-time. PeerJ 2019, 7, e6230. [Google Scholar] [CrossRef] [PubMed]
- Shafiei, H.; Khonsari, A.; Mousavi, P. Serverless Computing: A Survey of Opportunities, Challenges and Applications. arXiv Prepr. 2019, arXiv:1911.01296. [Google Scholar]
- Funk, C.; Baumgartner, W.; Garcia, B.; Roeder, C.; Bada, M.; Cohen, K.B.; Hunter, L.E.; Verspoor, K. Large-scale biomedical concept recognition: An evaluation of current automatic annotators and their parameters. BMC Bioinform. 2014, 15, 59. [Google Scholar] [CrossRef] [PubMed]
- Cuzzola, J.; Jovanović, J.; Bagheri, E.; Gašević, D. Evolutionary fine-tuning of automated semantic annotation systems. Expert Syst. Appl. 2015, 42, 6864–6877. [Google Scholar] [CrossRef]
- Svensson, D.; Sjögren, R.; Sundell, D.; Sjödin, A.; Trygg, J. doepipeline: A systematic approach to optimizing multi-level and multi-step data processing workflows. BMC Bioinform. 2019, 20, 498. [Google Scholar] [CrossRef]
- Jacobsen, A.; Kaliyaperumal, R.; da Silva Santos, L.O.B.; Mons, B.; Schultes, E.; Roos, M.; Thompson, M. A Generic Workflow for the Data FAIRification Process. Data Intell. 2020, 2, 56–65. [Google Scholar] [CrossRef]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.-W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef]
- Kanterakis, A.; Iatraki, G.; Pityanou, K.; Koumakis, L.; Kanakaris, N.; Karacapilidis, N.; Potamias, G. Towards Reproducible Bioinformatics: The OpenBio-C Scientific Workflow Environment. In Proceedings of the 2019 IEEE 19th International Conference on Bioinformatics and Bioengineering (BIBE), Athens, Greece, 28–30 October 2019; pp. 221–226. [Google Scholar]
- Wilkinson, M.D.; Verborgh, R.; da Silva Santos, L.O.B.; van Mulligen, E.M.; Bolleman, J.T.; Dumontier, M. Interoperability and FAIRness through a novel combination of Web technologies. PeerJ Comput. Sci. 2017, 3, e110. [Google Scholar] [CrossRef]
- Henninger, S. Using Iterative Refinement to Find Reusable Software. IEEE Softw. 1994, 11, 48–59. [Google Scholar] [CrossRef]
- Shade, A.; Teal, T.K. Computing Workflows for Biologists: A Roadmap. PLoS Biol. 2015, 13, e1002303. [Google Scholar] [CrossRef]
- Jackson, M.J.; Wallace, E.; Kavoussanakis, K. Using rapid prototyping to choose a bioinformatics workflow management system. bioRxiv 2020. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; van den Beek, M.; Blankenberg, D.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Eberhard, C.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016, 44, W3–W10. [Google Scholar] [CrossRef] [PubMed]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef]
- Wang, L.L.; Lo, K.; Chandrasekhar, Y.; Reas, R.; Yang, J.; Eide, D.; Funk, K.; Kinney, R.; Liu, Z.; Merrill, W.; et al. CORD-19: The COVID-19 open research dataset. arXiv 2020, arXiv:2004.10706v2. [Google Scholar]
- Van Auken, K.; Schaeffer, M.L.; McQuilton, P.; Laulederkind, S.J.F.; Li, D.; Wang, S.-J.; Hayman, G.T.; Tweedie, S.; Arighi, C.N.; Done, J.; et al. BC4GO: A full-text corpus for the BioCreative IV GO task. Database 2014, 2014, bau074. [Google Scholar] [CrossRef]
- Kors, J.A.; Clematide, S.; Akhondi, S.A.; van Mulligen, E.M.; Rebholz-Schuhmann, D. A multilingual gold-standard corpus for biomedical concept recognition: The Mantra GSC. J. Am. Med. Inform. Assoc. 2015, 22, 948–956. [Google Scholar] [CrossRef]
- Kim, J.-D.; Ohta, T.; Tateisi, Y.; Tsujii, J. GENIA corpus—A semantically annotated corpus for bio-textmining. Bioinformatics 2003, 19, i180–i182. [Google Scholar] [CrossRef]
- Doğan, R.I.; Leaman, R.; Lu, Z. NCBI disease corpus: A resource for disease name recognition and concept normalization. J. Biomed. Inform. 2014, 47, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.; Ashburner, M.; Rosse, C.; Bard, J.; Bug, W.; Ceusters, W.; Goldberg, L.J.; Eilbeck, K.; Ireland, A.; Mungall, C.J.; et al. The OBO Foundry: Coordinated evolution of ontologies to support biomedical data integration. Nat. Biotechnol. 2007, 25, 1251–1255. [Google Scholar] [CrossRef] [PubMed]
- Rector, A.L.; JE Rogers, J.E.; Pole, P. The GALEN High Level Ontology. Stud. Health Technol. Inform. 1996, 34, 174–176. [Google Scholar]
- Rector, A.L.; Rogers, J.E.; Zanstra, P.E.; Van Der Haring, E. OpenGALEN: Open source medical terminology and tools. AMIA Symp. 2003, 2003, 982. [Google Scholar]
- McCray, A.T.; Burgun, A.; Bodenreider, O. Aggregating UMLS semantic types for reducing conceptual complexity. Stud. Health Technol. Inform. 2001, 84, 216–220. [Google Scholar]
- Bodenreider, O.; McCray, A.T. Exploring semantic groups through visual approaches. J. Biomed. Inform. 2003, 36, 414–432. [Google Scholar] [CrossRef][Green Version]
- Lipscomb, C.E. Medical Subject Headings (MeSH). Bull. Med. Libr. Assoc. 2000, 88, 265–266. [Google Scholar]
- Rossanez, A.; dos Reis, J.C.; Torres, R.d.S.; de Ribaupierre, H. KGen: A knowledge graph generator from biomedical scientific literature. BMC Med. Inform. Decis. Mak. 2020, 20, 314. [Google Scholar] [CrossRef]
- Savova, G.K.; Masanz, J.J.; Ogren, P.V.; Zheng, J.; Sohn, S.; Kipper-Schuler, K.C.; Chute, C.G. Mayo clinical Text Analysis and Knowledge Extraction System (cTAKES): Architecture, component evaluation and applications. J. Am. Med. Inform. Assoc. 2010, 17, 507–513. [Google Scholar] [CrossRef]
- Aronson, A.R.; Lang, F.-M. An overview of MetaMap: Historical perspective and recent advances. J. Am. Med. Inform. Assoc. 2010, 17, 229–236. [Google Scholar] [CrossRef]
- Demner-Fushman, D.; Rogers, W.J.; Aronson, A.R. MetaMap Lite: An evaluation of a new Java implementation of MetaMap. J. Am. Med. Inform. Assoc. 2017, 24, 841–844. [Google Scholar] [CrossRef]
- Jonquet, C.; Toulet, A.; Arnaud, E.; Aubin, S.; Dzalé Yeumo, E.; Emonet, V.; Graybeal, J.; Laporte, M.-A.; Musen, M.A.; Pesce, V.; et al. AgroPortal: A vocabulary and ontology repository for agronomy. Comput. Electron. Agric. 2018, 144, 126–143. [Google Scholar] [CrossRef]
- Tchechmedjiev, A.; Abdaoui, A.; Emonet, V.; Zevio, S.; Jonquet, C. SIFR annotator: Ontology-based semantic annotation of French biomedical text and clinical notes. BMC Bioinform. 2018, 19, 405. [Google Scholar] [CrossRef]
- Shah, N.H.; Bhatia, N.; Jonquet, C.; Rubin, D.; Chiang, A.P.; Musen, M.A. Comparison of concept recognizers for building the Open Biomedical Annotator. BMC Bioinform. 2009, 10 (Suppl. 9), S14. [Google Scholar] [CrossRef]
- Harris, S.; Lamb, N.; Shadbolt, N. 4store: The design and implementation of a clustered RDF store. In Proceedings of the 5th International Workshop on Scalable Semantic Web Knowledge Base Systems (SSWS2009), Washington, DC, USA, 26 October 2009; pp. 94–109. [Google Scholar]
- Nunes, T.; Campos, D.; Matos, S.; Oliveira, J.L. BeCAS: Biomedical concept recognition services and visualization. Bioinformatics 2013, 29, 1915–1916. [Google Scholar] [CrossRef]
- Campos, D.; Matos, S.; Oliveira, J.L. A modular framework for biomedical concept recognition. BMC Bioinform. 2013, 14, 281. [Google Scholar] [CrossRef]
- Rebholz-Schuhmann, D.; Arregui, M.; Gaudan, S.; Kirsch, H.; Jimeno, A. Text processing through Web services: Calling Whatizit. Bioinformatics 2008, 24, 296–298. [Google Scholar] [CrossRef] [PubMed]
- Salgado, D.; Krallinger, M.; Depaule, M.; Drula, E.; Tendulkar, A.V.; Leitner, F.; Valencia, A.; Marcelle, C. MyMiner: A web application for computer-assisted biocuration and text annotation. Bioinformatics 2012, 28, 2285–2287. [Google Scholar] [CrossRef] [PubMed]
- Ison, J.; Kalaš, M.; Jonassen, I.; Bolser, D.; Uludag, M.; McWilliam, H.; Malone, J.; Lopez, R.; Pettifer, S.; Rice, P. EDAM: An ontology of bioinformatics operations, types of data and identifiers, topics and formats. Bioinformatics 2013, 29, 1325–1332. [Google Scholar] [CrossRef] [PubMed]
- Kyriakakis, A.; Koumakis, L.; Kanterakis, A.; Iatraki, G.; Tsiknakis, M.; Potamias, G. Enabling Ontology-Based Search: A Case Study in the Bioinformatics Domain. In Proceedings of the 2019 IEEE 19th International Conference on Bioinformatics and Bioengineering (BIBE), Athens, Greece, 28–30 October 2019; pp. 227–234. [Google Scholar]
- Ison, J.; Ienasescu, H.; Chmura, P.; Rydza, E.; Ménager, H.; Kalaš, M.; Schwämmle, V.; Grüning, B.; Beard, N.; Lopez, R.; et al. The bio.tools registry of software tools and data resources for the life sciences. Genome Biol. 2019, 20, 164. [Google Scholar] [CrossRef]
- Foster, E.D.; Deardorff, A. Open Science Framework (OSF). J. Med. Libr. Assoc. 2017, 105, 203–206. [Google Scholar]
- Kanterakis, A.; Karacapilidis, N.; Koumakis, L.; Potamias, G. On the development of an open and collaborative bioinformatics research environment. Procedia Comput. Sci. 2018, 126, 1062–1071. [Google Scholar] [CrossRef]
- Giardine, B.; Riemer, C.; Hardison, R.C.; Burhans, R.; Elnitski, L.; Shah, P.; Zhang, Y.; Blankenberg, D.; Albert, I.; Taylor, J.; et al. Galaxy: A platform for interactive large-scale genome analysis. Genome Res. 2005, 15, 1451–1455. [Google Scholar] [CrossRef] [PubMed]
- Köster, J.; Rahmann, S. Snakemake—a scalable bioinformatics workflow engine. Bioinformatics 2012, 28, 2520–2522. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, M.D.; Dumontier, M.; Sansone, S.-A.; Bonino da Silva Santos, L.O.; Prieto, M.; Batista, D.; McQuilton, P.; Kuhn, T.; Rocca-Serra, P.; Crosas, M.; et al. Evaluating FAIR maturity through a scalable, automated, community-governed framework. Sci. Data 2019, 6, 174. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| FAIR Principle [21] | How It Is Addressed in OpenBio.eu |
|---|---|
| F1. (Meta)data are assigned a globally unique and persistent identifier. | All ROs have a unique and permanent URL. |
| F2. Data are described with rich metadata (defined by R1 below). | Data/Tools/WFs can be downloaded in JSON including all metadata (i.e., description, dependencies). |
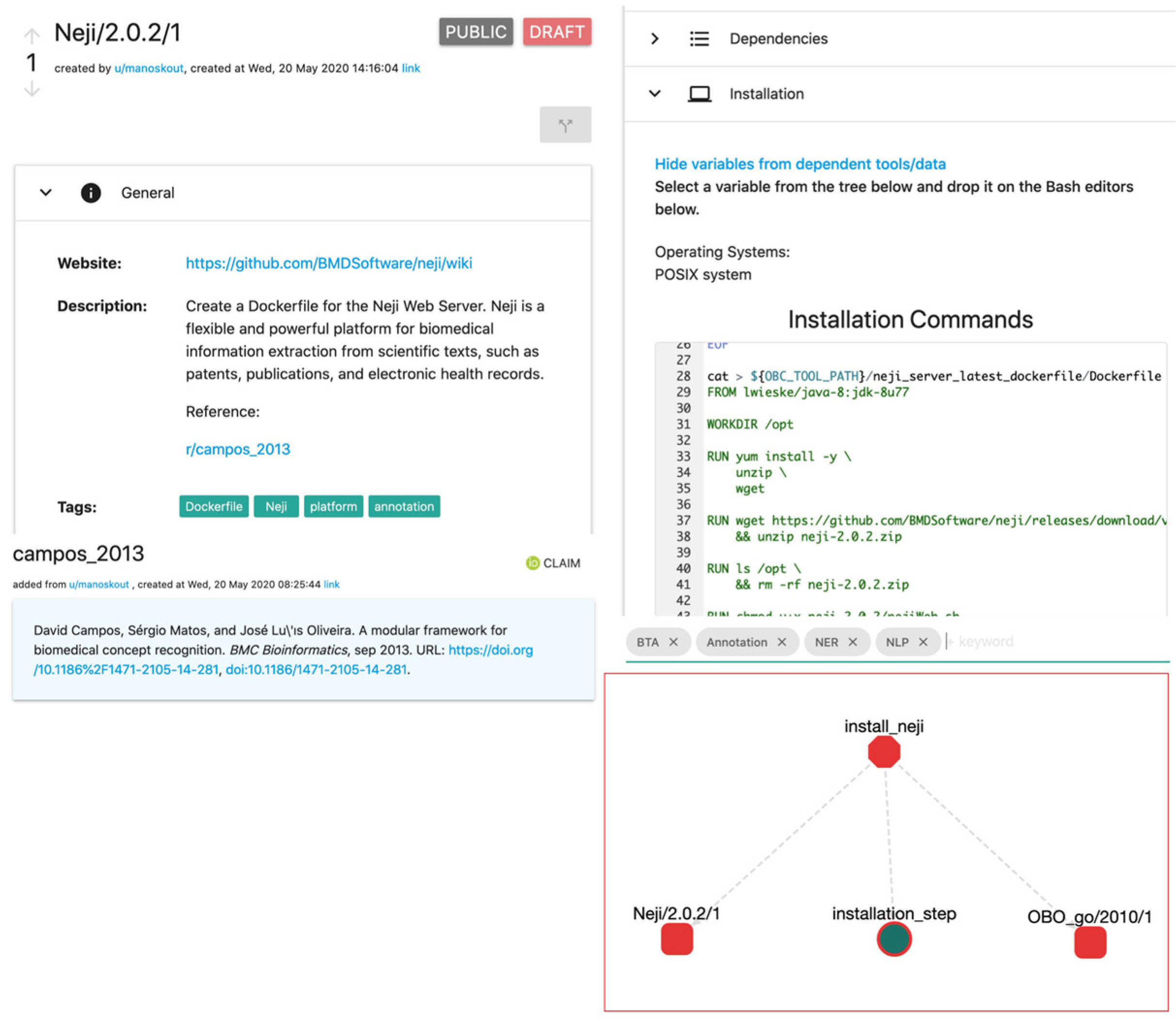
| F3. Metadata clearly and explicitly include the identifier of the data they describe. | Metadata reference other ROs in the format of a persistent identifier with a common pattern. For example: t/Neji/2.0.2/2 (tool, name, version, edit). |
| F4. (Meta)data are registered or indexed in a searchable resource. | All ROs are indexed and searched in a common UI. |
| A1. (Meta)data are retrievable by their identifier using a standardized communications protocol. | OpenBio.eu includes a documented REST-API with a standard gateway for accessing ROs + metadata. |
| A1.1 The protocol is open, free, and universally implementable. | Yes. Even unregistered users have access. The implementation is open source. |
| A1.2 The protocol allows for an authentication and authorization procedure, where necessary. | Not needed. All ROs are open and accessible for all users. |
| A2. Metadata are accessible, even when the data are no longer available. | True. ROs cannot be deleted. Even if the tools/data are deleted from their source, the metadata remain. |
| I1. (Meta)data use a formal, accessible, shared, and broadly applicable language for knowledge representation. | Data from the API are available in JSON format. The data model is available on the projects’ documentation page. |
| I2. (Meta)data use vocabularies that follow FAIR principles. | Currently, there is no predefined ontology or vocabulary used for tagging or describing Ros. |
| I3. (Meta)data include qualified references to other (meta)data. | Yes. All qualified references are OpenBio.eu IDs. |
| R1. Meta(data) are richly described with a plurality of accurate and relevant attributes. | Yes. Metadata include all information required to reproduce and run an RO. |
| R1.1. (Meta)data are released with a clear and accessible data usage license. | Yes. Both the Terms and Conditions and the Privacy Statement are listed. Terms include that all ROs also follow the BSD license. |
| R1.2. (Meta)data are associated with detailed provenance. | Yes. Metadata include edit number and are linked with previous edits for complete history tracking. |
| R1.3. (Meta)data meet domain-relevant community standards. | Yes, considering community standards for open-source scientific software. |
| Corpus | Inserted Corpus-Object | OpenBio.eu ID |
|---|---|---|
| COVID-19 | 63,000 research articles | CORD_19/01_03_2021/1 |
| BC4GO | 5000 passages in 200 articles | BC4GO/2013_08_02/1 |
| Mantra GSC | 5530 biomedical annotations | matra_gsc/latest/1 |
| GENIA | 3000 annotated abstracts | GENIA_annotation/latest/1 |
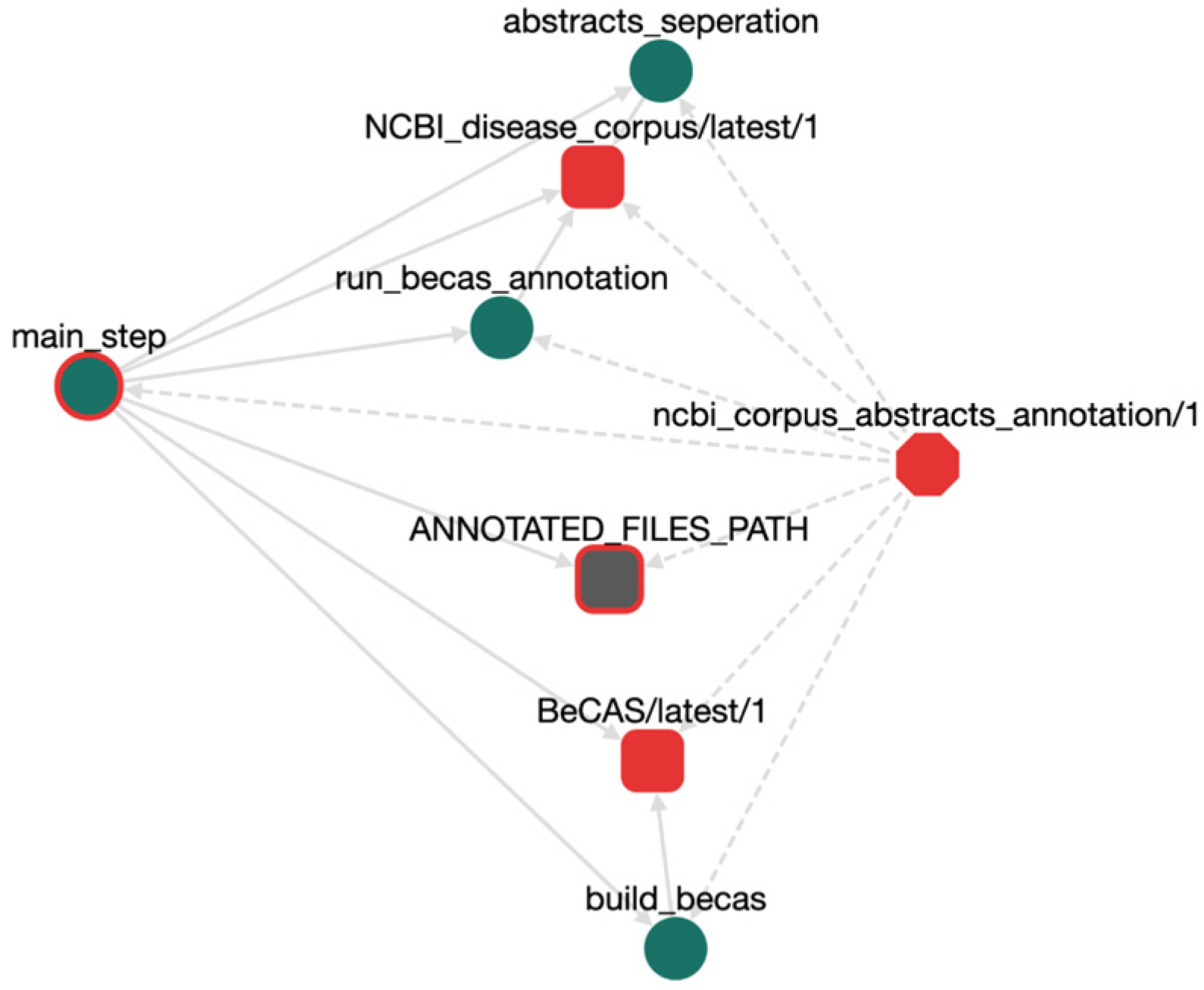
| NCBI DC | 793 abstracts (790 disease concepts) | NCBI_disease_corpus/latest/1 |
| Corpus | Inserted Object | OpenBio.eu ID |
|---|---|---|
| OBO Foundry | 167 ontologies with biomedical, chemical and social concepts in OWL format | OBO_obi/2013/1 |
| GALEN | Medical concepts in custom format | galen/01_09_2007/1 |
| MeSH | Controlled Vocabulary as RDF triplets | MeSH/2020/1 |
| Tool | Inserted Object | OpenBio ID |
|---|---|---|
| Apache cTAKES | Dockerfile with GUI | cTAKES/4.0.0/1 |
| Noble Coder | Dockerfile with CLI | noble_tools/latest/1 |
| MetaMap | Dockerfile with CLI | MetaMapLite/RELEASE3.6.2rc5/1 |
| NCBO-Annotator | Docker-compose | ncbo_bioportal/1/1 |
| Neji | Dockerfile with GUI | Neji/2.0.2/2 |
| BeCAS | Docker with API client | BeCAS/latest/1 |
| Characteristic | Action |
|---|---|
| Completely web-based; you do not have to install anything. | Use OpenBio.eu for your everyday science tasks from your browser. |
| No DSL (Domain Specific Language) required. | Simply describe your steps in BASH. Each workflow step is available as a BASH function. Do you have a BASH script that installs a TDW object (tool, dataset, or workflow)? Just copy/paste it! |
| Use Python, Perl, Java, or any language you like to conduct your analysis. | Call these tools the same way you would call them from the command line. |
| Fork existing tools/workflows to create your own versions. | Press the “Fork” button in the UI of any tool/data or workflow entry. |
| Simple workflow structure. | OpenBio.eu offers a simple GUI, just drag-and-drop an already imported TDW object to add it into a workflow and/or create dependencies between tools. Do the same for a workflow! Workflows can contain other workflows indefinitely. |
| Simple mental model. | Tools have variables (i.e., environment variables) such as: installation_path = path/to/executable. Workflows have input and output variables as well, such as: input_data = path/to/data, results = path/to/results. |
| Does it support iterations or conditional execution? | Yes. In fact, it supports anything that BASH supports (even recursion). |
| What name should I use for my tool/dataset/workflow? | Anything you like. Each TDW object is identified by a name (anything you want), a version (anything you want), and an ID provided by the system. The namespace is unique and global. |
| Add markdown descriptions for the objects. | Use t/tool_name/tool_version/id and w/ workflow_name/id to link to an object anywhere on the site. |
| Each object has a Questions and Answers section. | Navigate on the Discussion section of an RO. Contains a typical forum-like interface. |
| Add scientific references. | Link with r/reference_name. If you do not want to manually add all bibliographic details for a paper, just add the DOI; the system will do the rest. |
| Execute workflows in your own environment. | You do not have to share code or data with OpenBio.eu, only the BASH commands that install and run workflows. Monitor the execution during runtime via the provided graphical interface. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanterakis, A.; Kanakaris, N.; Koutoulakis, M.; Pitianou, K.; Karacapilidis, N.; Koumakis, L.; Potamias, G. Converting Biomedical Text Annotated Resources into FAIR Research Objects with an Open Science Platform. Appl. Sci. 2021, 11, 9648. https://doi.org/10.3390/app11209648
Kanterakis A, Kanakaris N, Koutoulakis M, Pitianou K, Karacapilidis N, Koumakis L, Potamias G. Converting Biomedical Text Annotated Resources into FAIR Research Objects with an Open Science Platform. Applied Sciences. 2021; 11(20):9648. https://doi.org/10.3390/app11209648
Chicago/Turabian StyleKanterakis, Alexandros, Nikos Kanakaris, Manos Koutoulakis, Konstantina Pitianou, Nikos Karacapilidis, Lefteris Koumakis, and George Potamias. 2021. "Converting Biomedical Text Annotated Resources into FAIR Research Objects with an Open Science Platform" Applied Sciences 11, no. 20: 9648. https://doi.org/10.3390/app11209648
APA StyleKanterakis, A., Kanakaris, N., Koutoulakis, M., Pitianou, K., Karacapilidis, N., Koumakis, L., & Potamias, G. (2021). Converting Biomedical Text Annotated Resources into FAIR Research Objects with an Open Science Platform. Applied Sciences, 11(20), 9648. https://doi.org/10.3390/app11209648