Abstract
Today, there are excellent resources for the semantic annotation of biomedical text. These resources span from ontologies, tools for NLP, annotators, and web services. Most of these are available either in the form of open source components (i.e., MetaMap) or as web services that offer free access (i.e., Whatizit). In order to use these resources in automatic text annotation pipelines, researchers face significant technical challenges. For open-source tools, the challenges include the setting up of the computational environment, the resolution of dependencies, as well as the compilation and installation of the software. For web services, the challenge is implementing clients to undertake communication with the respective web APIs. Even resources that are available as Docker containers (i.e., NCBO annotator) require significant technical skills for installation and setup. This work deals with the task of creating ready-to-install and run Research Objects (ROs) for a large collection of components in biomedical text analysis. These components include (a) tools such as cTAKES, NOBLE Coder, MetaMap, NCBO annotator, BeCAS, and Neji; (b) ontologies from BioPortal, NCBI BioSystems, and Open Biomedical Ontologies; and (c) text corpora such as BC4GO, Mantra Gold Standard Corpus, and the COVID-19 Open Research Dataset. We make these resources available in OpenBio.eu, an open-science RO repository and workflow management system. All ROs can be searched, shared, edited, downloaded, commented on, and rated. We also demonstrate how one can easily connect these ROs to form a large variety of text annotation pipelines.
1. Introduction
Annually, it is estimated that more than one million articles are added to PubMed, which is the most widespread public repository for biomedical papers. This extreme volume of curated scientific literature results in an information overload. Additionally, with the advent of scientific preprint servers such as bioRxiv (www.biorxiv.org, accessed on 11 October 2021) and medRxiv (www.medrxiv.org, accessed on 11 October 2021), thousands of papers in various stages of maturity are published daily. It is indicative that with the current COVID-19 pandemic, the number of relevant scientific articles edited by both bioRxiv and medRxiv during the period from October 2019 (beginning of the pandemic) to February 2021 is approximately 14,000 from 72,400 in total (~19%). In a situation where the global scientific output doubles every nine years, there is the question of whether this increase has a clear impact on “real” and actionable knowledge growth [1]. The majority of published papers are characterized as “background noise,” and researchers struggle to “separate the wheat from the chaff.” As an effect, keeping up with new information in this field is extremely difficult and time-consuming.
Besides the overwhelming quantity of papers, researchers also have to deal with multiple keywords to refer to a single given entity and the limited abilities of existing databases to perform complex semantic searches [2]. It is not an exaggeration to claim that science on that matter has not changed over the last 20 years. This is surprising, however, given that the results of most of the millions of published papers in experimental sciences were generated through an analysis pipeline of some kind. In fact, these pipelines are rarely publicly available. The temporal decay and eventual loss of these pipelines, in contrast to the resultant ever-preserved and well-indexed scientific papers, constitutes a major knowledge and cultural loss for our society. We argue that this unnatural separation between scientific reports and analysis pipelines is one of the origins of the reproducibility crisis that has been characterized as a plague for science, threatening funding and stigmatizing the community in general [3]. For instance, it has been estimated that the prevalence of irreproducible preclinical research exceeds 50% at a price of approximately $28.2 billion [4].
A potential way out and obvious remedy to the aforementioned problems is Biomedical Text Annotation (BTA), the process of identifying indicative terms that relate to and adequately capture the meaning of medical and biological entities occurring in scientific reports. Indeed, according to recent [5,6] and older [7] reviews in the field, the quality of existing resources for BTA has reached a very satisfactory level. Nevertheless, the same reviews, and especially [6], have pinpointed some limitations of existing resources. The first is the lack of semantic context that often guides annotators to “overfit” the extracted entities based on the specific ontology in which they are trained. A solution to this is to simply link existing ontologies, as outlined in [8,9]. Another challenge is sufficiently applying and adapting existing advances in deep neural networks for Natural Language Processing (NLP), so as to improve common BTA tasks such as Named Entity Recognition, translation, and question answering [10,11,12,13].
Another issue is that annotators face technical difficulties when it comes to scaling up for the purpose of annotating hundreds of millions of documents [14]. This is partly due to the fact that numerous existing solutions were introduced even decades ago, and either have not been tested with huge corpora or have limited scaling abilities. Modern technologies such as virtualized computing and hosting in “serverless” environments can help to resolve this issue [15,16]. Another important consideration is that existing benchmarks are application-specific [17]. Annotators work within many parameters that can have a detrimental effect on their efficacy. Tuning the parameters of an annotator for a specific test is a form of introducing bias when compared with other annotators. A remedy for this is to have independent third-party tools tune the parameters of the annotators during testing. There are already frameworks for automatic parameter tuning in large and complex workflows [18,19]. Of course, this would require the “wrapping” of the parameter space in a common format for all tested tools.
We argue that all approaches to the solution of the aforementioned problems and challenges are tightly connected with the FAIRification process [20], the process of making research (and not only) digital resources findable, accessible, interoperable, and reusable, for both humans and machines [21]. For example, making ontologies and lexicographic resources findable and accessible will help annotators to generalize their concepts, expand their domains, and support more languages. Making BTA tools interoperable will help with both benchmarking and scaling in modern computational environments. Moreover, making all workflows reusable will help with locating and correcting possible biases in comparison studies.
The work presented in this paper is about making BTA processes FAIR compliant. This is achieved by using the OpenBio.eu platform, an open science online environment where researchers can create, edit, share, re-combine, export, execute, rate, and collaborate on managing tools, data, and workflows (called hereafter Research Objects, or ROs) [22]. The purpose of the platform is to offer data stewardship services that will aid the reuse of data beyond their original scope. The code for ROs can be anything that is executable in a local computer. These can be simple scripts, source code, binaries, Docker, or any other containerization format. All information is imported into ROs through an intuitive UI without having to know any Domain Specific Language (DSL). A user only needs to insert the installation instructions that she would have to write in an execution environment; in other words, importing a tool in OpenBio.eu requires exactly the same steps as importing it in an operating system such as Linux [22].
In the context of the above-described framework, this work demonstrates the synthesis, orchestration, and deployment of BTA workflows via the combination of respective open-source ROs, all of which were appropriately imported and installed in OpenBio.eu. A total of five corpora, 169 ontologies, and six known tools for BTA were imported. The effort required to add some of these ROs in an open science environment such as OpenBio.eu is pointed out as a means of highlighting the challenges involved with the setup and configuration of BTA open-source computational resources. The remainder of this paper is organized as follows: Section 2 introduces the main architecture of OpenBio.eu and demonstrates how it enables the FAIRification of research objects; Section 3 presents, in detail, a set of ROs in BTA, their functionality, and other remarks from their FAIRification process; Section 4 presents how the FAIRified ROs can be combined in complex workflows, which can help with tasks such as comparison and scaling in high-performance computing environments; finally, Section 5 presents some concluding remarks, limitations, and future work directions.
OpenBio.eu is available at: https://www.openbio.eu (accessed on 11 October 2021). The source code is available at: https://github.com/kantale/OpenBio.eu (accessed on 11 October 2021) and detailed documentation is available at: https://kantale.github.io/OpenBio.eu/docs/ (accessed on 11 October 2021).
2. FAIRification of ROs with OpenBio.eu
Although FAIR guidelines concern data, we can very easily extend them to tools and workflows. For example, data may describe biomedical entities (i.e., gene expression), by defining appropriate formats. Similarly, we can easily create data structures to describe tools and workflows. Therefore, FAIR principles can and should be applied to all types of ROs that take part in scientific analysis. This last characteristic is demanding of another crucial requirement; that is, that the digital resource should also be embeddable, thus extending the conceptualization of the FAIR principles with an embeddable dimension, i.e., FAIR-E (“E” for embeddable).
In OpenBio.eu, the incorporated ROs are conceptualized as integrated constructs that contain both the code required for installation (or download in the case of a dataset RO) and the semantic description required for connecting them with other ROs. Coupled with semantic web technologies, the process allows for the exploitation of linked-data architectures as connections between FAIR ROs [23], and, at the same time, it hides the representation details from users, providing a more abstract level of interaction with the system. In this setting, FAIR-E overcomes the conceptual gap between the problem to be solved and its computational solution, since usually software is described in terms of functionality (how), and problems are formulated in terms of the task to be tackled (what) [24]. To state it in a more direct manner, the design philosophy underlying OpenBio.eu considers the data and the software that operates on the data as neighbor concepts, and, at least in terms of their semantic content, as identical concepts. On Table 1 we list all FAIR principles as they were originally published [21], along with how OpenBio.eu implements them. Generally, the top four FAIR principles are im-plemented as follows:

Table 1.
List of all official FAIR principles and how they are addressed in OpenBio.eu.
Findability in OpenBio.eu is achieved by having all ROs indexed in the same searchable repository. Moreover, OpenBio.eu follows an additive model, in the sense that ROs cannot be deleted and cannot be edited. Nevertheless, users can “fork” the RO of any other user and create an identical RO to which they can apply any edit that they wish. Each fork creates a new RO and is assigned a unique ID with a permanent URL.
Accessibility is an inherent feature of OpenBio.eu. All content is free and open source. All content is not only directly accessible but also installable on any modern computer. All content is available even to anonymous users. Moreover, content is available through a REST API.
Interoperability. ROs in OpenBio.eu can be exported into two basic formats: as a BASH executable or in Common Workflow Language (CWL) format. (a) BASH is the most standard command-line interface on Linux and OSX (with MS Windows support as well). Even though BASH is not a workflow language, per se, it offers a flexible enough framework to effectively code and arrange the individual steps of a workflow [25,26]. A key characteristic served by OpenBio.eu is to hide the complexity of BASH by pulling it at the front-end of the workflow orchestration process via its graphical workflow editing interface. This makes it unnecessary for the user to learn any domain-specific programming language (DSL), thus improving flexibility in workflow design, and at the same time easing the adaptation and sharing of workflows. (b) Workflows are exported in CWL through an algorithm that converts BASH function calls to directed acyclic graphs (DAGs). Through CWL, the interoperability of devised workflows between any contemporary operating system is achieved, allowing for the inclusion of ROs and workflows into a wide variety of Workflow Management Systems (WfMSs) such as CWLTool, Galaxy [27,28], Nextflow (www.nextflow.io, accessed on 11 October 2021) [29], Airflow, and Argo. Some of these WfMSs (i.e., CWL, Airflow, Nextflow) support execution in a virtualized environment (through Docker). This enhances reproducibility and alleviates potential problems such as system upgrades that might break compatibility between the system’s libraries and an installed RO.
Reusability. All the above features also contribute to the reusability guideline of FAIR. In OpenBio.eu, reusability is achieved with two mechanisms. The first, also common in other WfMSs, is by allowing the rich annotation of research objects. The second is by encouraging the community to provide reliable and accurate instructions to reproduce and reuse a RO in a real programming language. Reusability is measured objectively as the number of succesful uses of this RO from other users, as well as from the positive/negative criticism that it received from the community. In this context, an RO in OpenBio.eu is linked with a users’ profile and respective published material. This acts as a multi-directional connection between reproducible ROs, scientific publications, and user profiles. Users are awarded credit every time an RO that they “own” is reused, thus providing a previously missing incentive for RO sharing in academia. It is stressed here that ROs are not simple descriptions or annotations of “real-life” tools, data, or workflows; they are software components, directly downloadable and executable in any modern computation environment. Figure 1 shows how this functionality is realized and implemented in OpenBio.eu.
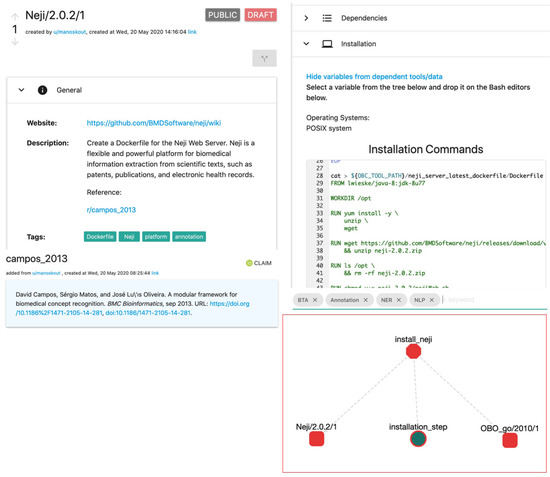
Figure 1.
Screenshots of various OpenBio.eu components. A tool (Neji) contains the description, installation instructions, and bibliographic references (top left). ROs are accompanied by directly executable source code (top right). The lower left shows the scientific paper linked with this tool and the lower right shows a graph with a workflow that uses this tool (Neji) with another RO which is an ontology (OBO Gene Ontology).
3. Imported Resources for Biomedical Text Annotation
Today, there exist many repositories with numerous resources for the task of BTA. Some representative examples in the area of biomedical research are ORBIT (orbit.nlm.nih.gov) and biotools (bio.tools). These repositories are very valuable, but they lack one important element: instructions and directions to install, download and compile them. Locating an RO in these repositories is usually the first step, but importing it into a “real” analysis pipeline is a cumbersome task that often requires advanced IT knowledge. As we have already described, in OpenBio.eu each imported RO is installable and executable.
3.1. Imported Corpora
For the purpose of the study presented in this paper, we have imported four corpora sources into OpenBio.eu, as summarized in Table 2.
Table 2.
Corpora imported into OpenBio.eu (with unique OpenBio.eu IDs).
The COVID-19 Open Research Dataset (CORD-19, www.semanticscholar.org/cord19) [30], is a free, weekly updated source that contains information on more than 800,000 research articles (last accessed on 1 March 2021) related to COVID-19, SARS-CoV-2, and other relevant literature. The articles in CORD-19 have been collected from popular scientific repositories and publishing houses, including Elsevier, bioRxiv, medRxiv, the World Health Organization (WHO), and PubMed Central (PMC). Each scientific article in CORD-19 has a list of specific attributes, namely “citations,” “publish time,” “title,” “abstract,” and “authors.” Furthermore, the majority of the articles (about 700,000) include a “full text” attribute. Although the CORD-19 dataset is the most important knowledge source related to COVID-19 and other coronavirus infections, its use may be constrained due to the fact that the provided data are mainly unstructured text references. So, the exploitation of NLP techniques and tools (from entity recognizers and annotators to text categorization) is the only way to extract the underlying knowledge.
The BC4GO dataset (www.biocreative.org/resources/corpora/bc-iv-go-task-corpus, accessed on 11 October 2021) [31] is a full-text corpus to be utilized as an assistant in the automatic identification of Gene Ontology (GO) terms. Each sample of the BC4GO dataset includes a GO annotated text, a gene or gene product, a GO term, and a GO evidence code. The authors evaluated the quality of the BC4GO dataset by comparing it with GO-annotated data that were generated by a group of experts. It includes about 5000 text passages in 200 articles related to 1356 unique GO terms. The evaluation results showed an increase in the accuracy of the GO annotation task. BC4GO enables the construction of a knowledge base, which in turn is able to improve various text mining tools related to the BioNLP research community [2].
The Mantra GSC (Mantra gold-standard corpus, biosemantics.erasmusmc.nl/index.php/resources/mantra-gsc) [32] aims to facilitate biomedical concept recognition. The Mantra GSC consists of five parallel textual corpora in five different languages (i.e., English, French, German, Spanish, and Dutch). The text segments in each textual corpus originate from MEDLINE abstract titles, drug labels, biomedical patent claims, scientific publications, and electronic health records. The total number of biomedical annotations is 5530.
GENIA (www.geniaproject.org, accessed on 11 October 2021) [33] is the most famous corpus for BTA, according to the ORBIT repository. Being also one of the most recent resources, it is very well studied and is considered a gold standard in the field. The most recent version contains 3000 abstracts from MEDLINE, annotated with information regarding biochemical substances and protein reactions.
NCBI DC (www.ncbi.nlm.nih.gov/CBBresearch/Dogan/DISEASE, accessed on 11 October 2021) [34] is perhaps the most carefully annotated corpus in the biomedical domain. Although it contains a relatively low number of abstracts (793), each abstract has been annotated by two professionals in a three-annotation phase. NCBI DC is considered a gold standard in corpus annotation.
3.2. Imported Ontologies
Ontologies, especially those of high importance, are usually well-maintained and easy to use, making them perhaps the only type of ROs in this area with adequate FAIR indications. For the purpose of the study presented in this paper, we have imported three ontology sources into OpenBio.eu, as summarized in Table 3.
Table 3.
Ontologies imported into OpenBio.eu (with unique OpenBio.eu IDs).
The Open Biological and Biomedical Ontology (OBO) Foundry (www.obofoundry.org, accessed on 11 October 2021) [35] contains 167 ontologies and is by far the most valuable biomedical ontology resource. Since its foundation, it has played a significant role in BTA, as it offers an open and collaborative environment for well-defined ontology models. What is exceptional about the OBO is that it marks obsolete or inactive ontologies that have not been actively maintained. As we will see later, this is a much-needed feature for tools involved in BTA. All OBO ontologies have been imported into OpenBio.eu.
The GALEN Common Reference Model (www.opengalen.org, accessed on 11 October 2021) [36,37] is a biomedical ontology with mainly historical value. It is influenced by the work in the UMLS Semantic Network (semanticnetwork.nlm.nih.gov, accessed on 11 October 2021) [38,39], and was defined and utilized as a system for modeling medical concepts. It contains a system for defining modifiers and modalities and represents concepts as substances, processes, and structures.
MeSH (Medical Subject Headings, www.nlm.nih.gov/mesh, accessed on 11 October 2021) [40] is a structured vocabulary maintained from the National Library of Medicine. From its foundation in the 1960s (www.nlm.nih.gov/hmd/collections/digital/MeSH/mesh.html, accessed on 11 October 2021), it has been actively used for annotating papers in the MEDLINE/PubMed databases, therefore it is one of the most successful and widely utilized ontologies for BTA. In OpenBio.eu, we have imported the ontology through a file in RDF T-triple (subject, predicate, object) format (www.w3.org/TR/rdf11-concepts/-section-triples, accessed on 11 October 2021).
An overview of the biomedical ontologies imported into OpenBio.eu is shown in Table 3. We are currently working to include meta-mappers, such as BioC (bioc.sourceforge.net) [9], that ease the task of merging existing ontologies into more generic knowledge constructs, such as knowledge graphs, that can be used to annotate and query large amounts of literature data [41].
3.3. Imported Tools for Biomedical Text Annotation
Here, we present the process of importing and installing BTA tools in OpenBio.eu and report on the experience. A very comprehensive review and description of these tools, along with a thorough comparison, can be found in [6]. Since most tools have a multitude of dependencies, we chose to add them as Docker files. Table 4 summarizes the various forms in which these tools were imported into OpenBio.eu.
Table 4.
BTA Tools imported into OpenBio.eu (with unique OpenBio.eu IDs); all tools were imported in various Docker forms.
The Apache cTAKES (ctakes.apache.org) [42] facilitates the extraction of medical information that can be found in online electronic clinical documents (e.g., clinical notes). It identifies the available types of named clinical entities (i.e., multi-token terms with nested structures), such as drugs, diseases, symptoms, and procedures. For each extracted entity, various attributes become available—for example, a text segment, an ontology, and other entity-specific attributes. In cTAKES, information extraction is founded on the Unstructured Information Management Architecture framework (UIMA, uima.apache.org) and the OpenNLP tool (opennlp.apache.org). cTAKES can be used to develop decision support systems for clinical research. In cTAKES, installation instructions are straightforward, and through these, we created a cTAKES Docker container in OpenBio.eu.
Noble Coder (ties.dbmi.pitt.edu/noble-coder) [14] is a tool for performing Named Entity Recognition (NER) in biomedical textual documents. Noble Coder is part of the Noble Tools Suite, which is a set of NLP tools written in Java. It includes an ontology API that supports the processing of Web Ontology Language (OWL) files. It also facilitates the use of various NLP methods, such as text normalization, n-gram extraction, and stemming. The last edit in the public repository for Noble Tools (github.com/dbmi-pitt/nobletools, accessed on 11 October 2021) is from August 2018; therefore, this project seems to be abandoned. After we forked this repository, we added the necessary changes so that we could rebuild it with currently available tools. Then, we created and imported the respective Docker configuration file into OpenBio.eu.
MetaMap (metamap.nlm.nih.gov) [43] is an easily adaptable tool that maps biomedical text to UMLS Metathesaurus concepts (www.nlm.nih.gov/research/umls/knowledge_sources/metathesaurus/index.html, accessed on 11 October 2021). It relies heavily on NLP and computational linguistic techniques. It supports complex text-specific tasks ranging from information retrieval (IR), text mining, text categorization, and text summarization to question answering and knowledge discovery. MetaMap is available under a special license called the UMLS Metathesaurus License Agreement (uts.nlm.nih.gov/license.html, accessed on 11 October 2021). As an effect, we could not import MetaMap into OpenBio.eu. Nevertheless, a lighter version of MetaMap, called MetaMapLite (metamap.nlm.nih.gov/MetaMapLite.shtml, accessed on 11 October 2021) [44], is available under the BSD license. MetaMapLite implements a very fast method for NER. Nevertheless, although it is faster than MetaMap, it is not as thorough in the matching of entities. In OpenBio.eu, we provide scripts to create the appropriate Docker files for this particular version of MetaMap.
NCBO annotator (bioportal.bioontology.org/annotator, accessed on 11 October 2021) [45] is a web service that provides text annotation utilities. By relying on textual biomedical metadata, NCBO annotates a given text with biomedical ontology concepts. Hence, it allows for the automation of the tag annotation process. The available ontologies and concepts can be retrieved from the UMLS Metatheseaurus and the NCBO repository (bioportal.bioontology.org, accessed on 11 October 2021). NCBO was by far the most difficult system to set up. NCBO maintains a GitHub repository (github.com/ncbo/ncbo_annotator, accessed on 11 October 2021) for its annotator, but lacks information regarding installation and configuration. Nevertheless, there are two projects that have built domain-specific annotation services based on NCBO’s annotator. The first is Argoportal (agroportal.lirmm.fr, accessed on 11 October 2021) [45] and the second is the SIFR annotator (bioportal.lirmm.fr/annotator, accessed on 11 October 2021) [46]. Argoportal is based on a fork of NCBO’s annotator and the SIFR annotator is based on a fork of Argoportal. Both repositories have been inactive for more than a year. On their respective repositories, they maintain a Docker Compose setup (github.com/sifrproject/docker-compose-bioportal, accessed on 11 October 2021) that configures mgrep [47] as a concept recognizer, 4store (github.com/4store/4store, accessed on 11 October 2021) [48] as an RDF database, Apache solR (ucene.apache.org/solr, accessed on 11 October 2021) as an indexing service, and nginx (nginx.org, accessed on 11 October 2021) as a reverse proxy. Despite their containerization, most of these services required significant changes before they were able to run and communicate correctly. In OpenBio.eu, we imported a new Docker Compose setup which is our fork on the SIFR annotator. Despite our efforts, some of the NCBO annotator functionality, such as concept mapping, is still missing; therefore, we consider the task of integrating this tool into OpenBio.eu a work in progress.
BeCAS, the Biomedical Concept Annotation System (bioinformatics.ua.pt/becas, accessed on 11 October 2021) [49], provides a web application, an API, and a widget aiming to facilitate the task of biomedical concept identification. It assists biomedical researchers in the annotation of about 1,200,000 biomedical concepts, which can be found in textual documents and PubMed abstracts. BeCAS is closed-source and freely available for non-commercial use. Nevertheless, it provides an API with which a client can access the entirety of its functionality. One of these clients is the becas-python library (tnunes.github.io/becas-python, accessed on 11 October 2021), which is based on Python v2. In OpenBio.eu, we created a Docker file that includes this client.
Neji (github.com/BMDSoftware/neji, accessed on 11 October 2021) [50] is a platform that aims to improve the extraction of biomedical information from the biomedical scientific literature, including patents, publications, and healthcare records. It provides a NER tool, founded on the utilization of both machine learning and dictionary-based approaches, to support various NLP tasks, including sentence splitting, dependency parsing, concept recognition, and text normalization. Neji was imported into OpenBio.eu as a Docker file.
As a final note, we should mention that some very well-known and widely cited tools, such as Whatizit (www.ebi.ac.uk/webservices/whatizit, accessed on 11 October 2021) [51] and MyMiner (myminer.armi.monash.edu.au, accessed on 11 October 2021) [52], which continue to be active, are of very limited use since they come with closed-source and dysfunctional or even absent APIs. Of course, there is a huge variety of very popular tools that we have not included, such as the NCBI/BioNLP collection of tools (www.ncbi.nlm.nih.gov/research/bionlp/Tools, accessed on 11 October 2021). It is our plan to add them in the future, and we also hope to incentivize the BTA community to create ROs in OpenBio.eu in order to create a live, long-term, and sustainable BTA infrastructure. Furthermore, we are working on OpenBio.eu’s inclusion of mature ontologies from the bioinformatics domain, such as EDAM (edamontology.org, accessed on 11 October 2021) [53], and their coupling with ontology-based search operations and tools in order to support reliable question answering (QA) operations in the biomedical domain [54].
Figure 2 shows an example workflow using two ROs that, as described above, were imported into OpenBio.eu; namely, BeCAS as the annotation tool and NCBI DC as the (disease) target corpus for annotation. The whole workflow was composed by drag-and-dropping the two BTA ROs in the online GUI provided by OpenBio.eu to compose and edit the workflows (see next section). Nodes indicate RO types: the hexagon (center) is the workflow; rounded squares are tools or data; circles are the steps followed to implement the functionality of a tool and/or a workflow; an arrow from a step A to step B means “A calls B;” and dotted-line arrows signify ownership. For example, the step “abstract_separation” belongs to the “ncbi_corpus_abstracts_ annotation/1” workflow. The circle with the red outline, the “main_step,” is the one that will be called first during execution.
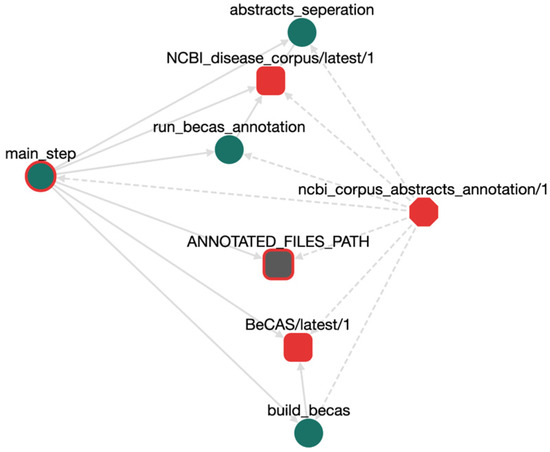
Figure 2.
An example workflow in OpenBio.eu for annotating the NCBI disease corpus with the BeCAS annotation tool.
4. Working with OpenBio.eu
Before presenting the basics of how to create, install, and/or import a new tool, data, or workflow in OpenBio.eu, we will highlight a few substantial features of the platform, aiming to reveal what differentiates OpenBio.eu from other similar platforms. A full list of features is available on the project’s documentation page.
Firstly, OpenBio.eu is able to “run” objects. In the biomedical domain, there are many repositories that store and give access to tools, data, and workflows. Examples are biotools [55] and OSF [56]. These repositories offer rich annotations of imported objects, but they lack one crucial component: the execution of the imported object (i.e., tool, workflow) on a computer that you have access to. OpenBio.eu is a repository for research objects that offers this ability. In OpenBio.eu, the importing of a new tool, data, or workflow is performed by providing explicit instructions on how to install, validate, and execute an object.
Secondly, the computer language that OpenBio.eu utilizes for object installation and execution is BASH (www.gnu.org/software/bash, accessed on 11 October 2021). Since we need to provide explicit instructions on how to install a tool, download data, or execute a workflow, we need a computer language to do so. OpenBio.eu uses BASH. Even if one may find it difficult or outdated, BASH is the defacto glue language of most Unix-like operating systems (the *nix universe), and by hosting code in BASH we ensure that it is directly executable in as many environments as possible. This does not mean that the hosting of code in BASH excludes other languages. On the contrary, BASH was chosen exactly because it enables the linkage of different computer languages, programs, and scripts into a common script. It is important to note that OpenBio.eu acts as a repository for user-provided BASH scripts. These scripts are self-dependent, meaning that other users who want to run these scripts do not have to install or download any other software. Moreover, OpenBio.eu offers a mechanism for linking these user-provided scripts together. For example, if user U1 submits tool T1, and user U2 submits tool T2, and declares that this tool has T1 as a dependency, OpenBio.eu makes sure that whenever T2 is downloaded, the scripts for T1 are also downloaded and both are executed in the right order. The same process occurs when different tools and data are combined through the graphical user interface to form workflows. In that sense, OpenBio.eu acts as a repository for BASH scripts, making sure that two or more scripts are compatible when linked in the same workflow. A fair criticism of the choice of BASH is that it is not a “write once run everywhere” language, such as Java or Python. However, BASH was not chosen as an implementation language (as in, “everything should be in BASH”), but as a gluing language (as in, “connect your components with BASH”). Finally, every script may be rated (upvoted or downvoted) by any user on the platform. This forces users to provide qualitative scripts that run on the majority of *nix systems.
Thirdly, in OpenBio.eu, tools, data, and workflows are the same type of object. Most WfMSs and open science environments distinguish between data, tools, and workflows. Users have to declare different properties for each, store them in different structures and tables, etc. OpenBio.eu does not make this distinction. Data, tools, and workflows are ROs of the same type (hereafter we use the term TDW to denote the common RO types: tools, data, and workflows). This is because, semantically, and in the context of a WfMS, there are no differences between Tools, Data and Workflows. Tools and workflows have dependencies, but data are useless without the presence of other data. We need commands to download, configure, compile, and install tools, but data need to be downloaded and, most of the time, they also need to be decompressed, pre-processed, and installed. Moreover, it is very common that data, tools, and workflows co-exist in a dependency tree of other tools, data and workflows. Table 5 lists some basic characteristics of the OpenBio.eu platform, accompanied by respective actions.
Table 5.
Basic characteristics of OpenBio.eu and actions to perform.
4.1. Importing ROs in OpenBio.eu
The OpenBio.eu platform is organized around four basic building-blocks (Figure 3): (a) the RO repository, which is equipped with free-text search operations to locate the TDW object most relevant to the task at hand, workflow execution reports, references, and (potential) query-answering discourse sessions (upper-left); (b) a module to define/describe a TDW (lower-left); (c) the platform’s core module where the installation (with validation) of TDW objects takes place, along with the definition of their dependencies (arrows between objects), as well as the composition of workflows (a GUI is provided to drag-and-drop the needed ROs); and (d) the module to define the execution environment and the actual execution of workflows (i.e., the Airflow execution manager that operates on CWL forms of workflows) accompanied by a resource manager (serviced by netdata) to monitor the execution process.
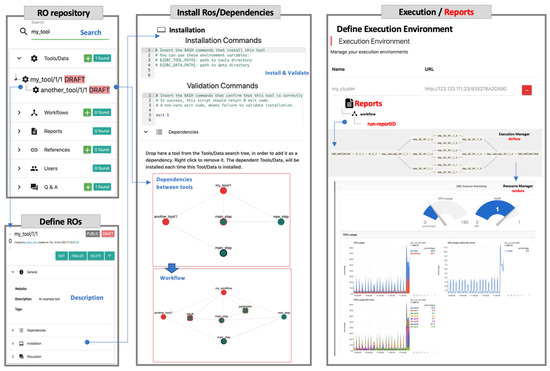
Figure 3.
The four basic building blocks of the OpenBio.eu platform.
OpenBio.eu users can create a TDW RO by simply importing the commands to install it. These commands are the same as those that install the tool on any computer that provides a BASH shell terminal. The same TDW object can have many versions. Different users can import TDWs that have the same name and version. Apart from the BASH commands, a TDW can be accompanied with rich-format text in markdown. In OpenBio.eu, users can define dependencies between TDWs with a simple drag-and-drop. TDW objects can be in the “Draft” or “Finalized” stage. Objects in the “Draft” stage may be edited in the future. Once users are confident that an imported object does not need any further refinements, they can choose to “Finalize” it. A “Finalized” object cannot be edited any further; it is now an immutable object that can take part in a reproducible, future-proof pipeline. In any case, users may “Fork” a “Draft” or “Finalized” version of an object. Forking is a concept borrowed from software development, where users can create an identical copy of a source code and edit it as they wish.
Moreover, a crucial factor that is largely absent from almost all WfMSs and BTA tools is social commenting and rating. Especially for students and newcomers in the field, it is difficult to discern the average sentiment toward an RO or whether there is any strong opinion about it (even from a small fraction of users). In addition, existing tools do not provide users with the means to express (and share) their own opinions. This affects not only the entry-level barrier in the domain but also prevents the elaboration of existing ROs and impedes the collective formulation of novel ideas that could improve research and highlight novel directions. OpenBio.eu fulfills this need by making every RO rate-able and comment-able; furthermore, it contains a Q&A collaborative module where any user can leave a question, request assistance, or make a generic remark. These functionalities are supported by a collaboration component, which supports the creation and elaboration of a semantically rich discourse graph about any RO appearing on the OpenBio.eu platform [57]. The nodes on this discourse graph may be of different types, including issues (a question that a user posts for discussion); solutions (a solution proposed by a user to solve an issue under consideration); positions in favor (an argument supporting a suggested solution); positions against (an argument aiming to defeat or challenge a solution); and notes (a comment usually added to bring in supplementary information without affecting the assessment of the discourse). The collaboration component also offers state-of-the-art search functionality, diverse options to augment the visualization of the graph, as well as the ability to rate the graph nodes (e.g., assignment of likes/dislikes). Overall, the collaboration component of the OpenBio.eu platform enables users to set up a highly interactive process, where they can easily decide which ROs should be considered, identify and discuss their strengths and weaknesses, and control the complexity of biomedical workflows.
4.2. Synthesizing Workflows in OpenBio.eu
Workflows are a central concept in computation biology and bioinformatics. Strictly speaking, a workflow is a series of computational steps, connected through dependencies; i.e., “a step gets to run only when these other steps are completed.” Common WfMSs in Bioinformatics, such as Galaxy [58], Nextflow [29], and Snakemake [59], exemplify this concept. Although this flow-centric construction of workflows has a long history in bioinformatics, it has been inherited from industrial design systems where rigidity is of paramount importance and is not perfectly suited to the versatility and flexibility of modern bioinformatics research. Moreover, the flow-centric construction of workflows requires above-average IT skills. In OpenBio.eu, users are able to construct workflows by simply importing the commands that execute a step. Again, these commands are the same as those that would be used in a BASH terminal. The main difference is that instead of implicitly defining step order through a dependency resolution algorithm, they can directly call steps from other steps. This abstraction follows the “function calls function” paradigm that is familiar to users with basic programming skills.
Workflows can be converted from “structure-centric” to “flow-centric” constructs with an algorithm that creates directed acyclic graphs (DAGs). These DAGs can be described in CWL and consequently imported to common workflow execution environments such as Galaxy, Nextflow, Snakemake, and Airflow. OpenBio.eu also provides a Docker container that is connected to the OpenBio.eu web server and acts as a personalized execution environment. Users can have as many Execution Environments as they wish. Every Workflow execution creates a separate object (report) that contains results and logs. These can be shared with other users.
A more detailed presentation and lengthy documentation of these mechanisms, along with examples, are available at the project’s documentation page: https://kantale.github.io/OpenBio.eu/docs/, accessed on 11 October 2021.
5. Discussion
Although BTA is a crucial component in modern science, we argue that many widely known ROs that comprise BTA workflows are far from being FAIR; namely, ontologies, tools, and corpora have serious shortcomings that prevent them from taking place in reproducible analyses. These shortcomings are associated with a lack of ease in downloading, installing, configuring, and using them. It is obvious that the degree to which one can perform these tasks significantly affects the ability of these ROs to take part in arbitrary analysis workflows. The utilization of untested ontologies, the use of computer languages from external (to the one used to build the workflow) execution environments, and scaling up in modern high-performance computational platforms are some of the challenges to be addressed [2].
This paper presents an effort to “FAIRify” some of the most well-known ROs in BTA through the use of the OpenBio.eu platform. This effort follows the principle that ROs should be rendered directly downloadable, installable, and re-usable. Moreover, tools, web services, corpora, and ontologies should all lie in a common repository, properly categorized and indexed. Researchers should be able to reuse these components by simply “plugging” them into an analysis workspace and should also be able to create personal copies (forks) of them to apply any change that they like. Finally, ROs should be rate-able and comment-able. Moreover, users should be able to perform these activities with simple web actions (such as drag-and-drop) without having to use any DSL. In Table 1, we present how the architecture of OpenBio.eu addresses and implements all official FAIR guidelines [21]. Of course, this is a subjective evaluation of the platform’s FAIR-ness status. Actual FAIR metrics can be achieved either by conducting user surveys or by using automatic FAIR validation mechanisms [60]. We intend to also provide these additional validations in a future study.
Perhaps the most significant challenge for open source and social platforms of this kind is achieving long-term survivability. Platforms that have achieved this (i.e., Galaxy and Nextflow) owe it to several qualitative characteristics such as a clear data model, solid implementation, and the use of modern underlying IT technologies. Here, we showcased how OpenBio.eu fulfills these characteristics. Moreover, these criteria are not adequate to guarantee widespread adoption unless the platform sufficiently tackles a “real and existing problem.” Here, we demonstrated that the current un-FAIR status of many ROs in BTA is a real problem that needs to be tackled. Finally, the permissive open-source license is another factor that can contribute to the long-term survivability of the project. Overall, there are more than 100 projects with similar functionality and scope (https://github.com/pditommaso/awesome-pipeline, accessed on 11 October 2021). We hope that these characteristics will help OpenBio.eu to stand out.
For the purpose of this study, we have added a variety of ROs for the task of BTA. These ROs are far from perfect and may need refinement. Moreover, crucial and widely used corpora, ontologies, and tools are missing. Our future work will include not only the expansion of this collection but also the creation of workflows that efficiently compare them. Toward this end, OpenBio.eu can be used to create meta-workflows that install, configure, and measure the efficiency of other workflows. Another future work is to compare the ability of modern high-performance environments to sufficiently scale these ROs, so as to process the entire corpus of open biomedical documents and measure the effect of managing this information overload. Of course, the most challenging future work is to persuade the open-source and bioinformatics community to contribute to this project, either by importing new ROs or by improving the platform. OpenBio.eu is an environment that, by design, welcomes users to import new ROs and perform edits to existing ROs, thus guiding biomedical text-mining communities to gradually improve these components toward achieving maximum FAIR status.
Author Contributions
Conceptualization, A.K., N.K. (Nikos Kanakaris), L.K., M.K., K.P., N.K. (Nikos Karacapilidis), and G.P.; software, A.K., M.K., K.P. and N.K. (Nikos Kanakaris); formal analysis, A.K. and L.K.; investigation, A.K. and M.K.; data curation, A.K., M.K., K.P. and N.K. (Nikos Kanakaris); writing—original draft preparation, A.K. and N.K. (Nikos Kanakaris); writing—review and editing, A.K. and N.K. (Nikos Kanakaris); supervision, G.P. and N.K. (Nikos Karacapilidis); project administration, G.P. All authors have read and agreed to the published version of the manuscript.
Funding
The work presented in this paper is supported by the OpenBio-C project (www.openbio.eu, accessed on 11 October 2021) which is co-financed by the European Union and Greek national funds through the Operational Program Competitiveness, Entrepreneurship and Innovation, under the call RESEARCH—CREATE—INNOVATE (project id: T1EDK-05275).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data and source code are available at https://github.com/kantale/OpenBio.eu (accessed on 11 October 2021).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bornmann, L.; Mutz, R. Growth rates of modern science: A bibliometric analysis based on the number of publications and cited references. J. Assoc. Inf. Sci. Technol. 2015, 66, 2215–2222. [Google Scholar] [CrossRef]
- Huang, C.-C.; Lu, Z. Community challenges in biomedical text mining over 10 years: Success, failure and the future. Brief. Bioinform. 2016, 17, 132–144. [Google Scholar] [CrossRef]
- Munafò, M.R.; Nosek, B.A.; Bishop, D.V.M.; Button, K.S.; Chambers, C.D.; Percie du Sert, N.; Simonsohn, U.; Wagenmakers, E.-J.; Ware, J.J.; Ioannidis, J.P.A. A manifesto for reproducible science. Nat. Hum. Behav. 2017, 1, 21. [Google Scholar] [CrossRef]
- Freedman, L.P.; Cockburn, I.M.; Simcoe, T.S. The economics of reproducibility in preclinical research. PLOS Biol. 2015, 13, 1–9. [Google Scholar] [CrossRef]
- Luque, C.; Luna, J.M.; Luque, M.; Ventura, S. An advanced review on text mining in medicine. WIREs Data Min. Knowl. Discov. 2019, 9, e1302. [Google Scholar] [CrossRef]
- Jovanović, J.; Bagheri, E. Semantic annotation in biomedicine: The current landscape. J. Biomed. Semantics 2017, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Neves, M.; Leser, U. A survey on annotation tools for the biomedical literature. Brief. Bioinform. 2014, 15, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.G.; Howsmon, D.; Zhang, B.; Hahn, J.; McGuinness, D.; Hendler, J.; Ji, H. Entity linking for biomedical literature. BMC Med. Inform. Decis. Mak. 2015, 15 (Suppl 1), S4. [Google Scholar] [CrossRef] [PubMed]
- Comeau, D.C.; Islamaj Doğan, R.; Ciccarese, P.; Cohen, K.B.; Krallinger, M.; Leitner, F.; Lu, Z.; Peng, Y.; Rinaldi, F.; Torii, M.; et al. BioC: A minimalist approach to interoperability for biomedical text processing. Database 2013, 2013, bat064. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, J.M.; Bader, G.D. Transfer learning for biomedical named entity recognition with neural networks. Bioinformatics 2018, 34, 4087–4094. [Google Scholar] [CrossRef] [PubMed]
- Tomori, S.; Ninomiya, T.; Mori, S. Domain Specific Named Entity Recognition Referring to the Real World by Deep Neural Networks. In Proceedings of the 54th Annual Meeting of the Association for Computational Linguistics (Volume 2: Short Papers), Berlin, Germany, 7–12 August 2016; Association for Computational Linguistics: Berlin, Germany, 2016; pp. 236–242. [Google Scholar]
- Maldonado, R.; Goodwin, T.R.; Skinner, M.A.; Harabagiu, S.M. Deep Learning Meets Biomedical Ontologies: Knowledge Embeddings for Epilepsy. AMIA Annu. Symp. Proc. AMIA Symp. 2018, 2017, 1233–1242. [Google Scholar] [PubMed]
- Sousa, D.; Couto, F.M. BiOnt: Deep Learning Using Multiple Biomedical Ontologies for Relation Extraction. Adv. Inf. Retr. 2020, 367–374. [Google Scholar]
- Tseytlin, E.; Mitchell, K.; Legowski, E.; Corrigan, J.; Chavan, G.; Jacobson, R.S. NOBLE—Flexible concept recognition for large-scale biomedical natural language processing. BMC Bioinform. 2016, 17, 32. [Google Scholar] [CrossRef]
- Almeida, J.S.; Hajagos, J.; Saltz, J.; Saltz, M. Serverless OpenHealth at data commons scale-traversing the 20 million patient records of New York’s SPARCS dataset in real-time. PeerJ 2019, 7, e6230. [Google Scholar] [CrossRef] [PubMed]
- Shafiei, H.; Khonsari, A.; Mousavi, P. Serverless Computing: A Survey of Opportunities, Challenges and Applications. arXiv Prepr. 2019, arXiv:1911.01296. [Google Scholar]
- Funk, C.; Baumgartner, W.; Garcia, B.; Roeder, C.; Bada, M.; Cohen, K.B.; Hunter, L.E.; Verspoor, K. Large-scale biomedical concept recognition: An evaluation of current automatic annotators and their parameters. BMC Bioinform. 2014, 15, 59. [Google Scholar] [CrossRef] [PubMed]
- Cuzzola, J.; Jovanović, J.; Bagheri, E.; Gašević, D. Evolutionary fine-tuning of automated semantic annotation systems. Expert Syst. Appl. 2015, 42, 6864–6877. [Google Scholar] [CrossRef]
- Svensson, D.; Sjögren, R.; Sundell, D.; Sjödin, A.; Trygg, J. doepipeline: A systematic approach to optimizing multi-level and multi-step data processing workflows. BMC Bioinform. 2019, 20, 498. [Google Scholar] [CrossRef]
- Jacobsen, A.; Kaliyaperumal, R.; da Silva Santos, L.O.B.; Mons, B.; Schultes, E.; Roos, M.; Thompson, M. A Generic Workflow for the Data FAIRification Process. Data Intell. 2020, 2, 56–65. [Google Scholar] [CrossRef]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.-W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef]
- Kanterakis, A.; Iatraki, G.; Pityanou, K.; Koumakis, L.; Kanakaris, N.; Karacapilidis, N.; Potamias, G. Towards Reproducible Bioinformatics: The OpenBio-C Scientific Workflow Environment. In Proceedings of the 2019 IEEE 19th International Conference on Bioinformatics and Bioengineering (BIBE), Athens, Greece, 28–30 October 2019; pp. 221–226. [Google Scholar]
- Wilkinson, M.D.; Verborgh, R.; da Silva Santos, L.O.B.; van Mulligen, E.M.; Bolleman, J.T.; Dumontier, M. Interoperability and FAIRness through a novel combination of Web technologies. PeerJ Comput. Sci. 2017, 3, e110. [Google Scholar] [CrossRef]
- Henninger, S. Using Iterative Refinement to Find Reusable Software. IEEE Softw. 1994, 11, 48–59. [Google Scholar] [CrossRef]
- Shade, A.; Teal, T.K. Computing Workflows for Biologists: A Roadmap. PLoS Biol. 2015, 13, e1002303. [Google Scholar] [CrossRef]
- Jackson, M.J.; Wallace, E.; Kavoussanakis, K. Using rapid prototyping to choose a bioinformatics workflow management system. bioRxiv 2020. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; van den Beek, M.; Blankenberg, D.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Eberhard, C.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016, 44, W3–W10. [Google Scholar] [CrossRef] [PubMed]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef]
- Wang, L.L.; Lo, K.; Chandrasekhar, Y.; Reas, R.; Yang, J.; Eide, D.; Funk, K.; Kinney, R.; Liu, Z.; Merrill, W.; et al. CORD-19: The COVID-19 open research dataset. arXiv 2020, arXiv:2004.10706v2. [Google Scholar]
- Van Auken, K.; Schaeffer, M.L.; McQuilton, P.; Laulederkind, S.J.F.; Li, D.; Wang, S.-J.; Hayman, G.T.; Tweedie, S.; Arighi, C.N.; Done, J.; et al. BC4GO: A full-text corpus for the BioCreative IV GO task. Database 2014, 2014, bau074. [Google Scholar] [CrossRef]
- Kors, J.A.; Clematide, S.; Akhondi, S.A.; van Mulligen, E.M.; Rebholz-Schuhmann, D. A multilingual gold-standard corpus for biomedical concept recognition: The Mantra GSC. J. Am. Med. Inform. Assoc. 2015, 22, 948–956. [Google Scholar] [CrossRef]
- Kim, J.-D.; Ohta, T.; Tateisi, Y.; Tsujii, J. GENIA corpus—A semantically annotated corpus for bio-textmining. Bioinformatics 2003, 19, i180–i182. [Google Scholar] [CrossRef]
- Doğan, R.I.; Leaman, R.; Lu, Z. NCBI disease corpus: A resource for disease name recognition and concept normalization. J. Biomed. Inform. 2014, 47, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.; Ashburner, M.; Rosse, C.; Bard, J.; Bug, W.; Ceusters, W.; Goldberg, L.J.; Eilbeck, K.; Ireland, A.; Mungall, C.J.; et al. The OBO Foundry: Coordinated evolution of ontologies to support biomedical data integration. Nat. Biotechnol. 2007, 25, 1251–1255. [Google Scholar] [CrossRef] [PubMed]
- Rector, A.L.; JE Rogers, J.E.; Pole, P. The GALEN High Level Ontology. Stud. Health Technol. Inform. 1996, 34, 174–176. [Google Scholar]
- Rector, A.L.; Rogers, J.E.; Zanstra, P.E.; Van Der Haring, E. OpenGALEN: Open source medical terminology and tools. AMIA Symp. 2003, 2003, 982. [Google Scholar]
- McCray, A.T.; Burgun, A.; Bodenreider, O. Aggregating UMLS semantic types for reducing conceptual complexity. Stud. Health Technol. Inform. 2001, 84, 216–220. [Google Scholar]
- Bodenreider, O.; McCray, A.T. Exploring semantic groups through visual approaches. J. Biomed. Inform. 2003, 36, 414–432. [Google Scholar] [CrossRef][Green Version]
- Lipscomb, C.E. Medical Subject Headings (MeSH). Bull. Med. Libr. Assoc. 2000, 88, 265–266. [Google Scholar]
- Rossanez, A.; dos Reis, J.C.; Torres, R.d.S.; de Ribaupierre, H. KGen: A knowledge graph generator from biomedical scientific literature. BMC Med. Inform. Decis. Mak. 2020, 20, 314. [Google Scholar] [CrossRef]
- Savova, G.K.; Masanz, J.J.; Ogren, P.V.; Zheng, J.; Sohn, S.; Kipper-Schuler, K.C.; Chute, C.G. Mayo clinical Text Analysis and Knowledge Extraction System (cTAKES): Architecture, component evaluation and applications. J. Am. Med. Inform. Assoc. 2010, 17, 507–513. [Google Scholar] [CrossRef]
- Aronson, A.R.; Lang, F.-M. An overview of MetaMap: Historical perspective and recent advances. J. Am. Med. Inform. Assoc. 2010, 17, 229–236. [Google Scholar] [CrossRef]
- Demner-Fushman, D.; Rogers, W.J.; Aronson, A.R. MetaMap Lite: An evaluation of a new Java implementation of MetaMap. J. Am. Med. Inform. Assoc. 2017, 24, 841–844. [Google Scholar] [CrossRef]
- Jonquet, C.; Toulet, A.; Arnaud, E.; Aubin, S.; Dzalé Yeumo, E.; Emonet, V.; Graybeal, J.; Laporte, M.-A.; Musen, M.A.; Pesce, V.; et al. AgroPortal: A vocabulary and ontology repository for agronomy. Comput. Electron. Agric. 2018, 144, 126–143. [Google Scholar] [CrossRef]
- Tchechmedjiev, A.; Abdaoui, A.; Emonet, V.; Zevio, S.; Jonquet, C. SIFR annotator: Ontology-based semantic annotation of French biomedical text and clinical notes. BMC Bioinform. 2018, 19, 405. [Google Scholar] [CrossRef]
- Shah, N.H.; Bhatia, N.; Jonquet, C.; Rubin, D.; Chiang, A.P.; Musen, M.A. Comparison of concept recognizers for building the Open Biomedical Annotator. BMC Bioinform. 2009, 10 (Suppl. 9), S14. [Google Scholar] [CrossRef]
- Harris, S.; Lamb, N.; Shadbolt, N. 4store: The design and implementation of a clustered RDF store. In Proceedings of the 5th International Workshop on Scalable Semantic Web Knowledge Base Systems (SSWS2009), Washington, DC, USA, 26 October 2009; pp. 94–109. [Google Scholar]
- Nunes, T.; Campos, D.; Matos, S.; Oliveira, J.L. BeCAS: Biomedical concept recognition services and visualization. Bioinformatics 2013, 29, 1915–1916. [Google Scholar] [CrossRef]
- Campos, D.; Matos, S.; Oliveira, J.L. A modular framework for biomedical concept recognition. BMC Bioinform. 2013, 14, 281. [Google Scholar] [CrossRef]
- Rebholz-Schuhmann, D.; Arregui, M.; Gaudan, S.; Kirsch, H.; Jimeno, A. Text processing through Web services: Calling Whatizit. Bioinformatics 2008, 24, 296–298. [Google Scholar] [CrossRef] [PubMed]
- Salgado, D.; Krallinger, M.; Depaule, M.; Drula, E.; Tendulkar, A.V.; Leitner, F.; Valencia, A.; Marcelle, C. MyMiner: A web application for computer-assisted biocuration and text annotation. Bioinformatics 2012, 28, 2285–2287. [Google Scholar] [CrossRef] [PubMed]
- Ison, J.; Kalaš, M.; Jonassen, I.; Bolser, D.; Uludag, M.; McWilliam, H.; Malone, J.; Lopez, R.; Pettifer, S.; Rice, P. EDAM: An ontology of bioinformatics operations, types of data and identifiers, topics and formats. Bioinformatics 2013, 29, 1325–1332. [Google Scholar] [CrossRef] [PubMed]
- Kyriakakis, A.; Koumakis, L.; Kanterakis, A.; Iatraki, G.; Tsiknakis, M.; Potamias, G. Enabling Ontology-Based Search: A Case Study in the Bioinformatics Domain. In Proceedings of the 2019 IEEE 19th International Conference on Bioinformatics and Bioengineering (BIBE), Athens, Greece, 28–30 October 2019; pp. 227–234. [Google Scholar]
- Ison, J.; Ienasescu, H.; Chmura, P.; Rydza, E.; Ménager, H.; Kalaš, M.; Schwämmle, V.; Grüning, B.; Beard, N.; Lopez, R.; et al. The bio.tools registry of software tools and data resources for the life sciences. Genome Biol. 2019, 20, 164. [Google Scholar] [CrossRef]
- Foster, E.D.; Deardorff, A. Open Science Framework (OSF). J. Med. Libr. Assoc. 2017, 105, 203–206. [Google Scholar]
- Kanterakis, A.; Karacapilidis, N.; Koumakis, L.; Potamias, G. On the development of an open and collaborative bioinformatics research environment. Procedia Comput. Sci. 2018, 126, 1062–1071. [Google Scholar] [CrossRef]
- Giardine, B.; Riemer, C.; Hardison, R.C.; Burhans, R.; Elnitski, L.; Shah, P.; Zhang, Y.; Blankenberg, D.; Albert, I.; Taylor, J.; et al. Galaxy: A platform for interactive large-scale genome analysis. Genome Res. 2005, 15, 1451–1455. [Google Scholar] [CrossRef] [PubMed]
- Köster, J.; Rahmann, S. Snakemake—a scalable bioinformatics workflow engine. Bioinformatics 2012, 28, 2520–2522. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, M.D.; Dumontier, M.; Sansone, S.-A.; Bonino da Silva Santos, L.O.; Prieto, M.; Batista, D.; McQuilton, P.; Kuhn, T.; Rocca-Serra, P.; Crosas, M.; et al. Evaluating FAIR maturity through a scalable, automated, community-governed framework. Sci. Data 2019, 6, 174. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).