Since they were discovered in 1890, X-rays and the techniques using X-ray diffraction [
1,
2] have shown great progress and have seen a variety of applications in industry because of their advantages over other nondestructive techniques. They can evaluate many mechanical properties including stress [
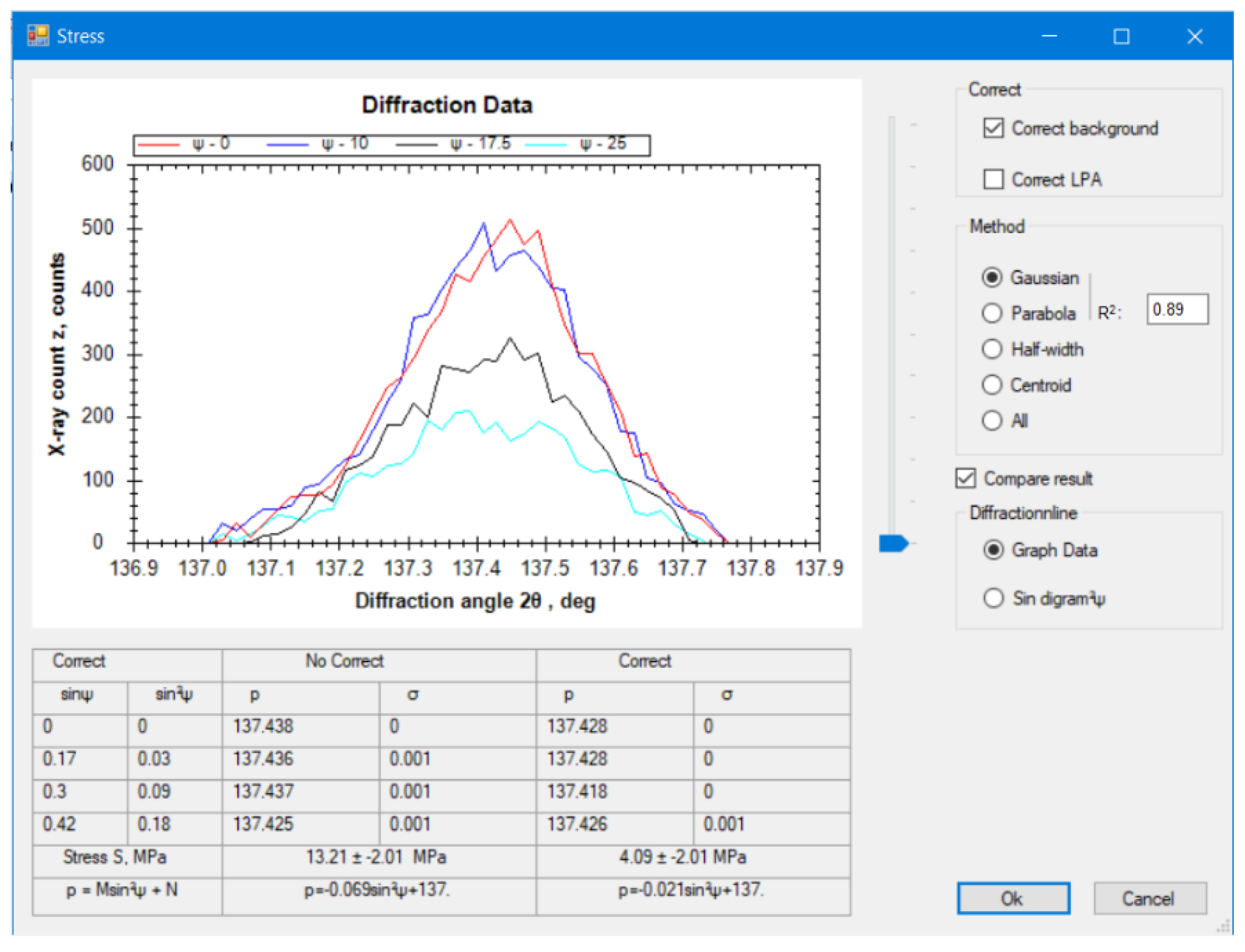
2,
3,
4], crystalline grain size [
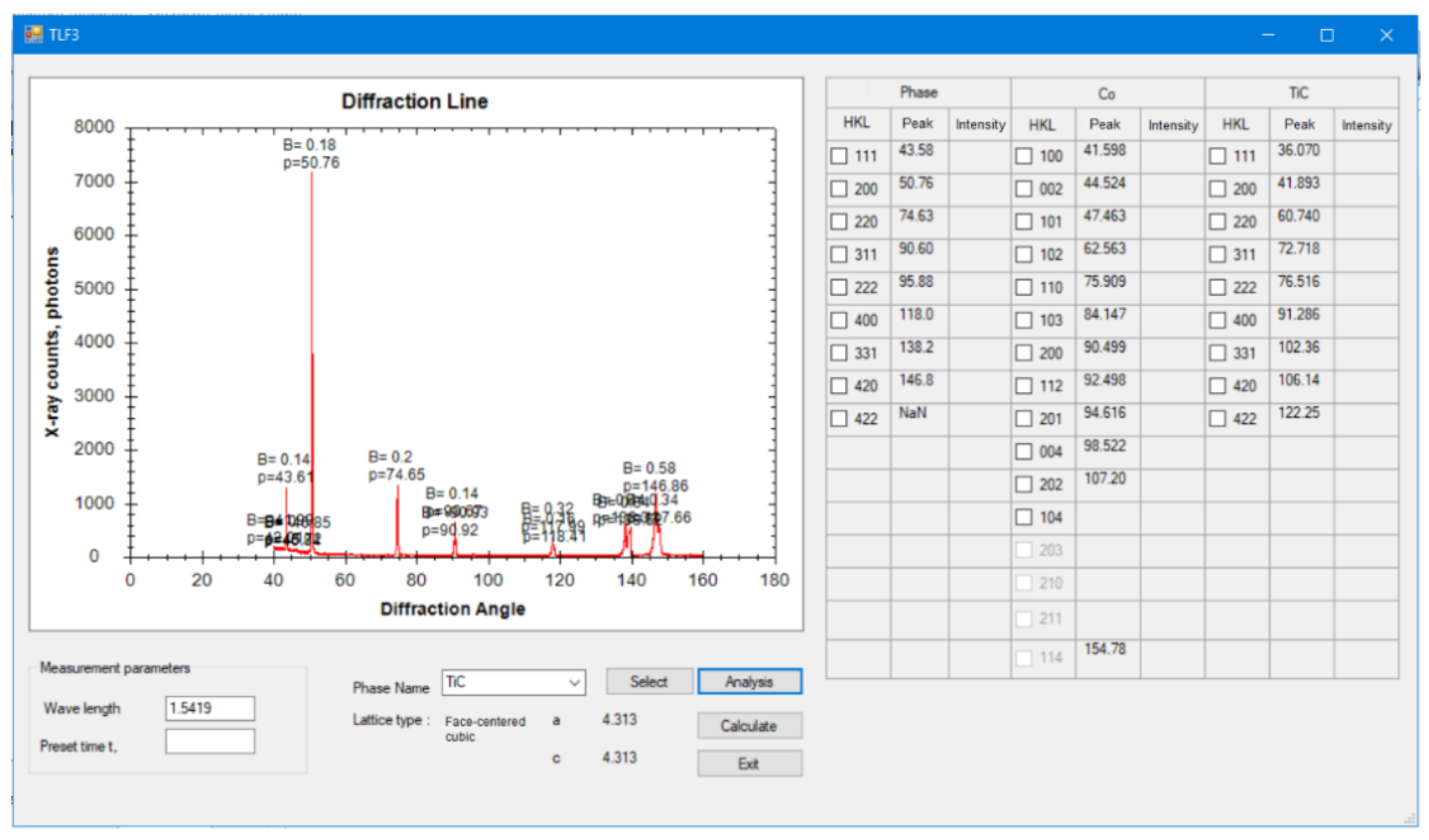
5], phase composition analysis [
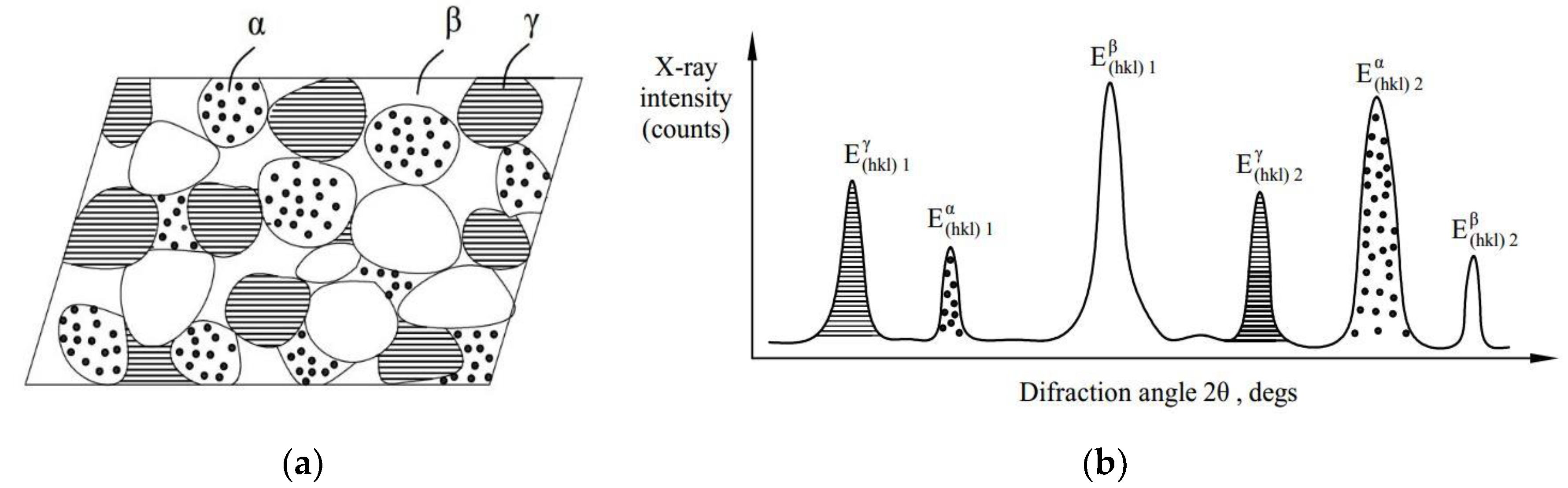
6,
7], hardness [
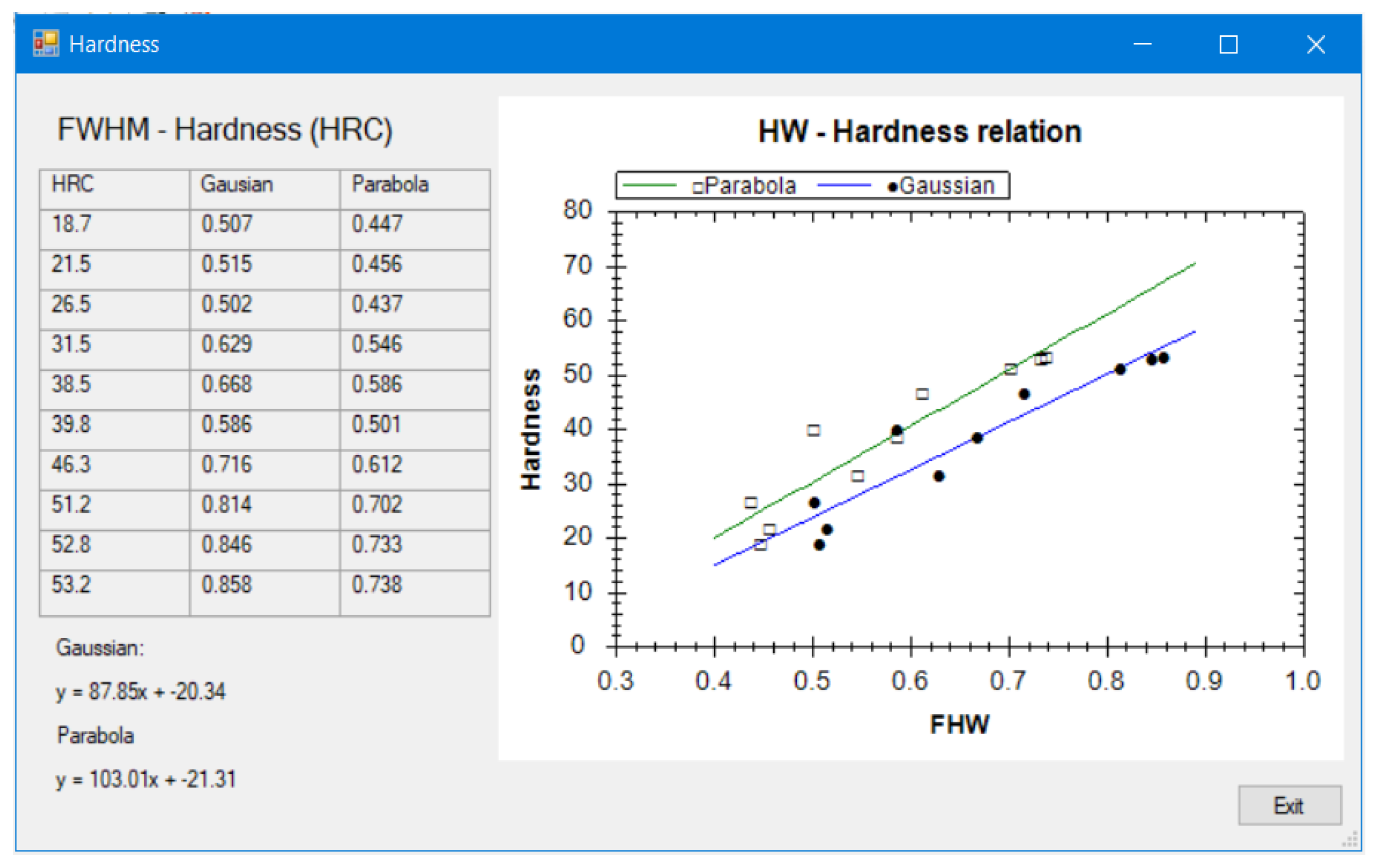
8], the thickness of plating or coating layer [
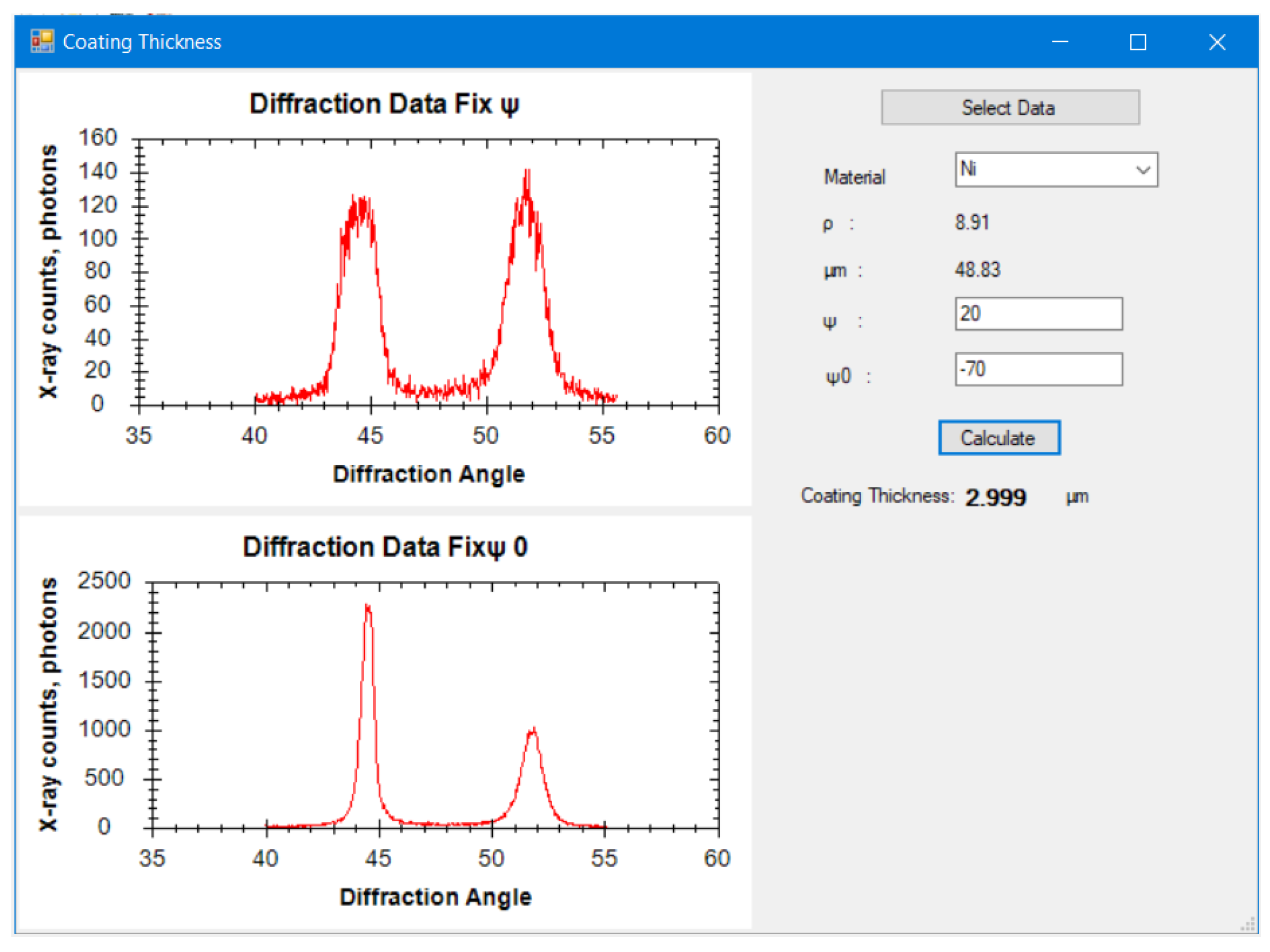
9,
10], etc. Moreover, X-ray diffraction has also been used to identify modifications of phase, texture, dislocation density, and mechanical twins of materials [
11]. Ultrasonic and magnetic techniques have many applications for many kinds of materials at the macro level to determine material inhomogeneity; however, they cannot determine the properties at the micro- and nano-level. The laser technique can only determine the outer profile of the sample surface such as roughness. The microscopic technique is used to observe crystalline microstructure, and when combined with image processing, it can determine crystal grain size, phase components in multi-phase materials, and layer thickness [
12]; however, it has difficulty in determining the state of materials such as stress, hardness, etc. Fortunately, X-rays with short wavelengths can detect the change or characteristics of a crystalline matrix from macro- to micro- and even nano-level, and it can therefore determine the hereabove parameters of crystalline materials. Moreover, a distinguished advantage of the XRD method over the other methods is that it can determine the standard deviations from a single measurement without replication of measurement. This is because the diffraction of photons in the crystal matrix occurs in all directions, and thus functions calculated from X-ray counting, such as peak position, stress, line width, and others, are random functions and contain coherent reproducibility of measured values [
13,
14].
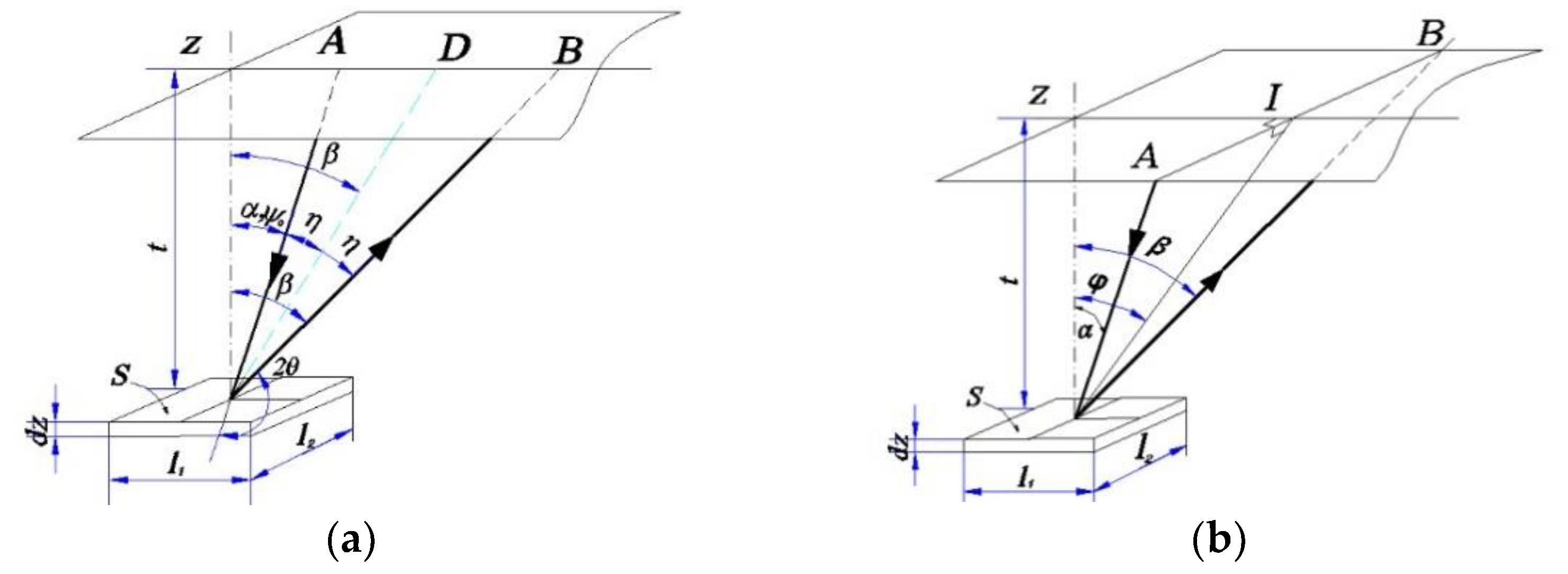
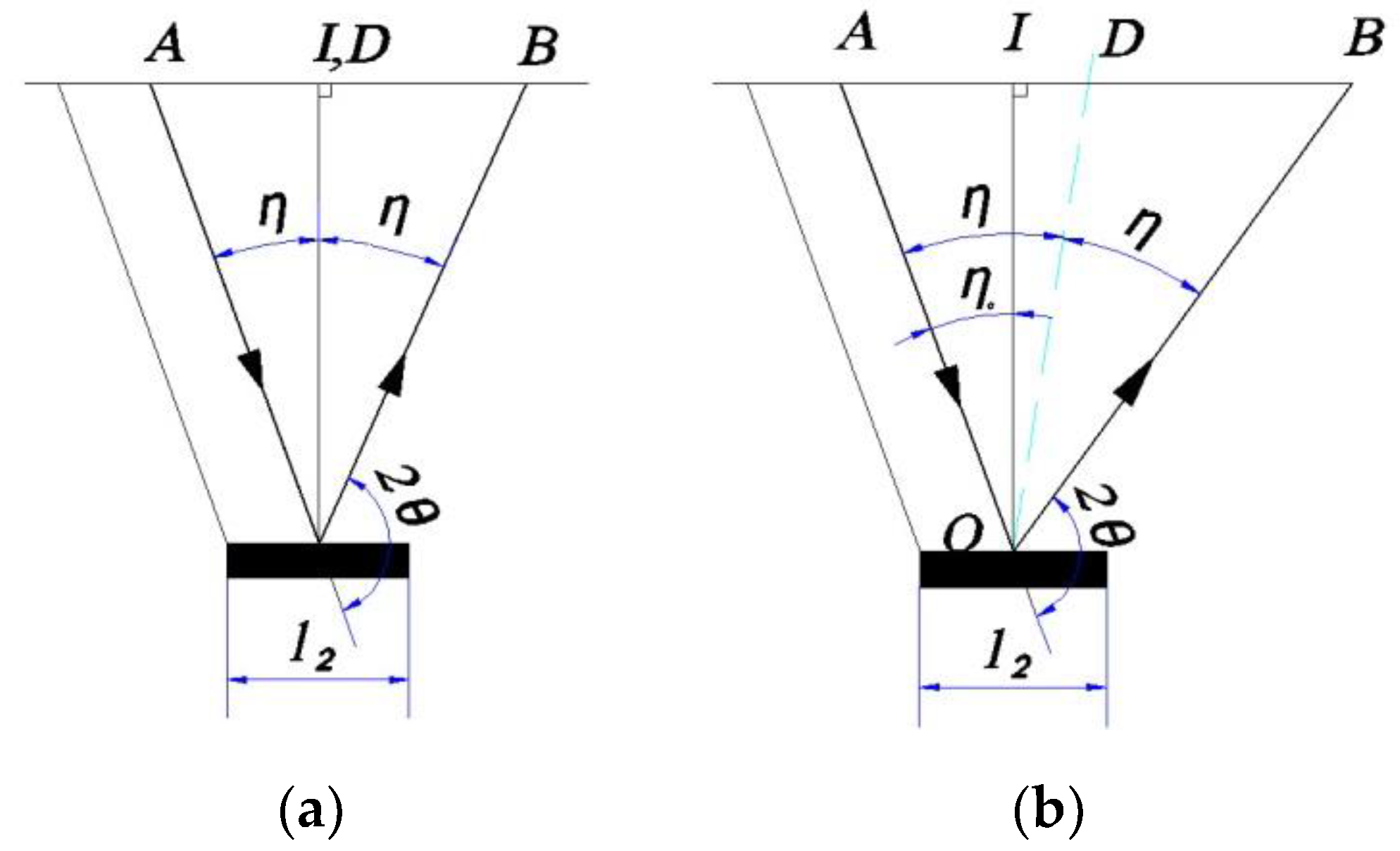
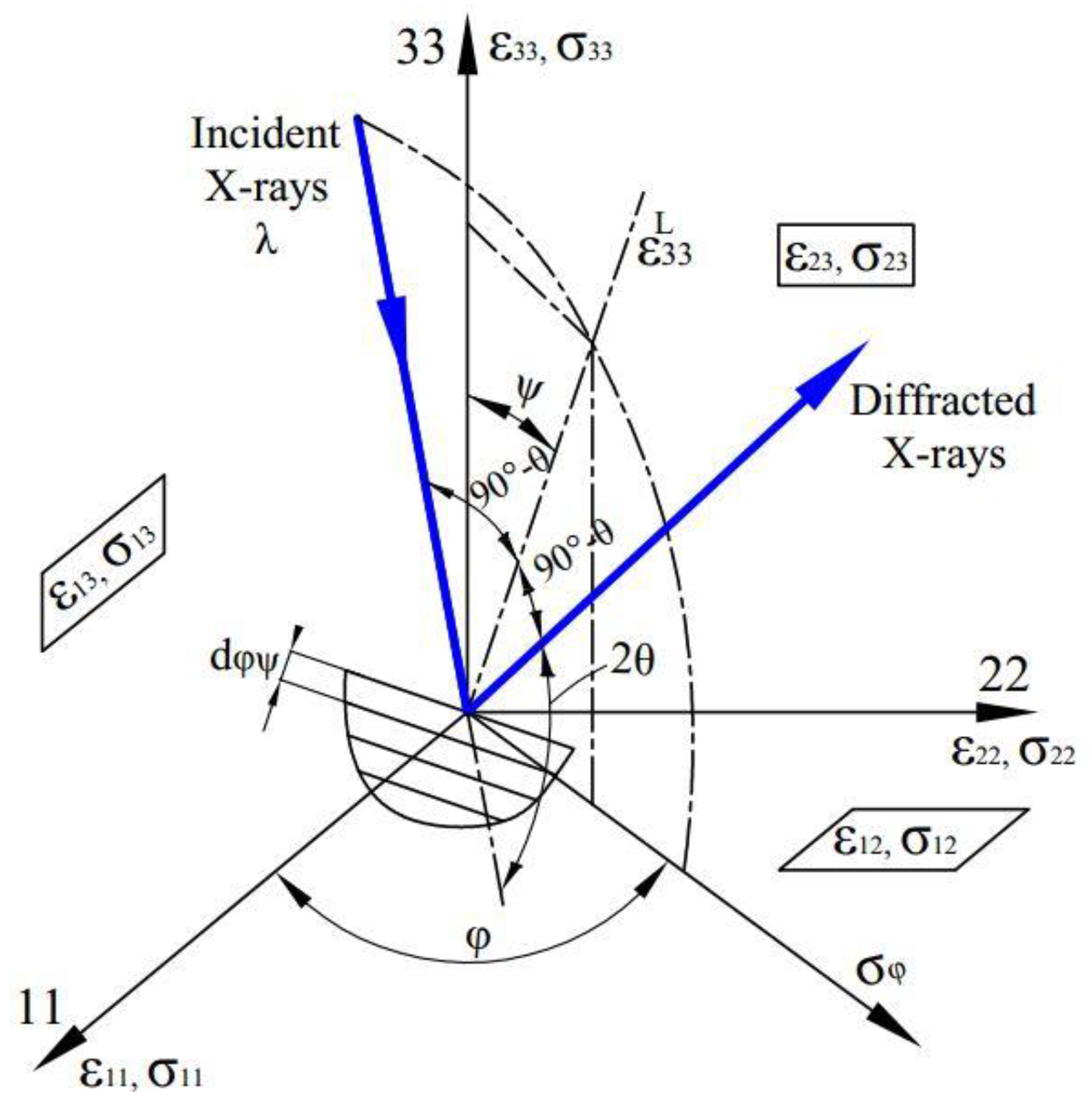
On the other hand, in order to obtain the correct value of X-ray intensity, and thus the accurate peak position and the full width at half of the diffraction line, the diffracted X-ray intensity must be corrected for the Lorentz-polarization and absorption factor [
2,
15,
16]. The absorption factor for only the iso-inclination method (Ω-type goniometer) using the fixed-ψ method was computed and widely used [
16]. However, the position-sensitive proportional counter (PSPC), in which the incident beams are fixed during the scanning process (fixed-ψ
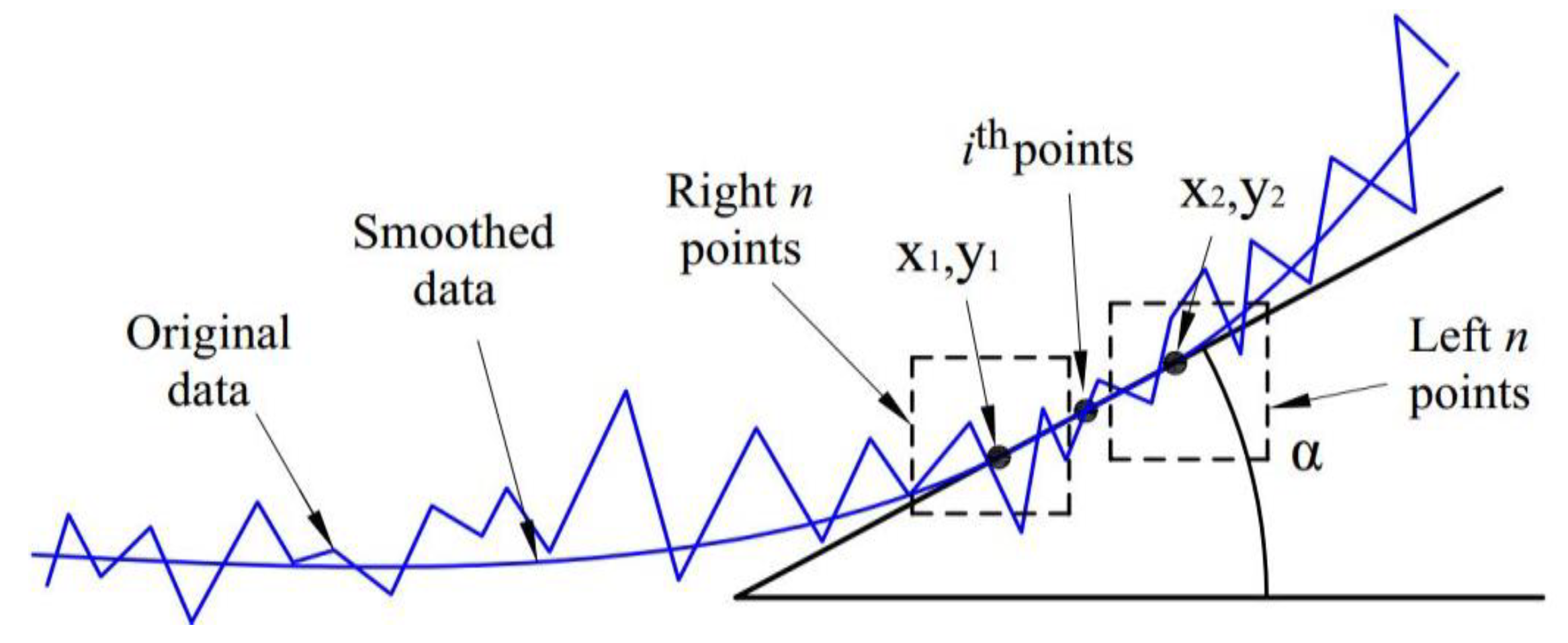
0 method), is used more widely. Moreover, to increase the diffraction angle 2θ up to approximately 170° and the inclination angle ψ up to approximately 165°, the side-inclination method (Ψ-type goniometer) is used. For these measurement methods, the absorption factor is still not fully derived and is not embedded in commercial X-ray systems. Furthermore, there are many methods of approximating the diffraction line to determine the diffraction peak. The choice of mathematical function to interpolate the peak diffraction greatly affected the accuracy of the measurement results. This has been reported in stress measurements using X-ray diffraction (XRD) [
17,
18]. To enlarge the scope of application of XRD for various measurements in a single device, there should be a computation system that can accurately determine many material properties. In a previous study, automated controlling and calculating software using C++ and commercial MS Excel was used for determining the residual stress of polycrystalline materials [
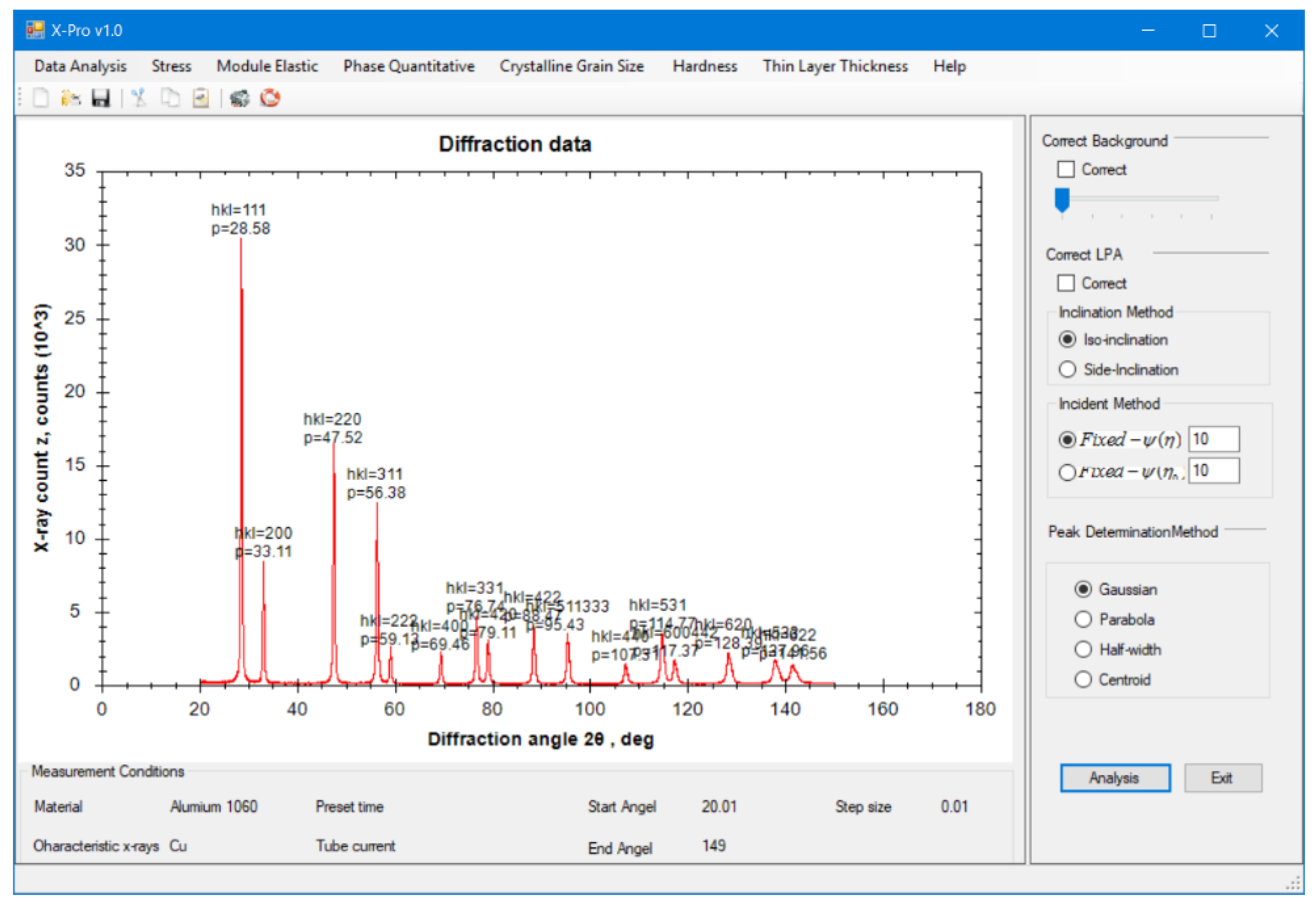
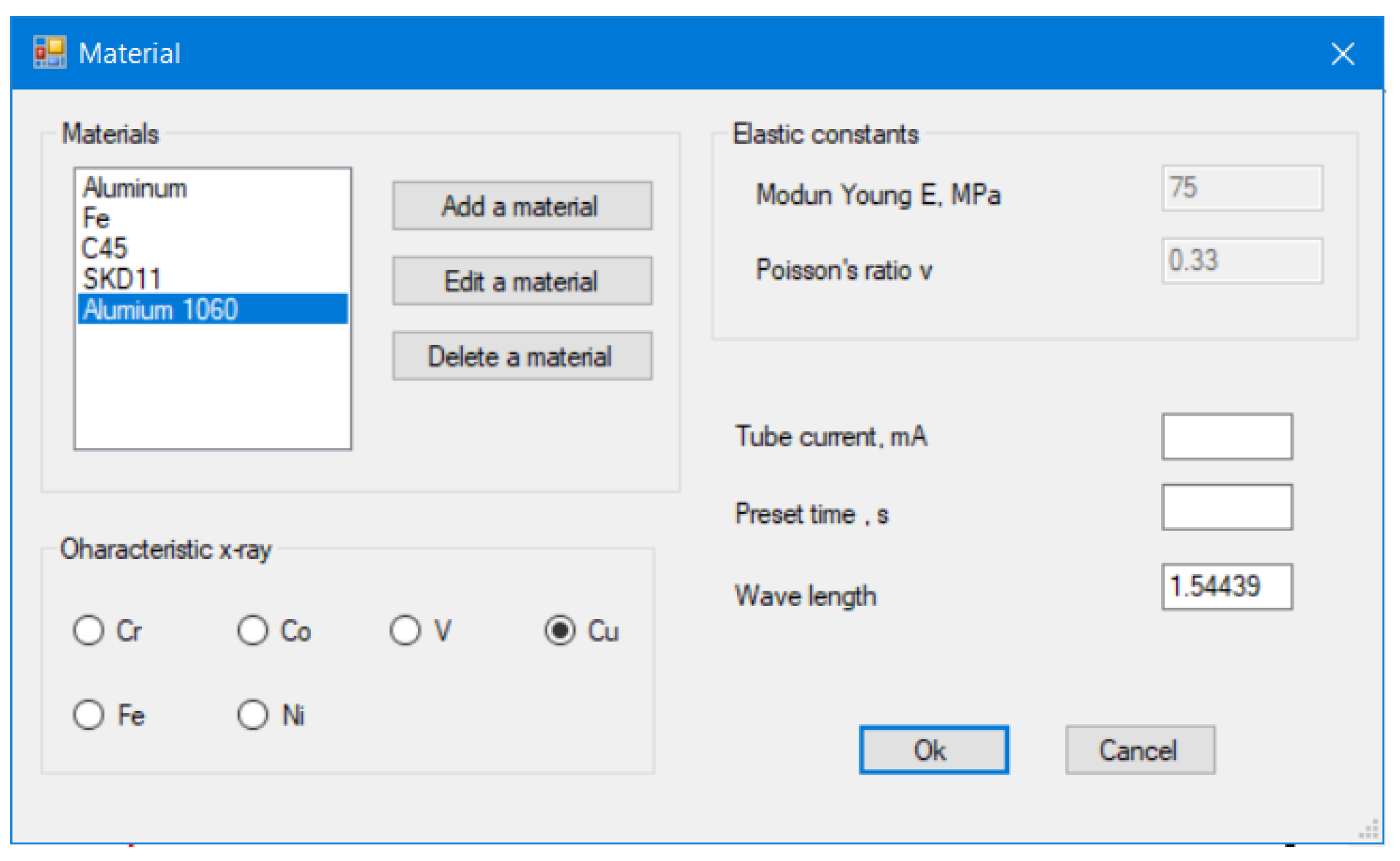
13]. This research represents a new computation program for polycrystalline materials that determines their properties using XRD, with total integration of absorption factors and mathematical function for the correction of diffraction lines. It can evaluate common mechanical properties in industry. The following measurements are carried out:
The demonstration measurements using X-rays and conventional methods were also compared to verify the validity of the computation and thus the applicability to the manufacturing site. All XRD measurements in this study use the X-ray characteristic CuKα with a wavelength λ of 1.5 Å and Ni foil filter. The use of a graphical user interface (GUI) also allows beginner-level programmers to easily reprogram resource codes for their individual computation.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}