
An Alternative Conceptual Model for the Spent Nuclear Fuel–Water Interaction in Deep Geologic Disposal Conditions


Abstract
:Featured Application
Abstract
1. Introduction
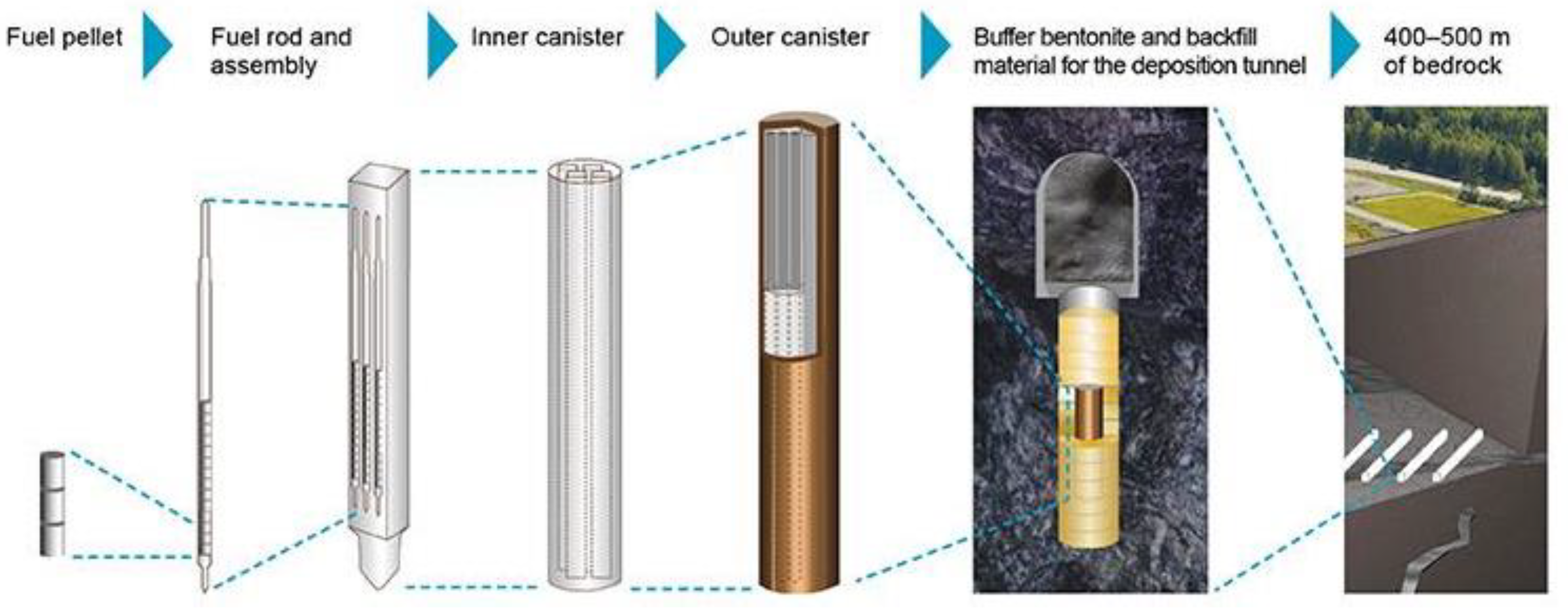
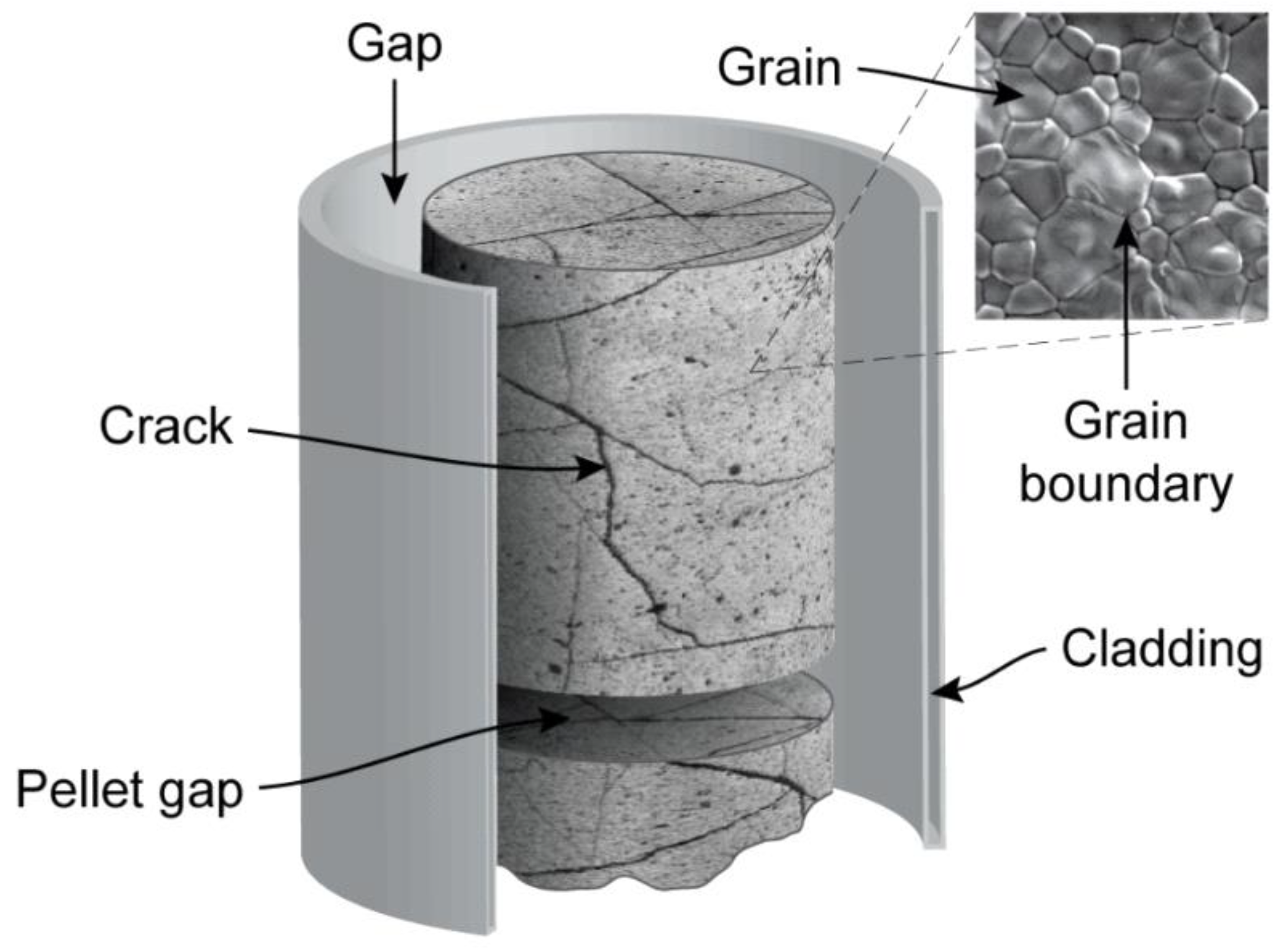
2. Spent Nuclear Fuel in Disposal Conditions
3. Observations from Leaching Tests in Reducing Conditions
4. Current Conceptual Models to Explain the Results in Reducing Conditions
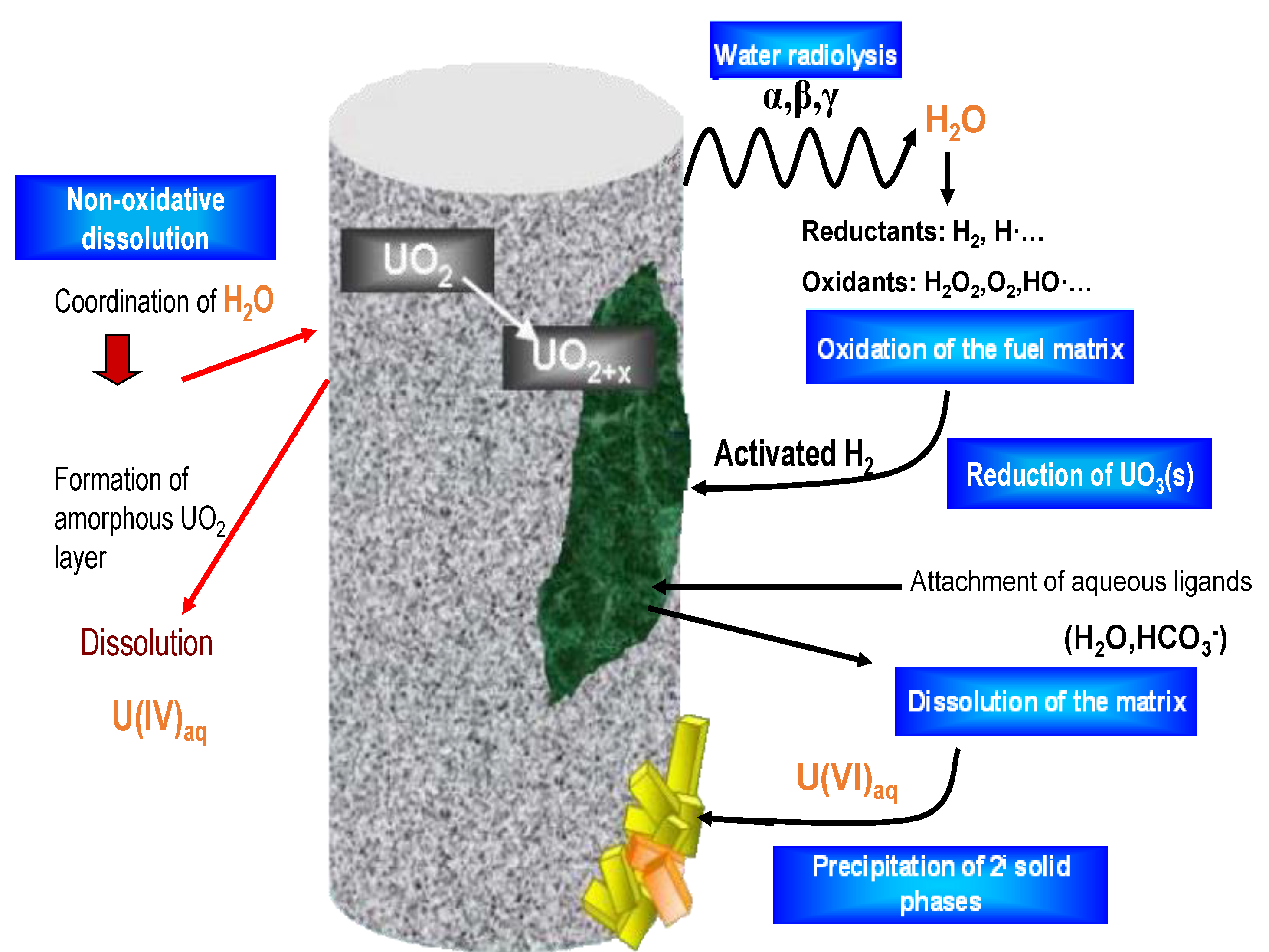
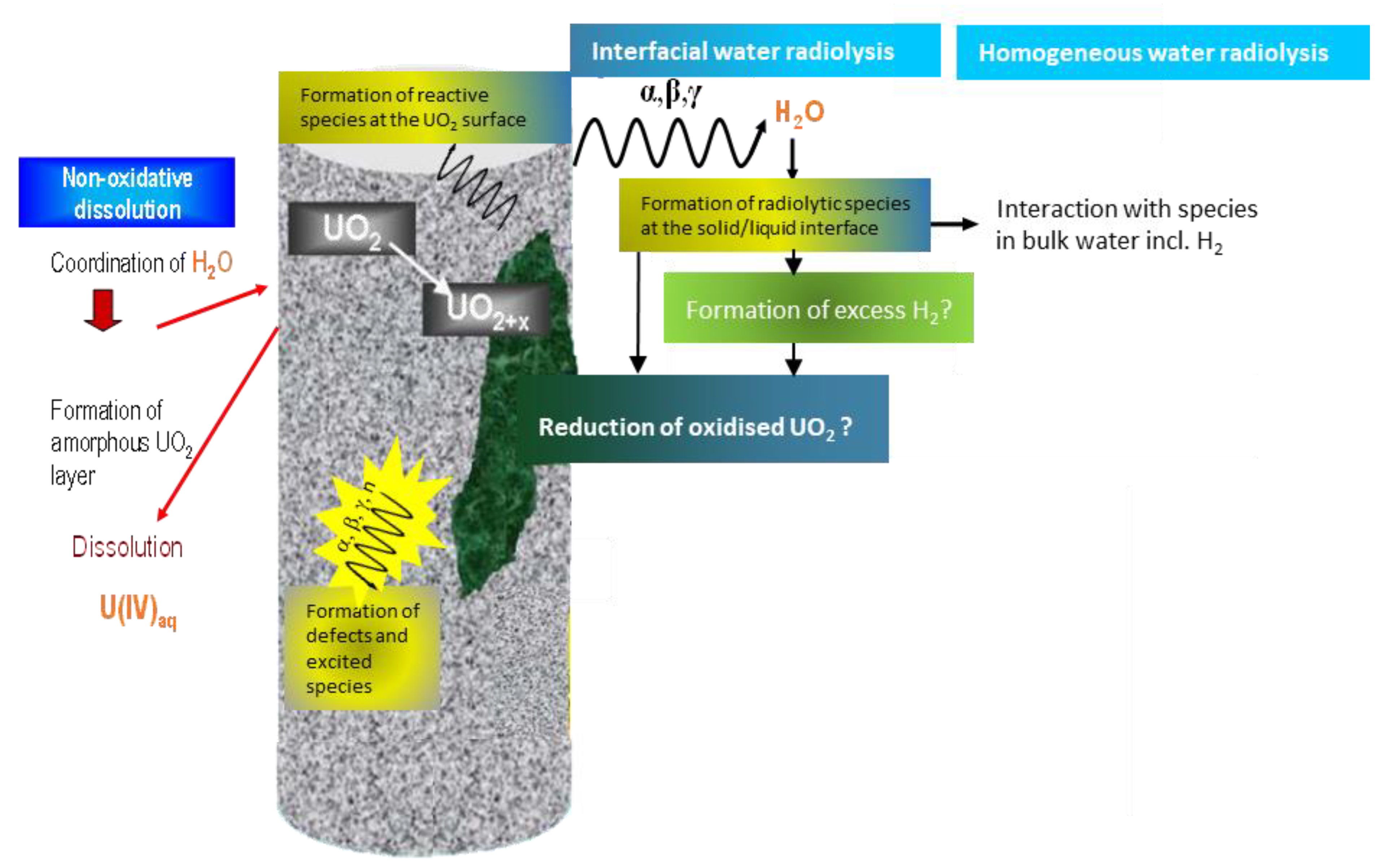
5. Proposed Alternative Conceptual Model
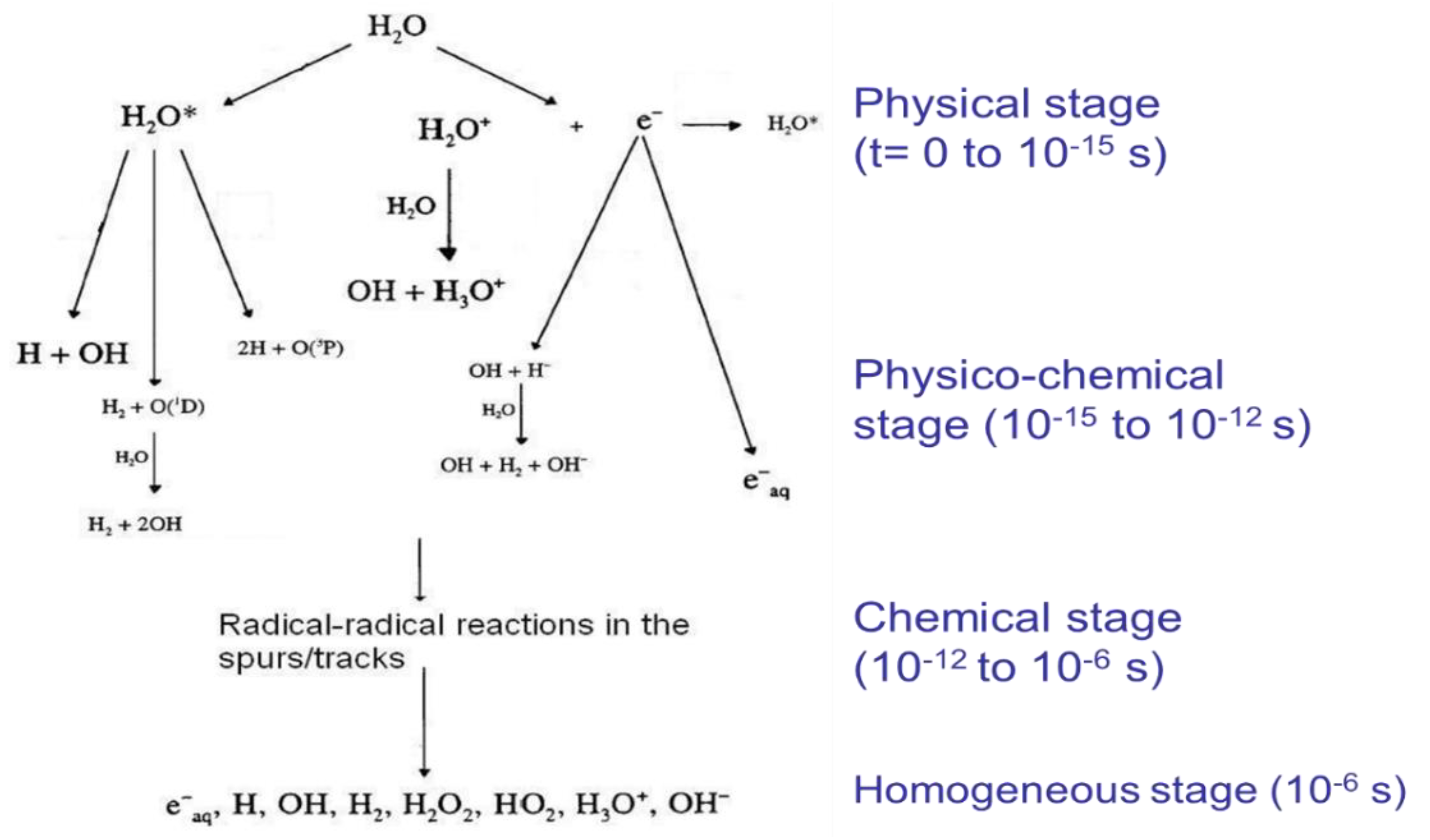
5.1. Processes Occurring at the Time of Interaction between Ionizing Radiation and Matter
5.2. Processes Occurring in the Bulk of UO2 (Direct Effect of Ionizing Radiation)
5.3. Processes Occurring at the Metal Oxides-Water Interface
5.4. Processes Occurring in Confined Water Volumes
5.5. Redox Conditions at the Spent Fuel Surface
6. Outlook on Developments of the Conceptual Model for Spent Fuel–Water Interaction
- Comparison of the effects of alpha particles produced in situ (for example, by using neutron capture on soluble compounds containing B-10) to those of external alpha sources;
- Further exploration of the field of radiolysis in confined water volumes and of water at the surface of metal oxides, in particular, fluorite-type solids;
- Investigation of whether excess H2 can be produced at the spent nuclear fuel–water interface;
- Modelling using Monte Carlo based techniques exploring physico-chemical processes at the spent fuel–water interface.
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Johnson, L.H.; Shoesmith, D.W. Spent Fuel. In Radioactive Waste Forms for the Future; Lutze, W., Ewing, R.C., Eds.; Elsevier: Amsterdam, The Netherlands, 1988; p. 635. [Google Scholar]
- Shoesmith, D.W. Fuel corrosion processes under waste disposal conditions. J. Nucl. Mater. 2000, 282, 1–31. [Google Scholar] [CrossRef]
- Posiva. Safety Case for the Disposal of Spent Nuclear Fuel at Olkiluoto—Performance Assessment 2012; Posiva Oy: Eurajoki, Finland, 2013; p. 520. [Google Scholar]
- Posiva Oy. Safety Case for the Disposal of Spent Nuclear Fuel at Olkiluoto—Formulation of Radionuclide Release Scenarios 2012; Posiva Oy: Eurajoki, Finland, 2013; p. 135. [Google Scholar]
- Fors, P.; Carbol, P.; Van Winckel, S.; Spahiu, K. Corrosion of high burn-up structured UO2 fuel in presence of dissolved H2. J. Nucl. Mater. 2009, 394, 1–8. [Google Scholar] [CrossRef]
- King, F.; Kolar, M.; Shoesmith, D.W. Modelling the oxidative dissolution of UO2. In Scientific Basis for Nuclear Waste Management Xxii; Wronkiewicz, D.J., Lee, J.H., Eds.; Materials Research Soc: Warrendale, PA, USA, 1999; Volume 556, pp. 463–470. [Google Scholar]
- Sunder, S.; Boyer, G.D.; Miller, N.H. XPS studies of UO2 oxidation by alpha-radiolysis of water at 100 degrees C. J. Nucl. Mater. 1990, 175, 163–169. [Google Scholar] [CrossRef]
- Carbol, P.; Fors, P.; Gouder, T.; Spahlu, K. Hydrogen suppresses UO2 corrosion. Geochim. Cosmochim. Acta 2009, 73, 4366–4375. [Google Scholar] [CrossRef]
- Ekeroth, E.; Granfors, M.; Schild, D.; Spahiu, K. The effect of temperature and fuel surface area on spent nuclear fuel dissolution kinetics under H2 atmosphere. J. Nucl. Mater. 2020, 531, 13. [Google Scholar] [CrossRef]
- Spahiu, K.; Devoy, J.; Cui, D.Q.; Lundstrom, M. The reduction of U(VI) by near field hydrogen in the presence of UO2(S). Radiochim. Acta 2004, 92, 597–601. [Google Scholar] [CrossRef]
- Carbol, P.; Cobos-Sabate, J.; Glatz, J.-P.; Ronchi, C.; Rondinella, V.; Wegen, D.H.; Wiss, T.; Loida, A.; Metz, V.; Kienzler, B.; et al. The effect of dissolved hydrogen on the dissolution of 233U doped UO2(s). In SKB Technical Report 05-09; Swedish Nuclear Fuel and Waste Management Co.: Stockholm, Sweden, 2005. [Google Scholar]
- Loida, A.; Grambow, B.; Geckeis, H. Anoxic corrosion of various high burnup spent fuel samples. J. Nucl. Mater. 1996, 238, 11–22. [Google Scholar] [CrossRef]
- Loida, A.; Grambow, B.; Geckeis, H. Spent fuel corrosion behavior is salt solution in the presence of hydrogen overpressure. In Proceedings of the ICEM’01, 8th International Conference on Radioactive Waste Management, Bruges, Belgium, 30 September –4 October 2001. [Google Scholar]
- Loida, A.; Metz, V.; Kienzler, B.; Geckeis, H. Radionuclide release from high burnup spent fuel during corrosion in salt brine in the presence of hydrogen overpressure. J. Nucl. Mater. 2005, 346, 24–31. [Google Scholar] [CrossRef]
- Spahiu, K.; Cui, D.Q.; Lundstrom, M. The fate of radiolytic oxidants during spent fuel leaching in the presence of dissolved near field hydrogen. Radiochim. Acta 2004, 92, 625–629. [Google Scholar] [CrossRef]
- Ollila, K.; Albinsson, Y.; Oversby, V.; Cowper, M. Dissolution Rates of Unirradiated UO2, UO2 Doped With 233U, and Spent Fuel under Normal Atmospheric Conditions and under Reducing Conditions Using An Isotope Dilution Method; SKB: Stockholm, Sweden, 2003. [Google Scholar]
- Spahiu, K.; Eklund, U.B.; Cui, D.Q.; Lundstrom, M. The influence of near field redox conditions on spent fuel leaching. In Scientific Basis for Nuclear Waste Management Xxv; McGrail, B.P., Cragnolino, G.A., Eds.; Materials Research Soc: Warrendale, PA, USA, 2002; Volume 713, pp. 633–638. [Google Scholar]
- Spahiu, K.; Werme, L.; Eklund, U.B. The influence of near field hydrogen on actinide solubilities and spent fuel leaching. Radiochim. Acta 2000, 88, 507–511. [Google Scholar] [CrossRef]
- Cera, E.; Bruno, J.; Duro, L.; Eriksen, T.E. Experimental Determination and Chemical Modelling of Radiolytic Processes at the Spent Fuel/Water Interface, Long Contact Time Experiments; Swedish Nuclear Fuel and Waste Management Compant Technical Report Stockholm, Sweden TR-06-07; Swedish Nuclear Fuel and Waste Management Co.: Stockholm, Sweden, 2006. [Google Scholar]
- Puranen, A.; Roth, O.; Evins, L.Z.; Spahiu, K. Aqueous leaching of high burnup UO2 fuel under hydrogen conditions. MRS Adv. 2018, 3, 1013–1018. [Google Scholar] [CrossRef]
- Odorowski, M. Etude de L’altération de la Matrice (U,Pu)2 du Combustible Irradié en Conditions de Stockage Géologique: Approche Expérimentale et Modélisation Géochimique. Ph.D. Thesis, Mines Paris Tech, Paris, France, 2015. [Google Scholar]
- Zetterström-Evins, L.; Juhola, P.; Vahanen, M. REDUPP Final Report; Posiva Oy: Eurajoki, Finland, 2014. [Google Scholar]
- Shoesmith, D.W. Used Fuel and Uranium Dioxide Dissolution Studies—A Review; Nuclear Waste Management Organization: Toronto, ON, Canada, 2007. [Google Scholar]
- Broczkowski, M.E.; Noel, J.J.; Shoesmith, D.W. The inhibiting effects of hydrogen on the corrosion of uranium dioxide under nuclear waste disposal conditions. J. Nucl. Mater. 2005, 346, 16–23. [Google Scholar] [CrossRef]
- Cui, D.; Low, J.; Sjostedt, C.J.; Spahiu, K. On Mo-Ru-Tc-Pd-Rh-Te alloy particles extracted from spent fuel and their leaching behavior under Ar and H2 atmospheres. Radiochim. Acta 2004, 92, 551–555. [Google Scholar] [CrossRef]
- Jonsson, M.; Nielsen, F.; Roth, O.; Ekeroth, E.; Nilsson, S.; Hossain, M.M. Radiation induced spent nuclear fuel dissolution under deep repository conditions. Environ. Sci. Technol. 2007, 41, 7087–7093. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.Z.; Qin, Z.; Noel, J.J.; Shoesmith, D.W. Modelling the radiolytic corrosion of alpha-doped UO2 and spent nuclear fuel. J. Nucl. Mater. 2017, 494, 87–94. [Google Scholar] [CrossRef]
- Liu, N.Z.; Wu, L.D.; Qin, Z.; Shoesmith, D.W. Roles of Radiolytic and Externally Generated H2 in the Corrosion of Fractured Spent Nuclear Fuel. Environ. Sci. Technol. 2016, 50, 12348–12355. [Google Scholar] [CrossRef]
- Trummer, M.; Roth, O.; Jonsson, M. H2 inhibition of radiation induced dissolution of spent nuclear fuel. J. Nucl. Mater. 2009, 383, 226–230. [Google Scholar] [CrossRef]
- Riba, O.; Coene, E.; Silva, O.; Duro, L. Spent fuel alteration model integrating processes of different time-scales. MRS Adv. 2020, 5, 159–166. [Google Scholar] [CrossRef]
- Martínez Esparza, A.; Cunado, M.A.; Gago, J.A.; Quinones, J.; Iglesia, E.; Cobos, J.; Gonzalez de la Huebra, A.; Cera, E.; Merino, J.; Bruno, J.; et al. Development of a Matrix Alteration Model (MAM); ENRESA Report 01; Empresa Nacional de Residuos Radiactivos, SA (Enresa): Madrid, Spain, 2005. [Google Scholar]
- Broczkowski, M.E.; Keech, P.G.; Noel, J.J.; Shoesmith, D.W. Corrosion of Uranium Dioxide Containing Simulated Fission Products in Dilute Hydrogen Peroxide and Dissolved Hydrogen. J. Electrochem. Soc. 2010, 157, C275–C281. [Google Scholar] [CrossRef]
- Lemmens, K.; Cachoir, C.; Mennecart, T. Dissolution Behavior of Spent Fuel at Highly Alkaline Conditions; Studiecentrum voor Kernenergie (SCK.CEN): Mol, Belgium, 2019. [Google Scholar]
- El Jamal, G.; Gouder, T.; Eloirdi, R.; Jonsson, M. Time-dependent surface modification of uranium oxides exposed to water plasma. Dalton Trans. 2021, 50, 4796–4804. [Google Scholar] [CrossRef]
- El Jamal, G.; Gouder, T.; Eloirdi, R.; Jonsson, M. X-Ray and ultraviolet photoelectron spectroscopy studies of Uranium (IV), (V) and (VI) exposed to H2O-plasma under UHV conditions. Dalton Trans. 2021, 50, 729–738. [Google Scholar] [CrossRef]
- Maier, A.C.; Kegler, P.; Klinkenberg, M.; Baena, A.; Finkeldei, S.; Brandt, F.; Jonsson, M. On the change in UO2 redox reactivity as a function of H2O2 exposure. Dalton Trans. 2020, 49, 1241–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mozumder, A. Charges particle tracks and their structure. In Advances in Radiation Chemistry; Burton, M., Magee, J.L., Eds.; Wiley-Interscience: New York, NY, USA, 1969; Volume 1, pp. 1–102. [Google Scholar]
- Mozumder, A. Early production of radicals from charged particle tracks in water. Radiat. Res. 1985, 104, S33–S39. [Google Scholar] [CrossRef]
- Swiatla-Wojcik, D.; Buxton, G.V. Modeling of radiation spur processes in water at temperatures up to 300-degrees-C. J. Phys. Chem. 1995, 99, 11464–11471. [Google Scholar] [CrossRef]
- Buxton, G.V. The Radiation Chemistry of the Liquid State: (1) Water and homogeneous Aqueous Solutions. In Radiation Chemistry: Principles and Applications; Farhataziz, Rodgers, M.A.J., Eds.; VCH Publishers, Inc.: New York, NY, USA, 1987; pp. 321–349. [Google Scholar]
- Allen, A.O. Radiation Chemistry of Aqueous Solutions. J. Phys. Colloid. Chem. 1948, 52, 479–490. [Google Scholar] [CrossRef]
- LaVerne, J.A.; Tandon, L. H2 Production in the Radiolysis of Water on UO2 and Other Oxides. J. Phys. Chem. B 2003, 107, 13623–13628. [Google Scholar] [CrossRef]
- Petrik, N.G.; Alexandrov, A.B.; Vall, A.I. Interfacial energy transfer during gamma radiolysis of water on the surface of ZrO2 and some other oxides. J. Phys. Chem. B 2001, 105, 5935–5944. [Google Scholar] [CrossRef]
- Bricout, M.; Onofri, C.; Debelle, A.; Pipon, Y.; Belin, R.C.; Garrido, F.; Lepretre, F.; Gutierrez, G. Radiation damage in uranium dioxide: Coupled effect between electronic and nuclear energy losses. J. Nucl. Mater. 2020, 531, 10. [Google Scholar] [CrossRef]
- De Bona, E.; Benedetti, A.; Dieste, O.; Staicu, D.; Wiss, T.; Konings, R.J.M. Radiation effects in alpha-doped UO2. Nucl. Instrum. Methods Phys. Res. Sect. B-Beam Interact. Mater. Atoms 2020, 468, 54–59. [Google Scholar] [CrossRef]
- Mohun, R.; Desgranges, L.; Canizares, A.; Raimboux, N.; Duval, F.; Omnee, R.; Jegou, C.; Miro, S.; Simon, P. Investigating the role of irradiation defects during UO2 oxidative dissolution. J. Nucl. Mater. 2018, 509, 305–312. [Google Scholar] [CrossRef] [Green Version]
- Tocino, F.; Szenknect, S.; Mesbah, A.; Clavier, N.; Dacheux, N. Dissolution of uranium mixed oxides: The role of oxygen vacancies vs. the redox reactions. Prog. Nucl. Energy 2014, 72, 101–106. [Google Scholar] [CrossRef]
- Corkhill, C.L.; Bailey, D.J.; Tocino, F.Y.; Stennett, M.C.; Miller, J.A.; Provis, J.L.; Travis, K.P.; Hyatt, N.C. Role of Microstructure and Surface Defects on the Dissolution Kinetics of CeO2, a UO2 Fuel Analogue. ACS Appl. Mater. Interfaces 2016, 8, 10562–10571. [Google Scholar] [CrossRef] [Green Version]
- LaVerne, J.A. H2 Formation from the Radiolysis of Liquid Water with Zirconia. J. Phys. Chem. B 2005, 109, 5395–5397. [Google Scholar] [CrossRef]
- LaVerne, J.A.; Tandon, L. H2 Production in the Radiolysis of Water on CeO2 and ZrO2. J. Phys. Chem. B 2002, 106, 380–386. [Google Scholar] [CrossRef]
- Boehm, H.P. Acidic and Basic Properties of Hydroxylated Metal Oxide Surfaces. Disc. Faraday Soc. 1971, 53, 264–289. [Google Scholar] [CrossRef]
- Schatz, T.; Cook, A.R.; Meisel, D. Capture of Charge Carriers at the Silica Nanoparticle-Water Interface. J. Phys. Chem. B 1999, 103, 10209–10213. [Google Scholar] [CrossRef]
- Reiff, S.C.; La Verne, J.A. Gamma and He Ion Radiolysis of Copper Oxides. J. Phys. Chem. C 2015, 119, 8821–8828. [Google Scholar] [CrossRef]
- Was, G.S. Fundamentals of Radiation Materials Science; Springer: Berlin/Heidelberg, Germany, 2007. [Google Scholar]
- Li, Z.; Huang, W. Reactivity of hydrogen species on oxide surfaces. Sci. China Chem. Open Access 2021, 64, 1076–1087. [Google Scholar] [CrossRef]
- Li, Z.R.; Werner, K.; Chen, L.; Jia, A.P.; Qian, K.; Zhong, J.Q.; You, R.; Wu, L.H.; Zhang, L.Y.; Pan, H.B.; et al. Interaction of Hydrogen with Ceria: Hydroxylation, Reduction, and Hydride Formation on the Surface and in the Bulk. Chem.-Eur. J. 2021, 27, 5268–5276. [Google Scholar] [CrossRef]
- Noutak, M.; Freyss, M.; Jomard, G.; Geneste, G. Electron polarons and donor point defects in americium dioxide AmO2. Phys. Rev. B 2020, 101, 024108. [Google Scholar] [CrossRef]
- Bo, T.; Lan, J.H.; Zhao, Y.L.; Zhang, Y.J.; He, C.H.; Chai, Z.F.; Shi, W.Q. First-principles study of water adsorption and dissociation on the UO2 (111), (110) and (100) surfaces. J. Nucl. Mater. 2014, 454, 446–454. [Google Scholar] [CrossRef]
- Wang, G.X.; Batista, E.R.; Yang, P. Excess Electrons on Reduced AnO2 (111) Surfaces (An = Th, U, Pu) and Their Impacts on Catalytic Water Splitting. J. Phys. Chem. C 2019, 123, 30245–30251. [Google Scholar] [CrossRef]
- Weck, P.F.; Kim, E.; Jove-Colon, C.F.; Sassani, D.C. On the role of strong electron correlations in the surface properties and chemistry of uranium dioxide. Dalton Trans. 2013, 42, 4570–4578. [Google Scholar] [CrossRef] [PubMed]
- Senanayake, S.D.; Idriss, H. Water reactions over stoichiometric and reduced UO2 (111) single crystal surfaces. Surf. Sci. 2004, 563, 135–144. [Google Scholar] [CrossRef]
- Stultz, J.; Paffett, M.T.; Joyce, S.A. Thermal evolution of hydrogen following water adsorption on defective UO2 (100). J. Phys. Chem. B 2004, 108, 2362–2364. [Google Scholar] [CrossRef]
- Le Caer, S. Water Radiolysis: Influence of Oxide Surfaces on H2 Production under Ionizing Radiation. Water 2011, 3, 235–253. [Google Scholar] [CrossRef] [Green Version]
- Reiff, S.C.; La Verne, J.A. Radiation-Induced Chemical Changes to Iron Oxides. J. Phys. Chem. B 2015, 119, 7358–7365. [Google Scholar] [CrossRef] [PubMed]
- Rotureau, P.; Renault, J.P.; Lebeau, B.; Patarin, J.; Mialocq, J.C. Radiolysis of Confined Water: Molecular Hydrogen Formation. Chem. Phys. Chem. 2005, 6, 1316–1323. [Google Scholar] [CrossRef]
- Le Caer, S.; Rotureau, P.; Brunet, F.; Charpentier, T.; Blain, G.; Renault, J.P.; Mialocq, J.C. Radiolysis of Confined water: Hydrogen Production at Low Dose Rate. Chem. Phys. Chem. 2005, 6, 2585–2596. [Google Scholar] [CrossRef]
- Traboulsi, A.; Vandenborre, J.; Blain, G.; Humbert, B.; Barbet, J.; Fattahi, M. Radiolytic Corrosion of Uranium Dioxide: Role of Molecular Species. J. Phys. Chem. C 2014, 118, 1071–1080. [Google Scholar] [CrossRef] [Green Version]
- Essehli, R.; Crumiere, F.; Blain, G.; Vandenborre, J.; Pottier, F.; Grambow, B.; Fattahi, M.; Mostafavi, M. H2 production by gamma and He ions water radiolysis, effect of presence TiO2 nanoparticles. Int. J. Hydrog. Energy 2011, 36, 14342–14348. [Google Scholar] [CrossRef]
- Fidalgo, A.B.; Kumagai, Y.; Jonsson, M. The role of surface-bound hydroxyl radicals in the reaction between H2O2 and UO2. J. Coord. Chem. 2018, 71, 1799–1807. [Google Scholar] [CrossRef] [Green Version]
- Hansson, N.L.; Tam, P.L.; Ekberg, C.; Spahiu, K. XPS study of external alpha-radiolytic oxidation of UO2 in the presence of argon or hydrogen. J. Nucl. Mater. 2021, 543, 10. [Google Scholar] [CrossRef]
- Allen, A.O.; Hochanadel, C.J.; Ghormley, J.A.; Davis, T.W. Decomposition of Water and Aqueous Solutions under Mixed Fast Neutron and Gamma Radiation. J. Phys. Chem. 1952, 56, 575–586. [Google Scholar] [CrossRef]
- Martins, P.F.B.D.; Lopes, P.P.; Ticianelli, E.A.; Stamenkovic, V.R.; Markovic, N.M.; Strmcnik, D. Hydrogen evolution reaction on copper: Promoting water dissociation by tuning the surface oxophilicity. Electrochem. Commun. 2019, 100, 30–33. [Google Scholar] [CrossRef]
- Strmcnik, D.; Lopes, P.P.; Genorio, B.; Stamenkovic, V.R.; Markovic, N.M. Design principles for hydrogen evolution reaction catalyst materials. Nano Energy 2016, 29, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Betova, I.; Bojinov, M.; Lilja, C. Influence of ionic strength on hydrogen generation during interaction of copper with deoxygenated neutral solution. Corrosion Sci. 2021, 188, 13. [Google Scholar] [CrossRef]
- Carrasco, J.; Hodgson, A.; Michaelides, A. A molecular perspective of water at metal interfaces. Nat. Mater. 2012, 11, 667–674. [Google Scholar] [CrossRef] [Green Version]
- Hodgson, A.; Haq, S. Water adsorption and the wetting of metal surfaces. Surf. Sci. Rep. 2009, 64, 381–451. [Google Scholar] [CrossRef]
- Pastina, B.; LaVerne, J.A. Hydrogen Peroxide Production in the Radiolysis of Water with Heavy Ions. J. Phys. Chem. A 1999, 103, 1592–1597. [Google Scholar] [CrossRef]
- Pastina, B.; LaVerne, J.A. Effect of Molecular Hydrogen on Hydrogen Peroxide in Water Radiolysis. J. Phys. Chem. A 2001, 105, 9316–9322. [Google Scholar] [CrossRef]
- Pastina, B.; Isabey, J.; Hickel, B. The influence of water chemistry on the radiolysis of the primary coolant water in pressurized water reactors. J. Nucl. Mater. 1999, 264, 309–318. [Google Scholar] [CrossRef]
- Posiva Oy. Safety Case for the Disposal of Spent Nuclear Fuel at Olkiluoto—Models and Data for the Repository System 2012 Parts 1 & 2; Posiva Oy: Eurajoki, Finland, 2013. [Google Scholar]
- SKB. Data Report for the Safety Assessment SR-Site; Swedish Nuclear Fuel and Waste Management Co. (SKB): Stockholm, Sweden, 2010; p. 457. [Google Scholar]
- Johnson, L. A Model for Radiolnuclide Release from Spent UO2 and MOX Fuel; NAGRA NAB 13-37; Nationale Genossenschaft für die Lagerung radioaktiver Abfälle: Wettingen, Switzerland, 2014; p. 20. [Google Scholar]
- Gobien, M.; Garisto, F.; Kremer, E.; Medri, C. Seventh Case Study: Reference Data and Codes; NWMO-TR-2018-10; Nuclear Waste Management Organization: Toronto, ON, Canada, 2018. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample (Reducing Agent) | Activity (Bq g−1 UO2) | Observations by the Authors | Reference |
|---|---|---|---|
| PWR segment (H2) 50 MWd kg−1 U | 5 × 108 a | Decreasing rates but propose a fractional release rate of 4 × 10−7 y−1 | Carbol et al. [11] citing Loida et al. [12,13,14] |
| PWR powder (H2) 43 MWd kg−1U | 4.3 × 108 a | 3 × 10−7 y−1 based on spent fuel powders | Spahiu et al. [15] |
| PWR fragment (Fe) 41 MWd kg−1 U | 4.1 × 108 a | Decreasing dissolution rates | Ollila et al. [16] |
| PWR segments (H2) 43 MWd kg−1 U | 4.3 × 108 a | Decreasing rates over >1 year | Spahiu et al. [17,18] |
| PWR segment (H2) 43 MWd kg−1 U | 4.3 × 108 | [U] concentrations 10−5 to 5 × 10−10 M practically constant over > 2 years | Ekeroth et al. [9] |
| PWR fragment (H2) 67 MWd kg−1 U | 1.2 × 109 b | Decreasing rates followed by steady-state after 502 days | Fors [5] |
| PWR fragments (H2) 40 MWd kg−1 U | 4 × 108 a | Inhibited dissolution rates over 3 years | Cera et al. [19] |
| PWR fragments (H2) 65 MWd kg−1 U | 1.3 × 109 a | Decreasing U concentrations reaching U solubility limits, inhibition of fuel dissolution | Puranen et al. [20] |
| MOX irradiated (H2) 48 MWd t−1 HM | 3 × 109 a | Decreasing U concentrations after 494 days, lower than U solubility limit | Carbol et al. [8] |
| Irradiated MOX (Fe) 47 MWd t−1 HM | 3 × 109 (beta, gamma 2.4 × 1010) b | No signs of oxidative dissolution | Odorowski 2015 [21] |
| UO2 un-doped (Fe) | 1 × 104 b | No measurable dissolution rate over 1 year | Odorowski 2015 [21] |
| UO2 Pu-doped (Fe) | 3.8 × 109 b | No measurable dissolution rate over 1 year | Odorowski 2015 [21] |
| UO2 Pu-doped (Fe) | 1.8 × 107 b | No measurable dissolution rate over 1 year | Odorowski 2015 [21] |
| Fresh MOX (7%Pu) (Fe) | 1.3 × 109 (beta, 9.1 × 109) b | No measurable dissolution rate over 1 year | Odorowski 2015 [21] |
| UO2 un-doped (Fe) | 1 × 104 b | 8.3 × 10−8 y−1 (min) 1.6 × 10−6 y−1 (max) | Zetterström Evins et al. [22] |
| UO2 U-233-doped 5% (Fe) | 1.6 × 107 b | 7.5 × 10−8 y−1 (min) 1.1 × 10−6 y−1 (max) | Zetterström Evins et al. [22] |
| UO2 U-233-doped 10% (Fe) | 3.1 × 107 b | 4.4 × 10−8 y−1 (min) 1.2 × 10−6 y−1 (max) | Zetterström Evins et al. [22] |
| UO2 U-233-doped 10% (H2) | 3.3 × 107 b | Decreasing total U concentrations over 328 days. No sign of surface oxidation observed. | Carbol et al. [8] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pastina, B.; LaVerne, J.A. An Alternative Conceptual Model for the Spent Nuclear Fuel–Water Interaction in Deep Geologic Disposal Conditions. Appl. Sci. 2021, 11, 8566. https://doi.org/10.3390/app11188566
Pastina B, LaVerne JA. An Alternative Conceptual Model for the Spent Nuclear Fuel–Water Interaction in Deep Geologic Disposal Conditions. Applied Sciences. 2021; 11(18):8566. https://doi.org/10.3390/app11188566
Chicago/Turabian StylePastina, Barbara, and Jay A. LaVerne. 2021. "An Alternative Conceptual Model for the Spent Nuclear Fuel–Water Interaction in Deep Geologic Disposal Conditions" Applied Sciences 11, no. 18: 8566. https://doi.org/10.3390/app11188566
APA StylePastina, B., & LaVerne, J. A. (2021). An Alternative Conceptual Model for the Spent Nuclear Fuel–Water Interaction in Deep Geologic Disposal Conditions. Applied Sciences, 11(18), 8566. https://doi.org/10.3390/app11188566