
Reference Mapping Considering Swaps of Adjacent Bases

 ,
,  ,
, 

Abstract
:1. Introduction
Previous Work
| Algorithm 1 Compute , , and . |
| 1: Assign tables I, D, and M of size |
| 2: Initialize(I, D, M) |
| 3: for do |
| 4: for do |
| 5: |
| 6: |
| 7: |
| 8: end for |
| 9: end for |
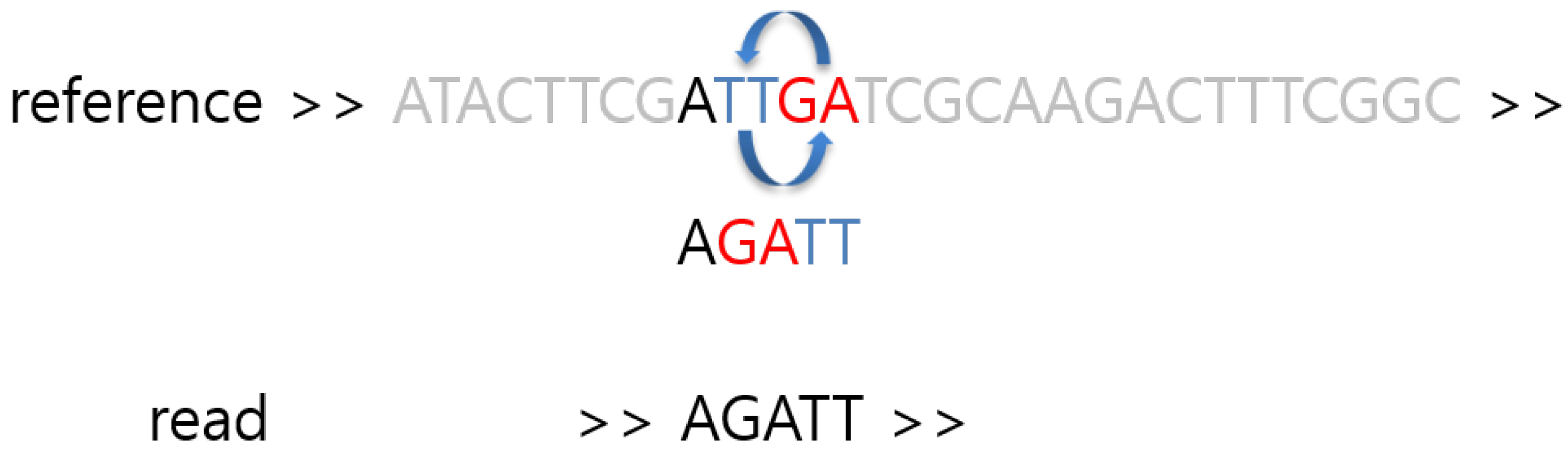
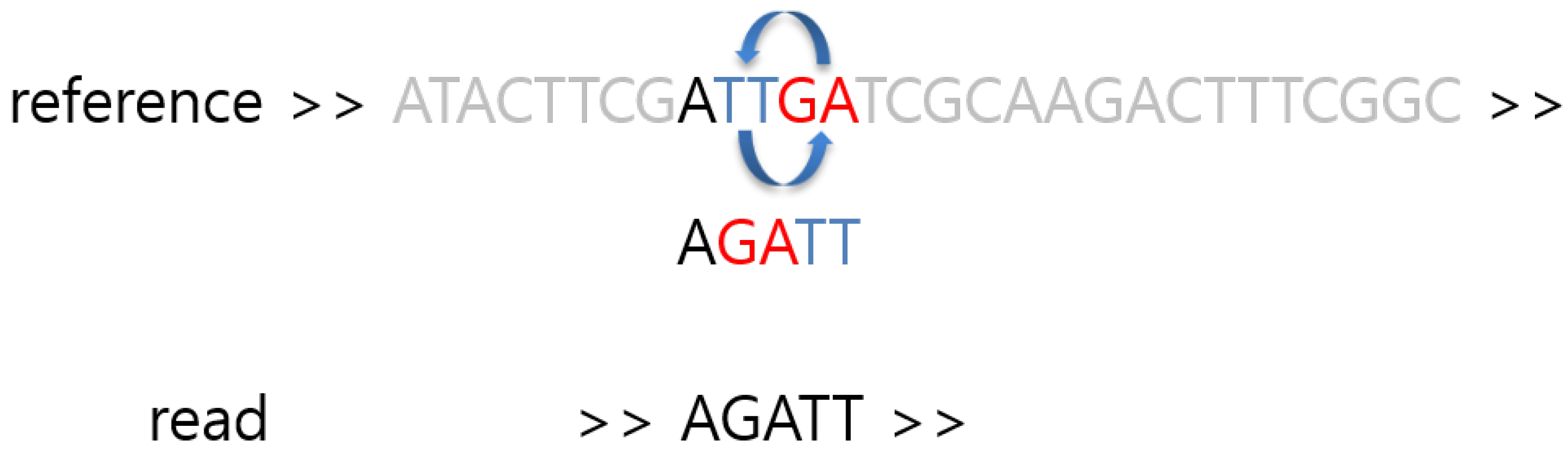
2. Materials and Methods
| Algorithm 2 Compute , , and considering swaps. |
| 1: Assign tables I, D, and M of size |
| 2: Initialize(I, D, M) |
| 3: for do |
| 4: for do |
| 5: |
| 6: |
| 7: |
| 8: if and and and then |
| 9: |
| 10: end if |
| 11: end for |
| 12: end for |
3. Results
- CPU and RAM: AMD Ryzen 9 3950X (3.5 GHz), 64 GB RAM (2666 MHz);
- OS: Fedora 27 (64 bit);
- GPU: GeForce RTX 2080 Ti (11 GB memory);
- Development tools and language: C++ (GCC 7.4.0), CUDA (SDK 9.1).
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| HGP | Human Genome Project |
| SNP | Single-nucleotide polymorphism |
| BWT | Burrows–Wheeler transform |
| BWA | Burrows–Wheeler aligner |
| GPU | Graphics processing unit |
| CUDA | Compute unified device architecture |
| CIGAR | Compact idiosyncratic gapped alignment report |
References
- Tilford, C.A.; Kuroda-Kawaguchi, T.; Skaletsky, H.; Rozen, S.; Brown, L.G.; Rosenberg, M.; McPherson, J.D.; Wylie, K.; Sekhon, M.; Kucaba, T.A.; et al. A physical map of the human Y chromosome. Nature 2001, 409, 943–945. [Google Scholar] [CrossRef] [Green Version]
- International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar]
- Metzker, M.L. Sequencing technologies—the next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [Green Version]
- Bao, S.; Jiang, R.; Kwan, W.; Wang, B.; Ma, X.; Song, Y.Q. Evaluation of next-generation sequencing software in mapping and assembly. J. Hum. Genet. 2011, 56, 406–414. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Li, H.; Ruan, J.; Durbin, R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008, 18, 1851–1858. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Yu, C.; Li, Y.; Lam, T.W.; Yiu, S.; Kristiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Ferragina, P.; Manzini, G. Opportunistic Data Structures with Applications. In Proceedings of the 41st Annual Symposium on Foundations of Computer Science, FOCS 2000, Redondo Beach, CA, USA, 12–14 November 2000; pp. 390–398. [Google Scholar] [CrossRef]
- Lam, T.W.; Li, R.; Tam, A.; Wong, S.C.K.; Wu, E.; Yiu, S. High Throughput Short Read Alignment via Bi-directional BWT. In Proceedings of the 2009 IEEE International Conference on Bioinformatics and Biomedicine, BIBM 2009, Washington, DC, USA, 1–4 November 2009; pp. 31–36. [Google Scholar] [CrossRef] [Green Version]
- Smith, T.F.; Waterman, M.S. Identification of Common Molecular Subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Wong, T.K.F.; Wu, E.; Luo, R.; Yiu, S.; Li, Y.; Wang, B.; Yu, C.; Chu, X.; Zhao, K.; et al. SOAP3: Ultra-fast GPU-based parallel alignment tool for short reads. Bioinformatics 2012, 28, 878–879. [Google Scholar] [CrossRef] [Green Version]
- Luo, R.; Wong, T.; Zhu, J.; Liu, C.M.; Zhu, X.; Wu, E.; Lee, L.K.; Lin, H.; Zhu, W.; Cheung, D.W.; et al. Correction: SOAP3-dp: Fast, accurate and sensitive GPU-based short read aligner. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Klus, P.; Lam, S.; Lyberg, D.; Cheung, M.S.; Pullan, G.; McFarlane, I.; Yeo, G.S.; Lam, B.Y. BarraCUDA-a fast short read sequence aligner using graphics processing units. BMC Res. Notes 2012, 5, 27. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Schmidt, B.; Maskell, D.L. CUSHAW: A CUDA compatible short read aligner to large genomes based on the Burrows-Wheeler transform. Bioinformatics 2012, 28, 1830–1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Schmidt, B. CUSHAW2-GPU: Empowering Faster Gapped Short-Read Alignment Using GPU Computing. IEEE Des. Test 2014, 31, 31–39. [Google Scholar] [CrossRef]
- Liu, Y.; Schmidt, B. Long read alignment based on maximal exact match seeds. Bioinformatics 2012, 28, 318–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gusfield, D. Algorithms on Strings, Trees, and Sequences—Computer Science and Computational Biology; Cambridge University Press: New York, NY, USA, 1997. [Google Scholar]
- Lowrance, R.; Wagner, R.A. An Extension of the String-to-String Correction Problem. J. ACM 1975, 22, 177–183. [Google Scholar] [CrossRef]
- Wagner, R.A. On the Complexity of the Extended String-to-String Correction Problem. In Proceedings of the 7th Annual ACM Symposium on Theory of Computing, Albuquerque, NM, USA, 5–7 May 1975; pp. 218–223. [Google Scholar] [CrossRef]
- Kim, D.K.; Lee, J.; Park, K.; Cho, Y. Efficient Algorithms for Approximate String Matching with Swaps. J. Complex. 1999, 15, 128–147. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.W.; Kim, Y.; Sim, J.S. Parallel Computation for Extended Edit Distances Including Swap Operations. J. KIISE Comput. Syst. Theory 2014, 41, 175–181. [Google Scholar]
- Lewin, B. Genes for SMA: Multum in parvo. Cell 1995, 80, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Amir, A.; Aumann, Y.; Landau, G.M.; Lewenstein, M.; Lewenstein, N. Pattern Matching with Swaps. J. Algorithms 2000, 37, 247–266. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.T.; Abecasis, G.R.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The SAM/BAM Format Specification Working Group. Sequence Alignment/Map Format Specification. Available online: https://samtools.github.io/hts-specs/SAMv1.pdf (accessed on 28 May 2021).
- Roelofs, G.; Gailly, J.L.; Adler, M. zlib. Available online: https://zlib.net/ (accessed on 28 May 2021).
- Deutsch, L.P.; Gailly, J.L. ZLIB Compressed Data Format Specification Version 3.3. Available online: https://datatracker.ietf.org/doc/html/rfc1950 (accessed on 28 May 2021).
- Deutsch, L.P. DEFLATE Compressed Data Format Specification Version 1.3. Available online: https://datatracker.ietf.org/doc/html/rfc1951 (accessed on 28 May 2021).
- Deutsch, L.P. GZIP File Format Specification Version 4.3. Available online: https://datatracker.ietf.org/doc/html/rfc1952 (accessed on 28 May 2021).

{kind=link}
| Tools | Paired-End Reads | GPU-Based | ||
|---|---|---|---|---|
| Runtime (s) | Number of Aligned Reads (Ratio) | Difference from SOAP3-dp | ||
| SOAP3-swap (Full SA, | 186.98 | 25,105,846 (98.58%) | Yes | |
| SOAP3-swap (Full SA, | 178.79 | 25,103,497 (98.57%) | Yes | |
| SOAP3-swap (Full SA, | 181.08 | 25,102,151 (98.56%) | Yes | |
| SOAP3-dp (Full SA) | 178.52 | 25,101,179 (98.56%) | ⋅ | Yes |
| SOAP3 | 212.22 | 22,613,051 (88.79%) | −2,488,128 | Yes |
| CUSHAW2-GPU | 599.94 | 24,957,932 (98.00%) | −143,247 | Yes |
| BarraCUDA | 947.67 | 24,551,512 (96.40%) | −549,667 | Yes |
| BWA | 2731.73 | 24,598,492 (96.59%) | −502,687 | No |
| Bowtie2 (Sensitive) | 1414.67 | 24,798,944 (97.37%) | −302,235 | No |
| CUSHAW2 | 2084.69 | 24,133,013 (94.76%) | −968,166 | No |
| Tools | Paired-End Reads | GPU-Based | ||
|---|---|---|---|---|
| Runtime (s) | Number of Aligned Reads (Ratio) | Difference from SOAP3-dp | ||
| SOAP3-swap (Full SA, | 641.47 | 31,514,424 (81.07%) | Yes | |
| SOAP3-swap (Full SA, | 624.95 | 31,513,625 (81.07%) | Yes | |
| SOAP3-swap (Full SA, | 623.67 | 31,513,300 (81.07%) | Yes | |
| SOAP3-dp (Full SA) | 563.83 | 31,513,171 (81.07%) | ⋅ | Yes |
| SOAP3 | 698.08 | 24,409,682 (62.79%) | −7,103,489 | Yes |
| BarraCUDA | 1131.08 | 30,380,166 (78.15%) | −1,133,005 | Yes |
| Bowtie2 (Sensitive) | 1261.55 | 30,602,667 (78.72%) | −910,504 | No |
| CUSHAW2 | 2697.97 | 30,099,066 (77.43%) | −1,414,105 | No |
| Swap Cost | Swapped Block Size | ||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | Total | ||
| Human genome | 265,727 | 7631 | 21 | 273,379 | |
| 202,850 | 117 | 31 | 202,998 | ||
| 145,054 | 57 | 6 | 145,117 | ||
| Drosophila genome | 304,209 | 13,919 | 215 | 318,343 | |
| 212,278 | 627 | 273 | 213,178 | ||
| 158,953 | 79 | 72 | 159,104 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.; Kang, M.; Jeong, J.-H.; Kang, D.W.; Park, S.J.; Sim, J.S. Reference Mapping Considering Swaps of Adjacent Bases. Appl. Sci. 2021, 11, 5038. https://doi.org/10.3390/app11115038
Kim Y, Kang M, Jeong J-H, Kang DW, Park SJ, Sim JS. Reference Mapping Considering Swaps of Adjacent Bases. Applied Sciences. 2021; 11(11):5038. https://doi.org/10.3390/app11115038
Chicago/Turabian StyleKim, Youngho, Munseong Kang, Ju-Hui Jeong, Dae Woong Kang, Soo Jun Park, and Jeong Seop Sim. 2021. "Reference Mapping Considering Swaps of Adjacent Bases" Applied Sciences 11, no. 11: 5038. https://doi.org/10.3390/app11115038
APA StyleKim, Y., Kang, M., Jeong, J.-H., Kang, D. W., Park, S. J., & Sim, J. S. (2021). Reference Mapping Considering Swaps of Adjacent Bases. Applied Sciences, 11(11), 5038. https://doi.org/10.3390/app11115038