Pyr1,xTFSI Ionic Liquids (x = 1–8): A Computational Chemistry Study


Abstract
:Featured Application
Abstract
1. Introduction
2. Materials and Methods
3. Results
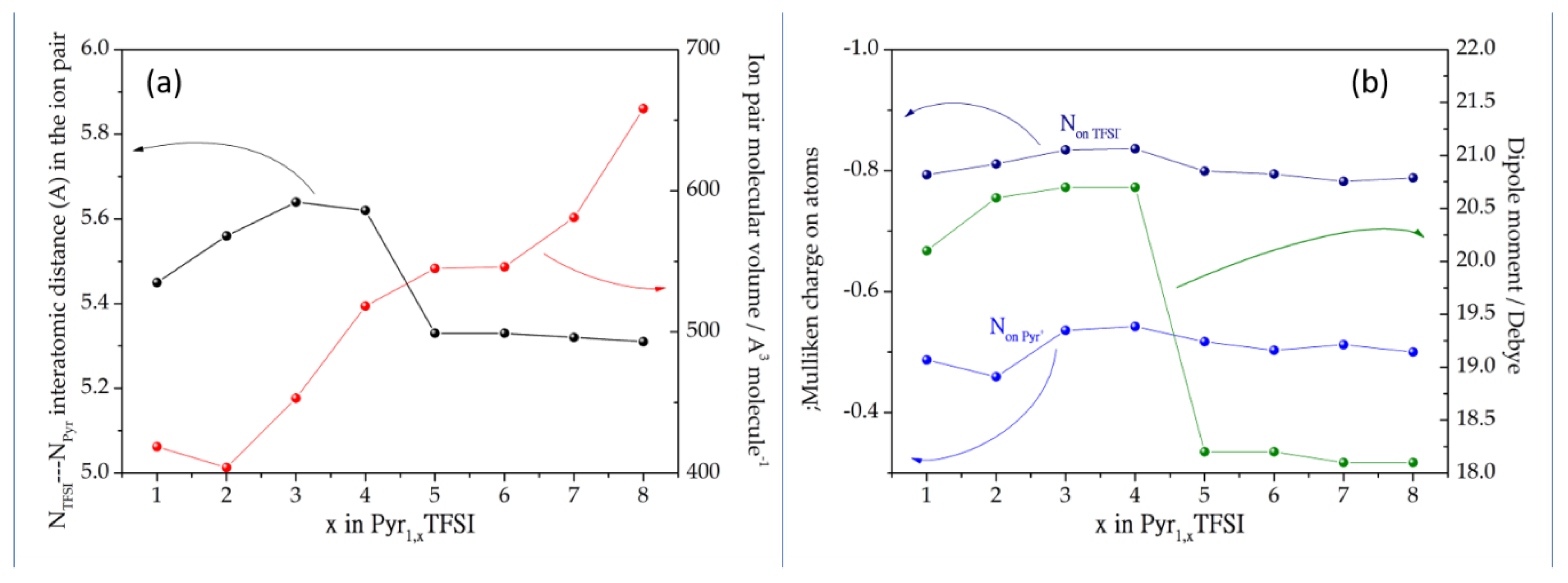
3.1. Ion Pairs Structure
3.2. Dissociation Thermodynamics
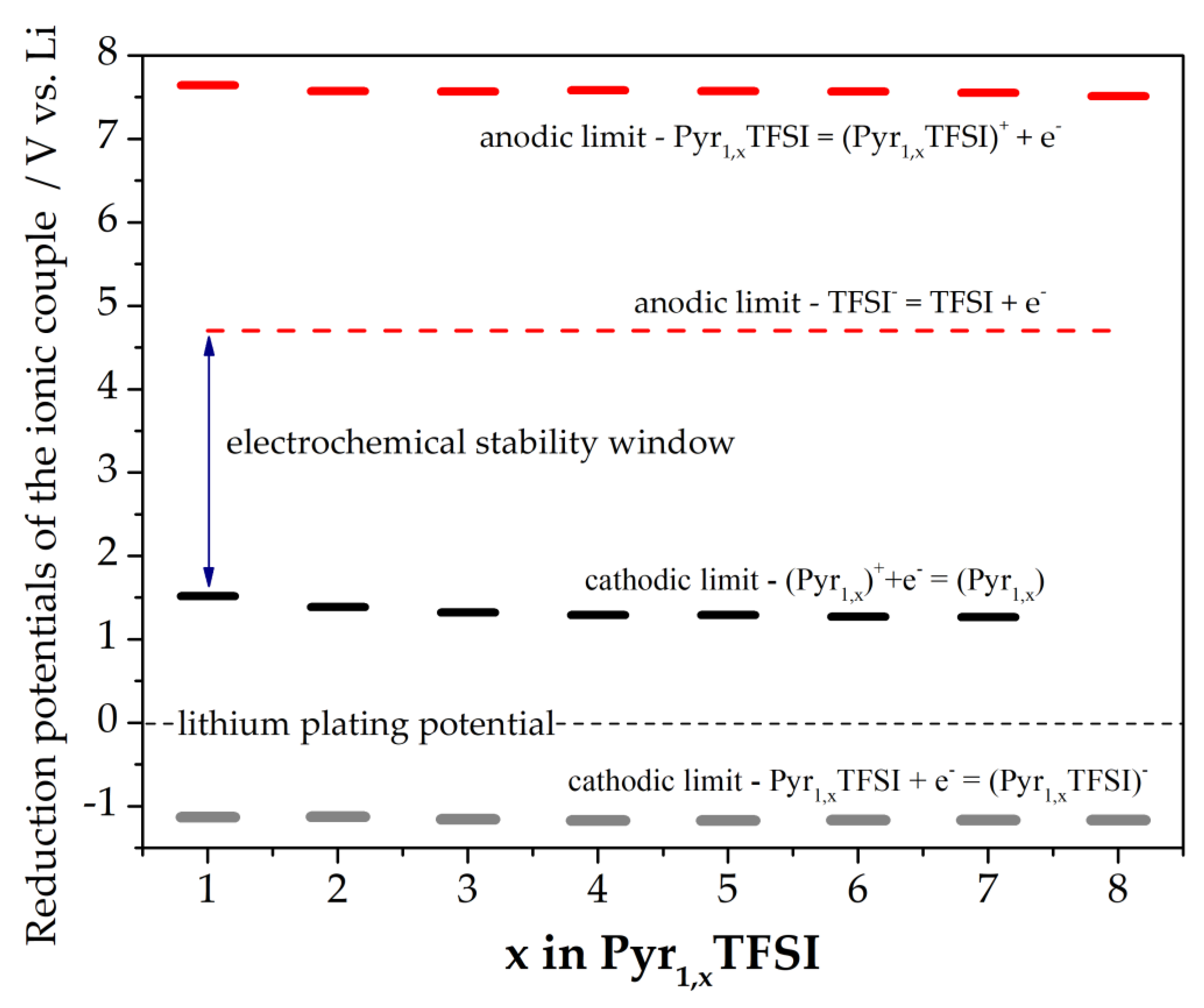
3.3. Electrochemical Stability
4. Conclusions
Supplementary Materials
Funding
Conflicts of Interest
References
- Sandhu, J.S. Recent advances in ionic liquids: Green unconventional solvents of this century: Part I. Green Chem. Lett. Rev. 2011, 4, 289–310. [Google Scholar] [CrossRef] [Green Version]
- Lei, Z.; Chen, B.; Koo, Y.M.; Macfarlane, D.R. Introduction: Ionic Liquids. Chem. Rev. 2017, 117, 6633–6635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewandowski, A.; Świderska-Mocek, A. Ionic liquids as electrolytes for Li-ion batteries—An overview of electrochemical studies. J. Power Sources 2009, 194, 601–609. [Google Scholar] [CrossRef]
- Kalhoff, J.; Eshetu, G.G.; Bresser, D.; Passerini, S. Safer electrolytes for lithium-ion batteries: State of the art and perspectives. ChemSusChem 2015, 8, 2154–2175. [Google Scholar] [CrossRef]
- Armand, M.; Endres, F.; MacFarlane, D.R.; Ohno, H.; Scrosati, B. Ionic-liquid materials for the electrochemical challenges of the future. Nat. Mater. 2009, 8, 621–629. [Google Scholar] [CrossRef]
- Suo, L.; Hu, Y.S.; Li, H.; Armand, M.; Chen, L. A new class of Solvent-in-Salt electrolyte for high-energy rechargeable metallic lithium batteries. Nat. Commun. 2013, 4, 1481. [Google Scholar] [CrossRef]
- Johansson, P.; Fast, L.E.; Matic, A.; Appetecchi, G.B.; Passerini, S. The conductivity of pyrrolidinium and sulfonylimide-based ionic liquids: A combined experimental and computational study. J. Power Sources 2010, 195, 2074–2076. [Google Scholar] [CrossRef]
- Brutti, S.; Navarra, M.A.; Maresca, G.; Panero, S.; Manzi, J.; Simonetti, E.; Appetecchi, G.B. Ionic liquid electrolytes for room temperature sodium battery systems. Electrochim. Acta 2019, 306, 317–326. [Google Scholar] [CrossRef]
- Lombardo, L.; Brutti, S.; Navarra, M.A.; Panero, S.; Reale, P. Mixtures of ionic liquid e Alkylcarbonates as electrolytes for safe lithium-ion batteries. J. Power Sources 2013, 227, 8–14. [Google Scholar] [CrossRef]
- Celeste, A.; Silvestri, L.; Agostini, M.; Sadd, M.; Palumbo, S.; Panda, J.K.; Matic, A.; Pellegrini, V.; Brutti, S. Enhancement of Functional Properties of Liquid Electrolytes for Lithium-Ion Batteries by Addition of Pyrrolidinium-Based Ionic Liquids with Long Alkyl-Chains. Batter. Supercaps 2020, 3, 1051–1068. [Google Scholar] [CrossRef]
- Arbizzani, C.; Gabrielli, G.; Mastragostino, M. Thermal stability and flammability of electrolytes for lithium-ion batteries. J. Power Sources 2011, 196, 4801–4805. [Google Scholar] [CrossRef]
- Johansson, P.; Jacobsson, P. Rational design of electrolyte components by ab initio calculations. J. Power Sources 2006, 153, 336–344. [Google Scholar] [CrossRef]
- Matsumoto, R.; Thompson, M.W.; Cummings, P.T. Ion Pairing Controls Physical Properties of Ionic Liquid-Solvent Mixtures. J. Phys. Chem. B 2019, 123, 9944–9955. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Russina, O.; Triolo, A. Ionic Liquids and Neutron Scattering. In Experimental Methods in the Physical Sciences; Academic Press: Cambridge, MA, USA, 2017; Volume 49, pp. 213–278. [Google Scholar]
- Russina, O.; lo Celso, F.; Plechkova, N.; Jafta, C.J.; Appetecchi, G.B.; Triolo, A. Mesoscopic organization in ionic liquids. Top. Curr. Chem. 2017, 375, 58. [Google Scholar] [CrossRef] [PubMed]
- Castiglione, F.; Moreno, M.; Raos, G.; Famulari, A.; Mele, A.; Appetecchi, G.B.; Passerini, S. Structural organization and transport properties of novel pyrrolidinium-based ionic liquids with perfluoroalkyl sulfonylimide anions. J. Phys. Chem. B 2009, 113, 10750–10759. [Google Scholar] [CrossRef]
- Carboni, M.; Spezia, R.; Brutti, S. Perfluoroalkyl-Fluorophosphate Anions for High Voltage Electrolytes in Lithium Cells: DFT Study. J. Phys. Chem. C 2014, 118, 24221–24230. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639. [Google Scholar]
- Angenendt, K.; Johansson, P. Ionic liquid structured from large density functional theory calculations using mindless configurations. J. Phys. Chem. C 2010, 114, 20577–20582. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Weingärtner, H. The static dielectric permittivity of ionic liquids. J. Mol. Liq. 2014, 192, 185–190. [Google Scholar] [CrossRef]
- Singh, T.; Kumar, A. Static dielectric constant of room temperature ionic liquids: Internal pressure and cohesive energy density approach. J. Phys. Chem. B 2008, 112, 12968–12972. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.; Coote, M.L.; Cramer, C.J.; Truhlar, D. Theoretical calculation of reduction potentials. In Organic Electrochemistry: Revised and Expanded; CRC Press: Boca Raton, FL, USA, 2015; Volume 6. [Google Scholar]
- Winget, P.; Cramer, C.J.; Truhlar, D.G. Computation of equilibrium oxidation and reduction potentials for reversible and dissociative electron-transfer reactions in solution. Theor. Chem. Acc. 2004, 112, 217–227. [Google Scholar] [CrossRef]
- Jónsson, E.; Johansson, P.; Xu, K. Electrochemical oxidation stability of anions for modern battery electrolytes: A CBS and DFT study. Phys. Chem. Chem. Phys. 2015, 17, 3697–3703. [Google Scholar] [CrossRef] [Green Version]
- Brutti, S.; Simonetti, E.; De Francesco, M.; Sarra, A.; Paolone, A.; Palumbo, O.; Fantini, S.; Lin, R.; Falagayrat, A.; Choi, H.; et al. Ionic liquid electrolytes for high-voltage lithium batteries. J. Sources 2020, 479, 238791. [Google Scholar]
- Bellusci, M.; Simonetti, E.; De Francesco, M.; Appetecchi, G.B. Ionic liquid electrolytes for safer and more reliable sodium battery systems. Appl. Sci. 2020, 10, 6323. [Google Scholar] [CrossRef]
- Howlett, P.C.; MacFarlane, D.R.; Hollenkamp, A.F. High lithium metal cycling efficiency in a room-temperature ionic liquid. Electrochem. Solid State Lett. 2004, 7, A97–A101. [Google Scholar] [CrossRef]
- Kim, G.T.; Appetecchi, G.B.; Montanarino, M.; Alessandrini, F.; Passerini, S. Long-term cyclability of lithium metal electrodes in ionic liquid-based electrolytes at room temperature. ECS Trans. 2010, 25, 12138. [Google Scholar] [CrossRef]
- Randstrom, S.; Appetecchi, G.B.; Lagergren, C.; Moreno, A.; Passerini, S. The Influence of aur and its components on the cathodic stability of N-butyl-N-methylpyrrolidinium bis(trifluoromethanesulphonyl)imide. Electrochim. Acta 2007, 53, 1837–1842. [Google Scholar] [CrossRef]
- Randstrom, S.; Montanino, M.; Appetecchi, G.B.; Legergren, C.; Moreno, A.; Passerini, S. Effect of water and oxygen traces on the cathodic stability of N-alkyl-N-methylpyrrolidinium bis(trifluoromethanesulphonyl9imide. Electrochim. Acta 2008, 53, 6397–6401. [Google Scholar] [CrossRef]
- Appetecchi, G.B.; Montanino, M.; Passerini, S. Ionic liquid-based electrolytes for high-energy, safer lithium batteries. ACS Symp. Ser. 2012, 1117, 67–128. [Google Scholar]
- Lee, W.; Muhammad, S.; Sergey, C.; Lee, H.; Yoon, J.; Kang, Y.; Yoon, W. Advances in the Cathode Materials for Lithium Rechargeable Batteries. Angew. Chem. Int. Ed. 2020, 59, 2578–2605. [Google Scholar] [CrossRef] [PubMed]
- Sun, X. Advances in spinel Li4Ti5O12 anode materials for lithium-ion batteries. New J. Chem. 2014, 39, 38–63. [Google Scholar] [CrossRef]
- Hassoun, J.; Scrosati, B. Review—Advances in Anode and Electrolyte Materials for the Progress of Lithium-Ion and beyond Lithium-Ion Batteries. J. Electrochem. Soc. 2015, 162, A2582–A2588. [Google Scholar] [CrossRef]
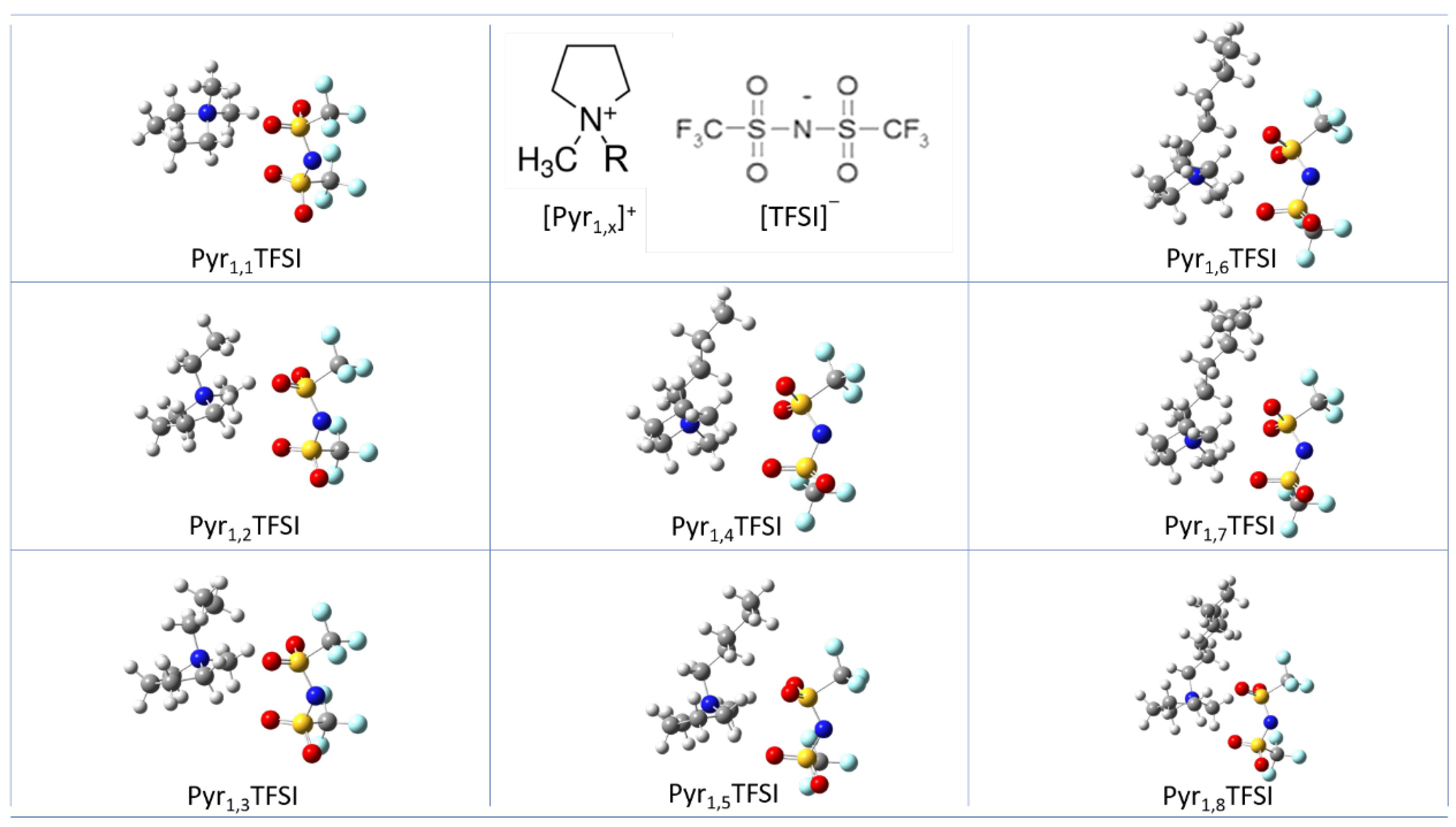
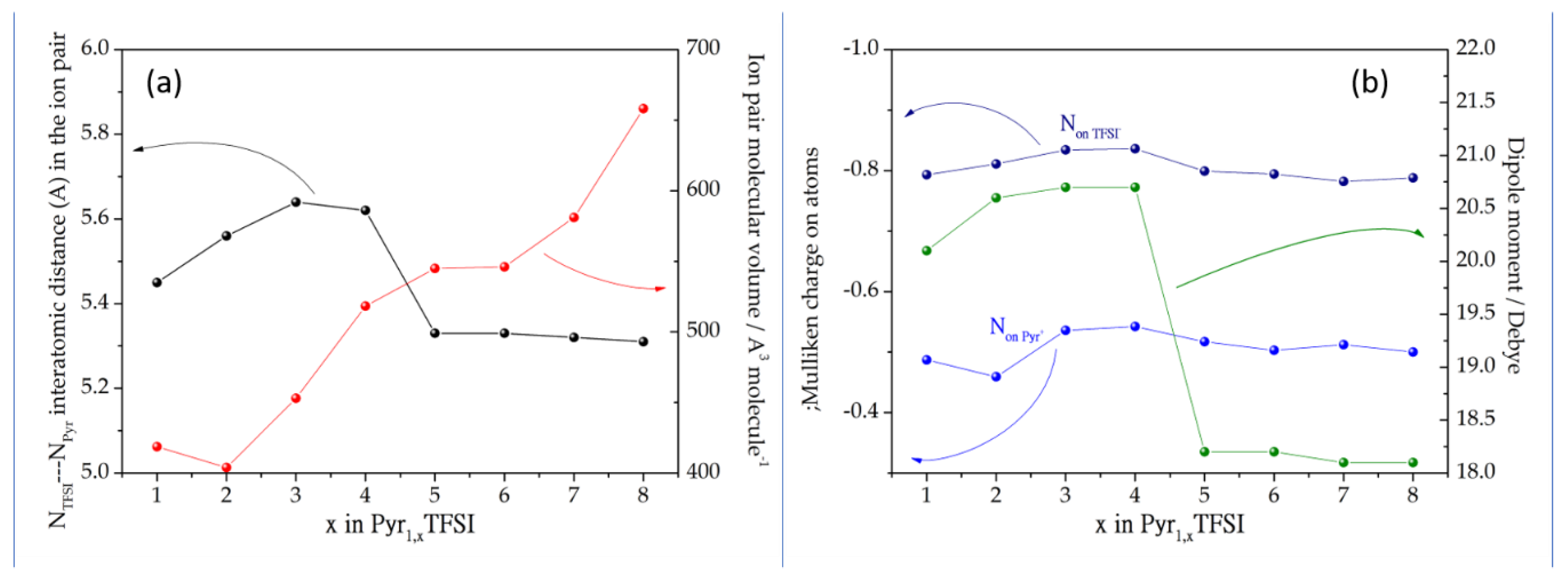
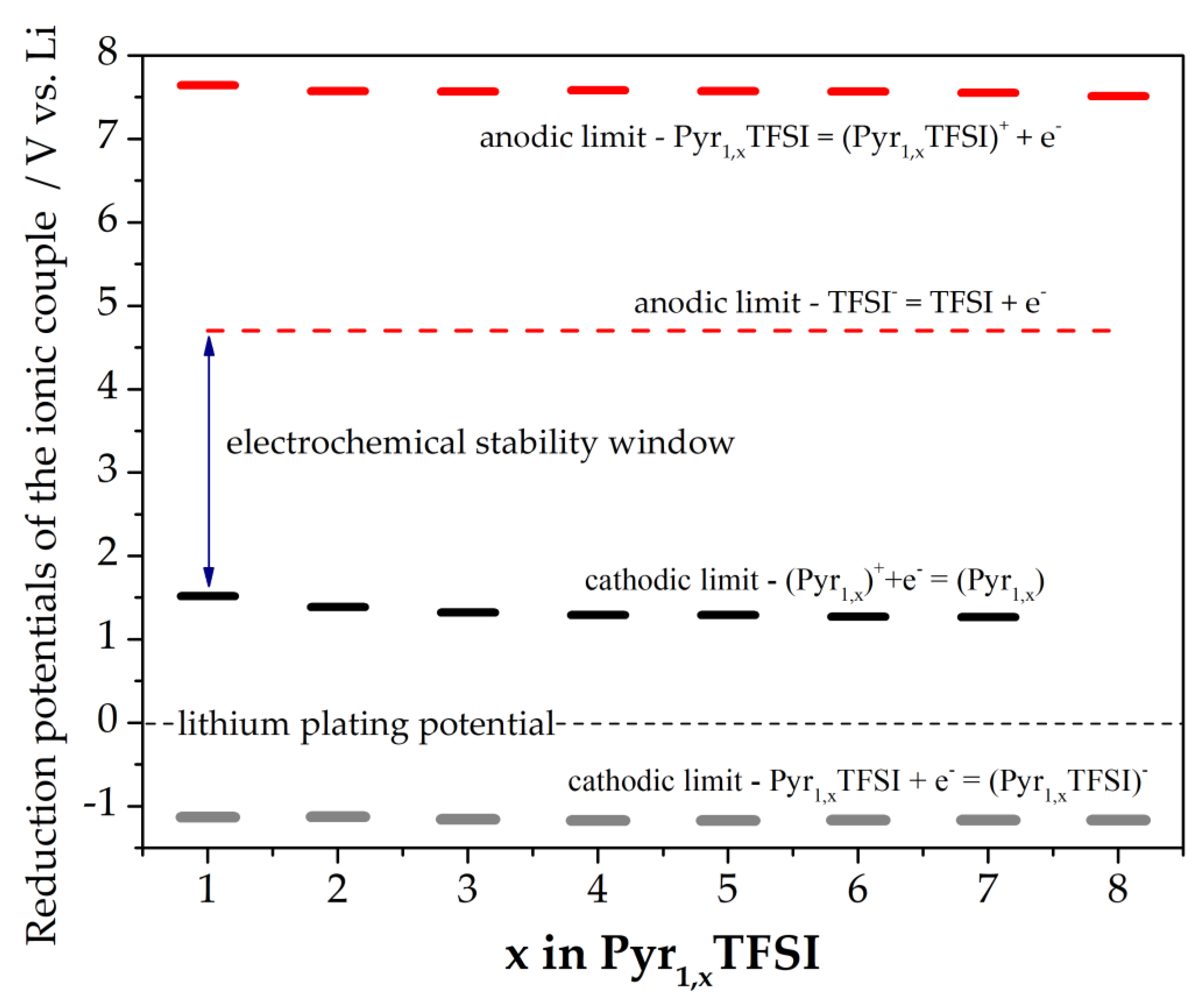

{kind=link}
{kind=link}
{kind=link}
| Dissociation Reaction | Gibbs Energy of Dissociation at 298 K |
|---|---|
| Pyr1,1TFSI → Pyr1,1+ + TFSI− | 2.6 |
| Pyr1,2TFSI → Pyr1,1+ + TFSI− | 4.7 |
| Pyr1,3TFSI → Pyr1,1+ + TFSI− | 5.9 |
| Pyr1,4TFSI → Pyr1,1+ + TFSI− | 8.7 |
| Pyr1,5TFSI → Pyr1,1+ + TFSI− | 3.0 |
| Pyr1,6TFSI → Pyr1,1+ + TFSI− | 1.8 |
| Pyr1,7TFSI → Pyr1,1+ + TFSI− | 0.3 |
| Pyr1,8TFSI → Pyr1,1+ + TFSI− | 0.4 |
| LiTFSI → Li+ + TFSI− | 540 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brutti, S. Pyr1,xTFSI Ionic Liquids (x = 1–8): A Computational Chemistry Study. Appl. Sci. 2020, 10, 8552. https://doi.org/10.3390/app10238552
Brutti S. Pyr1,xTFSI Ionic Liquids (x = 1–8): A Computational Chemistry Study. Applied Sciences. 2020; 10(23):8552. https://doi.org/10.3390/app10238552
Chicago/Turabian StyleBrutti, Sergio. 2020. "Pyr1,xTFSI Ionic Liquids (x = 1–8): A Computational Chemistry Study" Applied Sciences 10, no. 23: 8552. https://doi.org/10.3390/app10238552
APA StyleBrutti, S. (2020). Pyr1,xTFSI Ionic Liquids (x = 1–8): A Computational Chemistry Study. Applied Sciences, 10(23), 8552. https://doi.org/10.3390/app10238552