Pyr1,xTFSI Ionic Liquids (x = 1–8): A Computational Chemistry Study


Abstract
Featured Application
Abstract
1. Introduction
2. Materials and Methods
3. Results
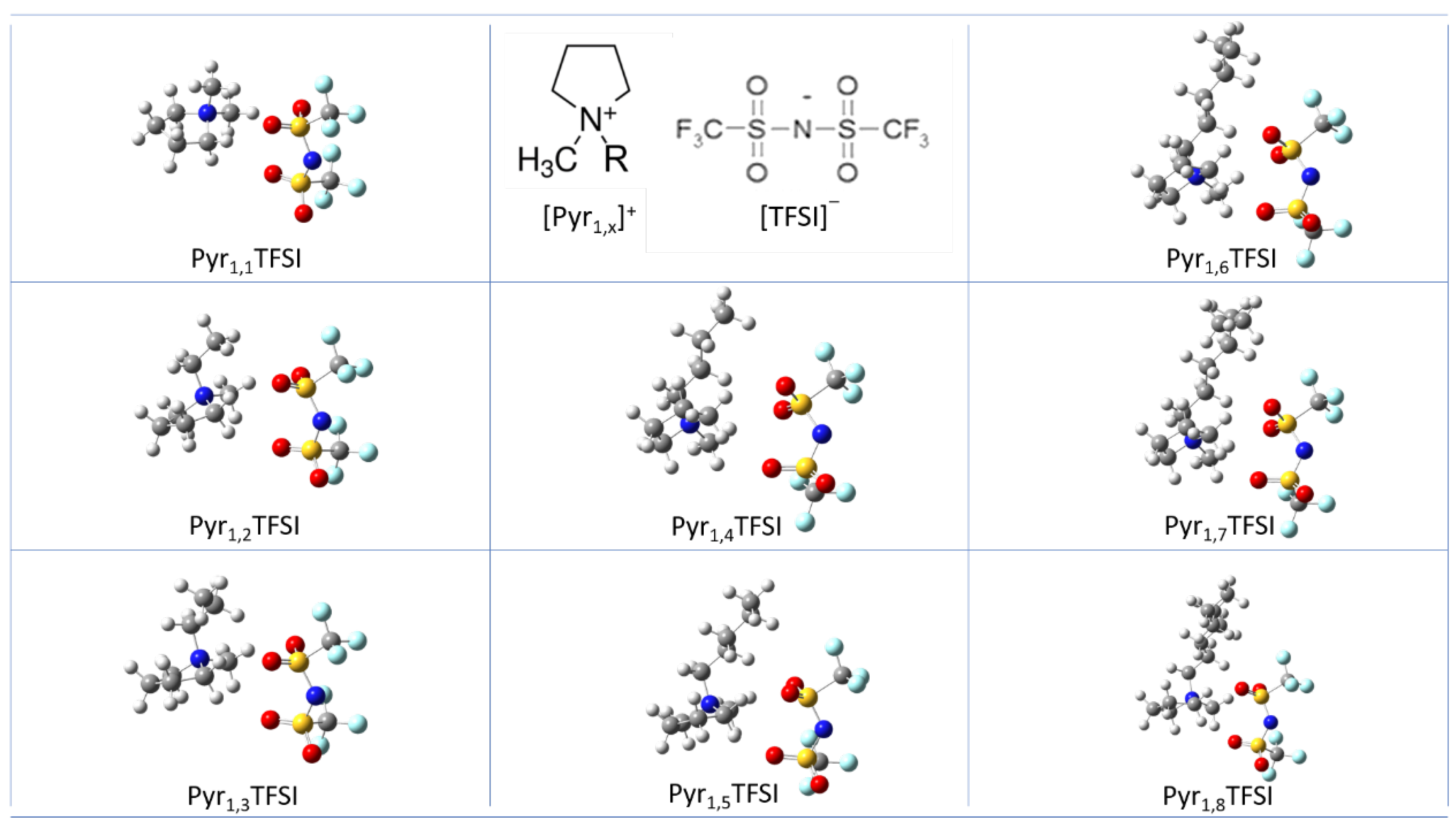
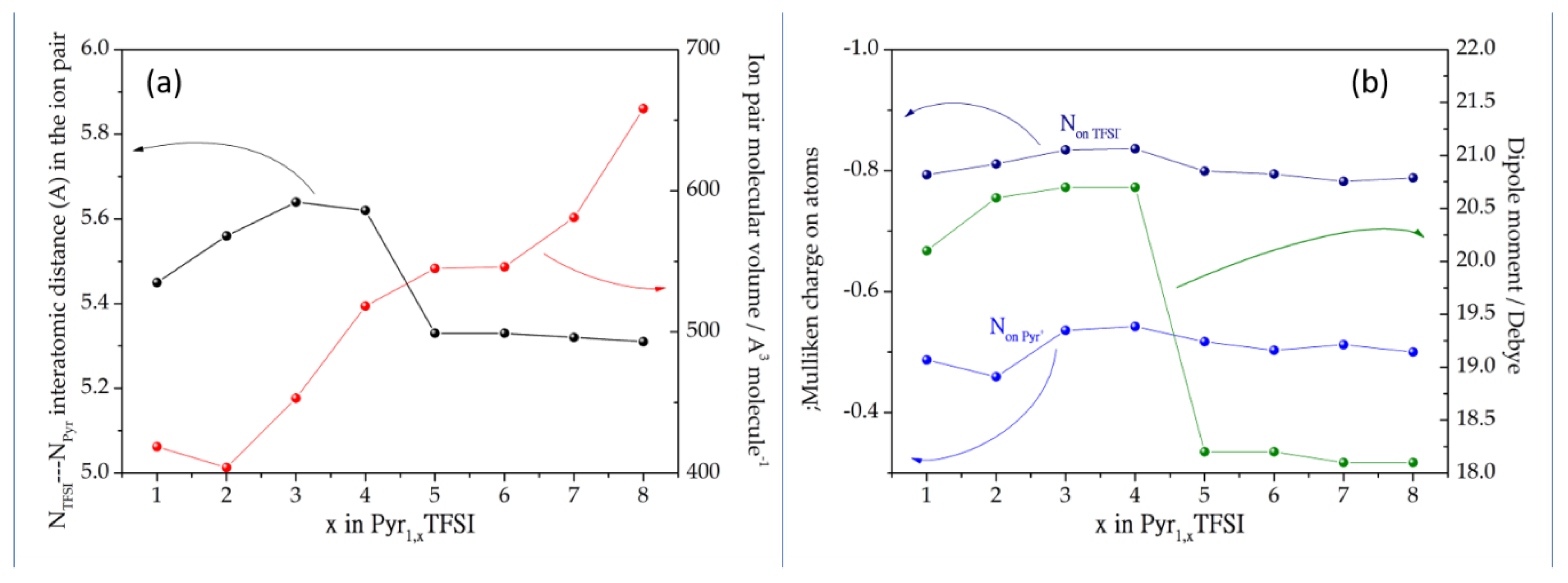
3.1. Ion Pairs Structure
3.2. Dissociation Thermodynamics
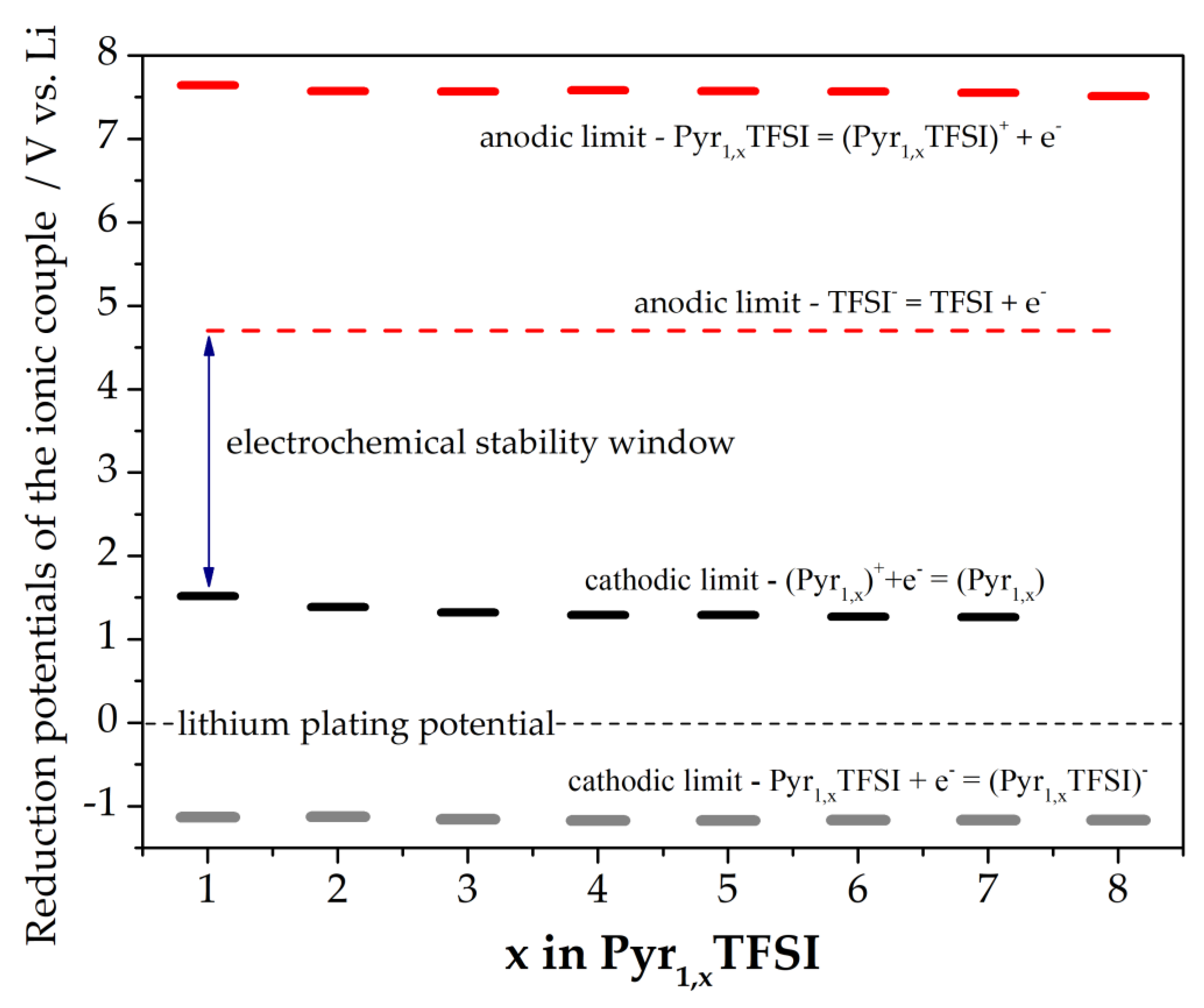
3.3. Electrochemical Stability
4. Conclusions
Supplementary Materials
Funding
Conflicts of Interest
References
- Sandhu, J.S. Recent advances in ionic liquids: Green unconventional solvents of this century: Part I. Green Chem. Lett. Rev. 2011, 4, 289–310. [Google Scholar] [CrossRef]
- Lei, Z.; Chen, B.; Koo, Y.M.; Macfarlane, D.R. Introduction: Ionic Liquids. Chem. Rev. 2017, 117, 6633–6635. [Google Scholar] [CrossRef] [PubMed]
- Lewandowski, A.; Świderska-Mocek, A. Ionic liquids as electrolytes for Li-ion batteries—An overview of electrochemical studies. J. Power Sources 2009, 194, 601–609. [Google Scholar] [CrossRef]
- Kalhoff, J.; Eshetu, G.G.; Bresser, D.; Passerini, S. Safer electrolytes for lithium-ion batteries: State of the art and perspectives. ChemSusChem 2015, 8, 2154–2175. [Google Scholar] [CrossRef]
- Armand, M.; Endres, F.; MacFarlane, D.R.; Ohno, H.; Scrosati, B. Ionic-liquid materials for the electrochemical challenges of the future. Nat. Mater. 2009, 8, 621–629. [Google Scholar] [CrossRef]
- Suo, L.; Hu, Y.S.; Li, H.; Armand, M.; Chen, L. A new class of Solvent-in-Salt electrolyte for high-energy rechargeable metallic lithium batteries. Nat. Commun. 2013, 4, 1481. [Google Scholar] [CrossRef]
- Johansson, P.; Fast, L.E.; Matic, A.; Appetecchi, G.B.; Passerini, S. The conductivity of pyrrolidinium and sulfonylimide-based ionic liquids: A combined experimental and computational study. J. Power Sources 2010, 195, 2074–2076. [Google Scholar] [CrossRef]
- Brutti, S.; Navarra, M.A.; Maresca, G.; Panero, S.; Manzi, J.; Simonetti, E.; Appetecchi, G.B. Ionic liquid electrolytes for room temperature sodium battery systems. Electrochim. Acta 2019, 306, 317–326. [Google Scholar] [CrossRef]
- Lombardo, L.; Brutti, S.; Navarra, M.A.; Panero, S.; Reale, P. Mixtures of ionic liquid e Alkylcarbonates as electrolytes for safe lithium-ion batteries. J. Power Sources 2013, 227, 8–14. [Google Scholar] [CrossRef]
- Celeste, A.; Silvestri, L.; Agostini, M.; Sadd, M.; Palumbo, S.; Panda, J.K.; Matic, A.; Pellegrini, V.; Brutti, S. Enhancement of Functional Properties of Liquid Electrolytes for Lithium-Ion Batteries by Addition of Pyrrolidinium-Based Ionic Liquids with Long Alkyl-Chains. Batter. Supercaps 2020, 3, 1051–1068. [Google Scholar] [CrossRef]
- Arbizzani, C.; Gabrielli, G.; Mastragostino, M. Thermal stability and flammability of electrolytes for lithium-ion batteries. J. Power Sources 2011, 196, 4801–4805. [Google Scholar] [CrossRef]
- Johansson, P.; Jacobsson, P. Rational design of electrolyte components by ab initio calculations. J. Power Sources 2006, 153, 336–344. [Google Scholar] [CrossRef]
- Matsumoto, R.; Thompson, M.W.; Cummings, P.T. Ion Pairing Controls Physical Properties of Ionic Liquid-Solvent Mixtures. J. Phys. Chem. B 2019, 123, 9944–9955. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Russina, O.; Triolo, A. Ionic Liquids and Neutron Scattering. In Experimental Methods in the Physical Sciences; Academic Press: Cambridge, MA, USA, 2017; Volume 49, pp. 213–278. [Google Scholar]
- Russina, O.; lo Celso, F.; Plechkova, N.; Jafta, C.J.; Appetecchi, G.B.; Triolo, A. Mesoscopic organization in ionic liquids. Top. Curr. Chem. 2017, 375, 58. [Google Scholar] [CrossRef] [PubMed]
- Castiglione, F.; Moreno, M.; Raos, G.; Famulari, A.; Mele, A.; Appetecchi, G.B.; Passerini, S. Structural organization and transport properties of novel pyrrolidinium-based ionic liquids with perfluoroalkyl sulfonylimide anions. J. Phys. Chem. B 2009, 113, 10750–10759. [Google Scholar] [CrossRef]
- Carboni, M.; Spezia, R.; Brutti, S. Perfluoroalkyl-Fluorophosphate Anions for High Voltage Electrolytes in Lithium Cells: DFT Study. J. Phys. Chem. C 2014, 118, 24221–24230. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639. [Google Scholar]
- Angenendt, K.; Johansson, P. Ionic liquid structured from large density functional theory calculations using mindless configurations. J. Phys. Chem. C 2010, 114, 20577–20582. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Weingärtner, H. The static dielectric permittivity of ionic liquids. J. Mol. Liq. 2014, 192, 185–190. [Google Scholar] [CrossRef]
- Singh, T.; Kumar, A. Static dielectric constant of room temperature ionic liquids: Internal pressure and cohesive energy density approach. J. Phys. Chem. B 2008, 112, 12968–12972. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.; Coote, M.L.; Cramer, C.J.; Truhlar, D. Theoretical calculation of reduction potentials. In Organic Electrochemistry: Revised and Expanded; CRC Press: Boca Raton, FL, USA, 2015; Volume 6. [Google Scholar]
- Winget, P.; Cramer, C.J.; Truhlar, D.G. Computation of equilibrium oxidation and reduction potentials for reversible and dissociative electron-transfer reactions in solution. Theor. Chem. Acc. 2004, 112, 217–227. [Google Scholar] [CrossRef]
- Jónsson, E.; Johansson, P.; Xu, K. Electrochemical oxidation stability of anions for modern battery electrolytes: A CBS and DFT study. Phys. Chem. Chem. Phys. 2015, 17, 3697–3703. [Google Scholar] [CrossRef]
- Brutti, S.; Simonetti, E.; De Francesco, M.; Sarra, A.; Paolone, A.; Palumbo, O.; Fantini, S.; Lin, R.; Falagayrat, A.; Choi, H.; et al. Ionic liquid electrolytes for high-voltage lithium batteries. J. Sources 2020, 479, 238791. [Google Scholar]
- Bellusci, M.; Simonetti, E.; De Francesco, M.; Appetecchi, G.B. Ionic liquid electrolytes for safer and more reliable sodium battery systems. Appl. Sci. 2020, 10, 6323. [Google Scholar] [CrossRef]
- Howlett, P.C.; MacFarlane, D.R.; Hollenkamp, A.F. High lithium metal cycling efficiency in a room-temperature ionic liquid. Electrochem. Solid State Lett. 2004, 7, A97–A101. [Google Scholar] [CrossRef]
- Kim, G.T.; Appetecchi, G.B.; Montanarino, M.; Alessandrini, F.; Passerini, S. Long-term cyclability of lithium metal electrodes in ionic liquid-based electrolytes at room temperature. ECS Trans. 2010, 25, 12138. [Google Scholar] [CrossRef]
- Randstrom, S.; Appetecchi, G.B.; Lagergren, C.; Moreno, A.; Passerini, S. The Influence of aur and its components on the cathodic stability of N-butyl-N-methylpyrrolidinium bis(trifluoromethanesulphonyl)imide. Electrochim. Acta 2007, 53, 1837–1842. [Google Scholar] [CrossRef]
- Randstrom, S.; Montanino, M.; Appetecchi, G.B.; Legergren, C.; Moreno, A.; Passerini, S. Effect of water and oxygen traces on the cathodic stability of N-alkyl-N-methylpyrrolidinium bis(trifluoromethanesulphonyl9imide. Electrochim. Acta 2008, 53, 6397–6401. [Google Scholar] [CrossRef]
- Appetecchi, G.B.; Montanino, M.; Passerini, S. Ionic liquid-based electrolytes for high-energy, safer lithium batteries. ACS Symp. Ser. 2012, 1117, 67–128. [Google Scholar]
- Lee, W.; Muhammad, S.; Sergey, C.; Lee, H.; Yoon, J.; Kang, Y.; Yoon, W. Advances in the Cathode Materials for Lithium Rechargeable Batteries. Angew. Chem. Int. Ed. 2020, 59, 2578–2605. [Google Scholar] [CrossRef] [PubMed]
- Sun, X. Advances in spinel Li4Ti5O12 anode materials for lithium-ion batteries. New J. Chem. 2014, 39, 38–63. [Google Scholar] [CrossRef]
- Hassoun, J.; Scrosati, B. Review—Advances in Anode and Electrolyte Materials for the Progress of Lithium-Ion and beyond Lithium-Ion Batteries. J. Electrochem. Soc. 2015, 162, A2582–A2588. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Dissociation Reaction | Gibbs Energy of Dissociation at 298 K |
|---|---|
| Pyr1,1TFSI → Pyr1,1+ + TFSI− | 2.6 |
| Pyr1,2TFSI → Pyr1,1+ + TFSI− | 4.7 |
| Pyr1,3TFSI → Pyr1,1+ + TFSI− | 5.9 |
| Pyr1,4TFSI → Pyr1,1+ + TFSI− | 8.7 |
| Pyr1,5TFSI → Pyr1,1+ + TFSI− | 3.0 |
| Pyr1,6TFSI → Pyr1,1+ + TFSI− | 1.8 |
| Pyr1,7TFSI → Pyr1,1+ + TFSI− | 0.3 |
| Pyr1,8TFSI → Pyr1,1+ + TFSI− | 0.4 |
| LiTFSI → Li+ + TFSI− | 540 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brutti, S. Pyr1,xTFSI Ionic Liquids (x = 1–8): A Computational Chemistry Study. Appl. Sci. 2020, 10, 8552. https://doi.org/10.3390/app10238552
Brutti S. Pyr1,xTFSI Ionic Liquids (x = 1–8): A Computational Chemistry Study. Applied Sciences. 2020; 10(23):8552. https://doi.org/10.3390/app10238552
Chicago/Turabian StyleBrutti, Sergio. 2020. "Pyr1,xTFSI Ionic Liquids (x = 1–8): A Computational Chemistry Study" Applied Sciences 10, no. 23: 8552. https://doi.org/10.3390/app10238552
APA StyleBrutti, S. (2020). Pyr1,xTFSI Ionic Liquids (x = 1–8): A Computational Chemistry Study. Applied Sciences, 10(23), 8552. https://doi.org/10.3390/app10238552