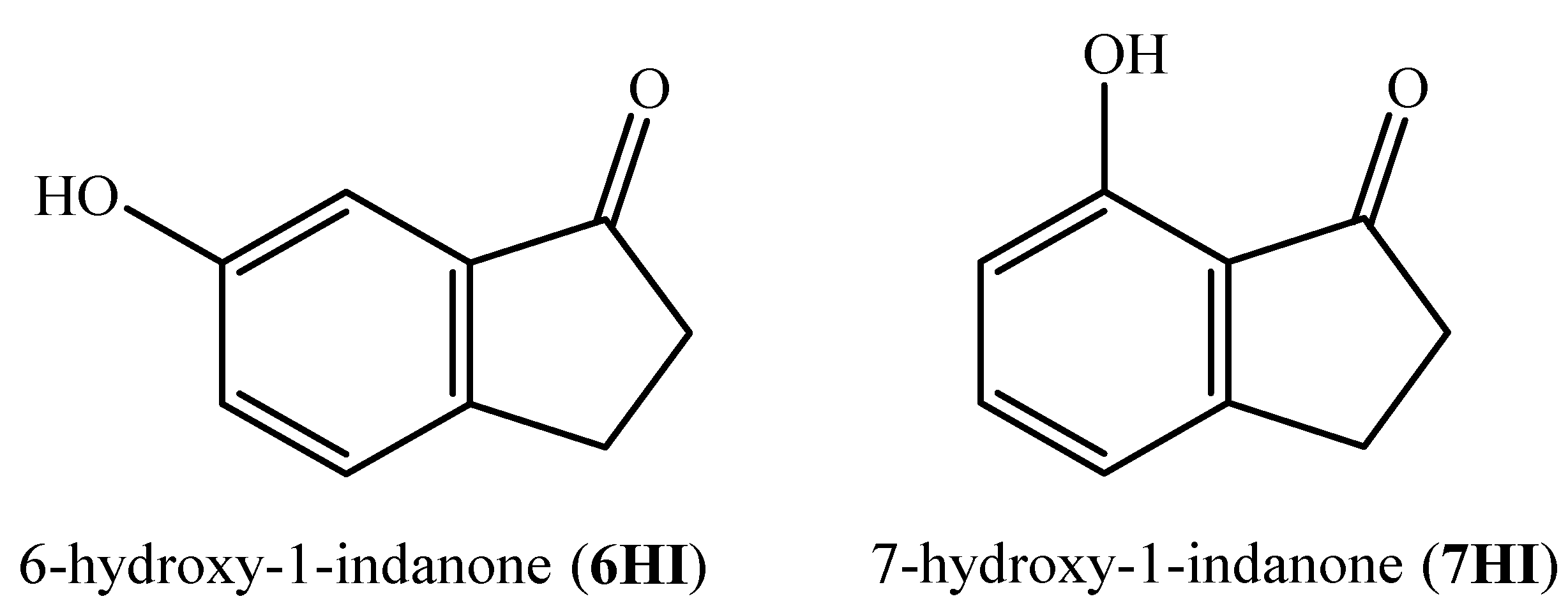
Energetic and Structural Studies of Two Biomass-Derived Compounds: 6- and 7-hydroxy-1-indanones



Abstract
Featured Application
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Differential Scanning Calorimetry
2.2.2. Combustion Calorimetry
2.2.3. Calvet Microcalorimetry
2.2.4. Computational Approach
3. Results and Discussion
3.1. Temperatures and Enthalpies of Fusion
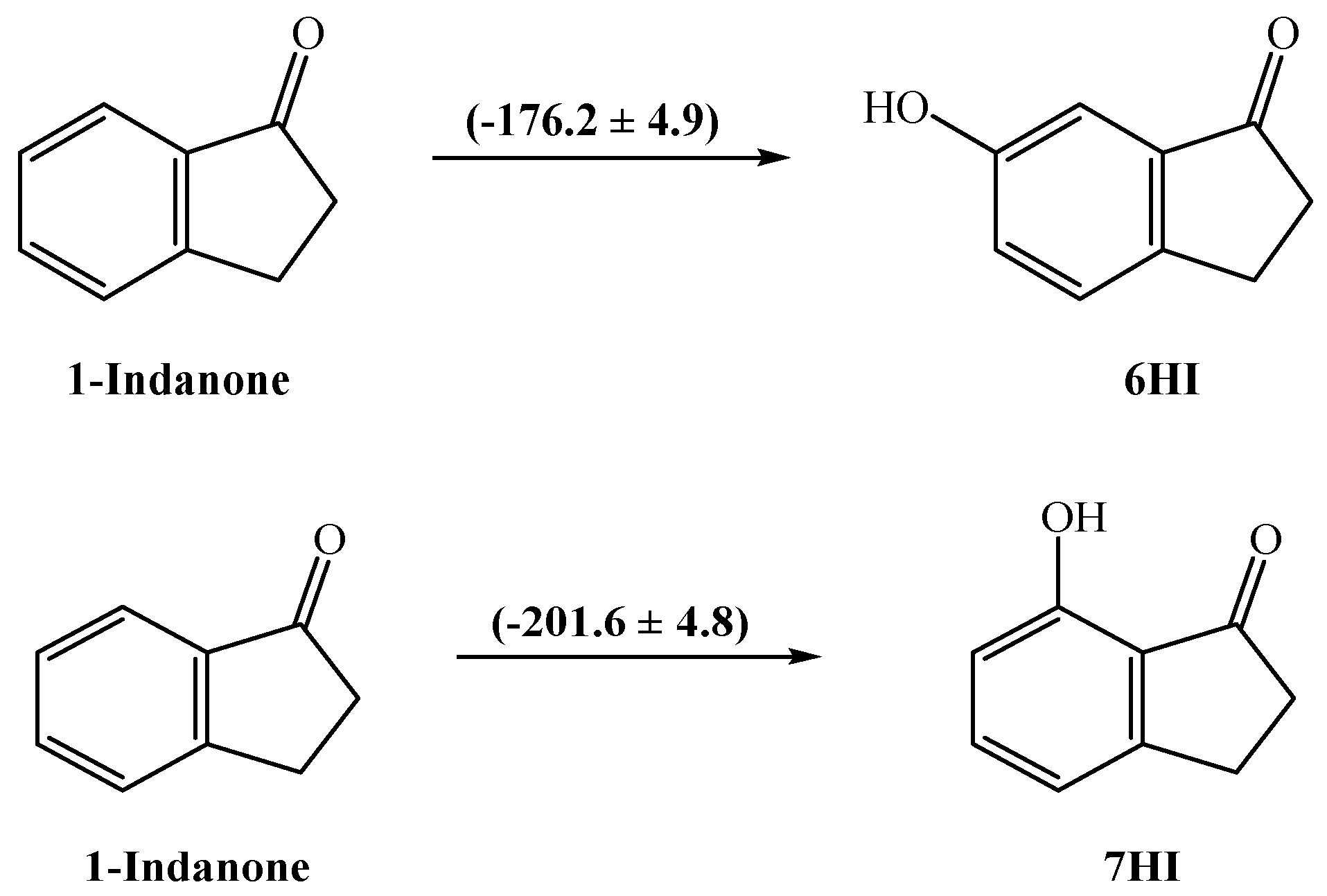
3.2. Enthalpies of Formation in the Crystalline Phase
3.3. Enthalpies of Sublimation
3.4. Conformational Analysis
3.5. Experimental and Computational Enthalpies of Formation in the Gaseous Phase
3.6. Calculated Enthalpic Increments (H → OH) and Analysis of the Intramolecular H-Bond Interaction


4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Freitas, V.L.S.; Lima, A.C.M.O.; Sapei, E.; Ribeiro da Silva, M.D.M.C. Comprehensive thermophysical and thermochemical studies of vanillyl alcohol. J. Chem. Thermodyn. 2016, 102, 287–292. [Google Scholar] [CrossRef]
- Silva, A.L.R.; Lima, A.C.M.O.; Ribeiro da Silva, M.D.M.C. Energetic characterization of indanone derivatives involved in biomass degradation. J. Therm. Anal. Calorim. 2018, 134, 1267–1276. [Google Scholar] [CrossRef]
- Kohl, T.; Sapei, E.; Rocha, I.M.; Galvão, T.L.P.; Ribeiro da Silva, M.D.M.C.; Ribeiro da Silva, M.A.V. Modeling of fast pyrolysis of wood for prediction of bio-oil composition. In Proceedings of the Asia Pacific Confederation of Chemical Engineering Congress 2015: APCChE 2015, Incorporating CHEMECA, Melbourne, Australia, 26 September–1 October 2015; Engineers Australia: Melbourne, Australia; p. 2410. [Google Scholar]
- Rocha, I.M.; Galvão, T.L.P.; Sapei, E.; Ribeiro da Silva, M.D.M.C.; Ribeiro da Silva, M.A.V. Levoglucosan: A calorimetric, thermodynamic, spectroscopic, and computational investigation. J. Chem. Eng. Data. 2013, 58, 1813–1821. [Google Scholar] [CrossRef]
- Goldberg, R.N.; Schliesser, J.; Mittal, A.; Decker, S.R.; Santos, A.F.L.O.M.; Freitas, V.L.S.; Urbas, A.; Lang, B.E.; Heiss, C.; Ribeiro da Silva, M.D.M.C. A thermodynamic investigation of the cellulose allomorphs: Cellulose(am), cellulose Ib(cr), cellulose II(cr), and cellulose III(cr). J. Chem. Thermodyn. 2015, 81, 184–226. [Google Scholar] [CrossRef]
- Goldberg, R.N.; Ribeiro da Silva, M.A.V.; Ribeiro da Silva, M.D.M.C.; Ferreira, A.I.L.; Shi, Q.; Woodfield, B.F. Thermochemistry of α-D-xylose(cr). J. Chem. Thermodyn. 2013, 58, 20–28. [Google Scholar]
- Silva, A.L.R.; Moura, C.; Ribeiro da Silva, M.D.M.C. (2019) Energetic vs structural study of two biomass degradation derivatives: 2-cyclopentenone and 3-methyl-2-cyclopentenone. J. Chem. Thermodyn. 2019, 132, 390–396. [Google Scholar] [CrossRef]
- Chou, P.-T.; Martinez, M.L.; Shannon, L. Studer Role of Triplet States in the Reverse Proton Transfer Mechanism of 7-Hydroxy-1-indanone. J. Phys. Chem. 1991, 95, 10306–10310. [Google Scholar] [CrossRef]
- Graña, A.M.; Rios, M.A.; Rodriguez, J. Ab initio study of the structure and tautomerism in 7-hydroxy-14ndanone. J. Mol. Struct. (Theochem) 1991, 226, 303–306. [Google Scholar] [CrossRef]
- Sobolewski, A.L.; Domcke, W. Ab initio potential-energy functions for excited state intramolecular proton transfer: A comparative study of o-hydroxybenzaldehyde, salicylic acid and 7-hydroxy-1-indanone. Phys. Chem. Chem. Phys. 1999, 1, 3065–3072. [Google Scholar] [CrossRef]
- Tang, K.C.; Chang, M.J.; Lin, T.Y.; Pan, H.A.; Fang, T.C.; Chen, K.Y.; Chou, P.T. Fine tuning the energetics of excited-state intramolecular proton transfer (ESIPT): White light generation in a single ESIPT system. J. Am. Chem. Soc. 2011, 133, 17738–17745. [Google Scholar] [CrossRef]
- Liu, H.; Cheng, X.; Zhang, H.; Wang, Y.; Zhang, H.; Yamaguchi, S. ESIPT-active organic compounds with white luminescence based on crystallization-induced keto emission (CIKE). Chem. Comm. 2017, 53, 7832–7835. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, R.; Xu-wu, A.; Chickos, J.S.; Leitão, M.L.P.; Roux, M.V.; Torres, L.A. Reference materials for calorimetry and differential thermal analysis. Thermochim. Acta 1999, 331, 93–204. [Google Scholar] [CrossRef]
- Ribeiro da Silva, M.A.V.; Ribeiro da Silva, M.D.M.C.; Pilcher, G. The construction, calibration and use of a new high-precision static-bomb calorimeter. Rev. Port. Quím. 1984, 26, 163–172. [Google Scholar]
- Ribeiro da Silva, M.A.V.; Ribeiro da Silva, M.D.M.C.; Pilcher, G. Enthalpies of combustion of 1, 2-dihydroxybenzene and of six alkylsubstituted 1, 2-dihydroxybenzenes. J. Chem. Thermodyn. 1984, 16, 1149–1155. [Google Scholar] [CrossRef]
- Hubbard, W.N.; Scott, D.W.; Waddington, G. Standard states and corrections for combustions in a bomb at constant volume. In Experimental Thermochemistry; Rossini, F.D., Ed.; Interscience: New York, NY, USA, 1956; Volume 1, pp. 75–128. [Google Scholar]
- Paulechka, E.; Riccardi, D. NIST Standard Reference Database 206: Combustion Calorimetry Tool; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2020. Available online: https://trc.nist.gov/cctool/ (accessed on 1 June 2020).
- Adedeji, F.A.; Brown, D.L.S.; Connor, J.A.; Leung, W.L.; Paz-Andrade, I.M.; Skinner, H.A. Thermochemistry of arene chromium tricarbonyls and the strenghts of arene-chromium bonds. J. Organomet. Chem. 1975, 97, 221–228. [Google Scholar] [CrossRef]
- Santos, L.M.N.B.F.; Schröder, B.; Fernandes, O.O.P.; Ribeiro da Silva, M.A.V. Measurement of enthalpies of sublimation by drop method in a Calvet type calorimeter: Design and test of a new system. Thermochim. Acta 2004, 415, 15–20. [Google Scholar] [CrossRef]
- Baboul, A.G.; Curtiss, L.A.; Redfern, P.C.; Raghavachari, K. Gaussian-3 theory using density functional geometries and zero-point energies. J. Chem. Phys. 1999, 110, 7650–7657. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.A.J.; Stratmann, R.E.; Burant, J.C. Gaussian03, revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Rossini, F.D. (Ed.) Assignment of uncertainties to thermochemical data. In Experimental Thermochemistry; Interscience: New York, NY, USA, 1956; Volume 1, pp. 297–325. [Google Scholar]
- Olofson, G. Assignment of uncertainties. In Combustion Calorimetry; Sunner, S., Mansson, M., Eds.; Pergamon Press: Oxford, UK, 1979; Volume 1, pp. 137–159. [Google Scholar]
- Cox, J.D.; Wagman, D.D.; Medvedev, V.A. CODATA Key Values for Thermodynamics; Hemisphere: New York, NY, USA, 1979. [Google Scholar]
- Merrick, J.P.; Moran, D.; Radom, L. An evaluation of harmonic vibrational frequency scale factors. J. Phys. Chem. A 2007, 111, 11683–11700. [Google Scholar] [CrossRef]
- Matos, M.A.R.; Sousa, C.C.S.; Morais, V.M.F. Energetics of Hydroxytetralones: A Calorimetric and Computational Thermochemical Study. J. Chem. Eng. Data 2009, 54, 2189–2194. [Google Scholar] [CrossRef]
- Pedley, J.P. Thermochemical Data and Structures of Organic Compounds; Thermodynamics Research Centre: College Station, TX, USA, 1994.
- Kovács, A.; Szabó, A.; Hargittai, I. Structural Characteristics of Intramolecular Hydrogen Bonding in Benzene Derivatives. Acc. Chem. Res. 2002, 35, 887–894. [Google Scholar] [CrossRef]
- Silva, A.L.R.; Matos, M.A.R.; Morais, V.M.F.; Ribeiro da Silva, M.D.M.C. Thermochemical and conformational study of optical active phenylbenzazole derivatives. J. Chem. Thermodyn. 2018, 116, 7–20. [Google Scholar] [CrossRef]
- Estácio, S.G.; Couto, P.C.; Cabral, B.J.C.; Minas da Piedade, M.E.; Simões, M.J.A. Energetics of Intramolecular Hydrogen Bonding in Di-substituted Benzenes by the ortho-para Method. J. Phys. Chem. A 2004, 108, 10834–10843. [Google Scholar] [CrossRef]
- Korth, H.G.; Heer, M.I.; Mulder, P. A DFT study on intramolecular hydrogen bonding in 2-substituted phenols: Conformations, enthalpies, and correlation with solute parameters. J. Phys. Chem. A 2002, 106, 8779–8789. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | |||
|---|---|---|---|
| 6HI | 426.8 ± 0.9 | 430.0 a | 21.7 ± 1.0 |
| 7HI | 382.8 ± 0.8 | 384.8 a | 25.6 ± 0.5 |
| Compound | ||||
|---|---|---|---|---|
| 6HI | −29,211.1 ± 6.8 | −4327.9 ± 2.3 | −4330.3 ± 2.3 | −354.6 ± 2.6 |
| 7HI | −29,192.6 ± 5.6 | −4325.1 ± 2.0 | −4327.6 ± 2.0 | −357.3 ± 2.3 |
| Compound | No. Exp. | Texp/(K) | |||
|---|---|---|---|---|---|
| 6HI | 6 | 416.87 ± 0.04 | 136.58 ± 0.52 | 22.18 ± 0.01 | 114.4 ± 1.7 |
| 7HI | 6 | 375.50 ± 0.05 | 105.05 ± 0.48 | 13.39 ± 0.01 | 91.7 ± 2.0 |
| Compound | |||
|---|---|---|---|
| 6HI | −354.6 ± 2.6 | 114.4 ± 1.7 | −240.2 ± 3.1 |
| 7HI | −357.3 ± 2.3 | 91.7 ± 2.0 | −265.6 ± 3.0 |
| Reaction | ||||
|---|---|---|---|---|
 | (7) | 6HI | 8933.89 | −241.5 |
| 7HI | 8959.97 | −267.6 | ||
 | (8) | 6HI | −0.96 | −242.0 |
| 7HI | 25.12 | −268.1 | ||
 | (9) | 6HI | 35.08 | −239.8 |
| 7HI | 61.16 | −265.9 | ||
 | (10) | 6HI | −1.80 | −242.9 |
| 7HI | 24.28 | −269.0 | ||
 | (11) | 6HI | 0.11 | −240.1 |
| 7HI | 26.19 | −266.2 | ||
 | (12) | 6HI | −1.47 | −241.7 |
| 7HI | 24.61 | −267.8 | ||
 | (13) | 6HI | −1.80 | −237.9 |
| 7HI | 24.28 | −264.0 | ||
| <(6HI, g)> = −(240.9 ± 1.3) kJ·mol−1 <(7HI, g)> = −(266.9 ± 1.3) kJ·mol−1 | ||||
| Gas-Phase Enthalpy of Formation | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | Atom. | Iso. (8) | Iso. (9) | Iso. (10) | Iso. (11) | Iso. (12) | Iso. (13) | Calculated Mean-Value | Experimental |
| 6HI | −241.5 | −242.0 | −239.8 | −242.9 | −240.1 | −241.7 | −237.9 | −240.9 ± 1.3 | −240.2 ± 3.1 |
| 7HI | −267.6 | −268.1 | −265.9 | −269.0 | −266.2 | −267.8 | −264.0 | −266.9 ± 1.3 | −265.6 ± 3.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro da Silva, A.L.; Ribeiro da Silva, M.D.M.C. Energetic and Structural Studies of Two Biomass-Derived Compounds: 6- and 7-hydroxy-1-indanones. Appl. Sci. 2020, 10, 8512. https://doi.org/10.3390/app10238512
Ribeiro da Silva AL, Ribeiro da Silva MDMC. Energetic and Structural Studies of Two Biomass-Derived Compounds: 6- and 7-hydroxy-1-indanones. Applied Sciences. 2020; 10(23):8512. https://doi.org/10.3390/app10238512
Chicago/Turabian StyleRibeiro da Silva, Ana Luisa, and Maria D. M. C. Ribeiro da Silva. 2020. "Energetic and Structural Studies of Two Biomass-Derived Compounds: 6- and 7-hydroxy-1-indanones" Applied Sciences 10, no. 23: 8512. https://doi.org/10.3390/app10238512
APA StyleRibeiro da Silva, A. L., & Ribeiro da Silva, M. D. M. C. (2020). Energetic and Structural Studies of Two Biomass-Derived Compounds: 6- and 7-hydroxy-1-indanones. Applied Sciences, 10(23), 8512. https://doi.org/10.3390/app10238512