An Empirical Evaluation of Nuclei Segmentation from H&E Images in a Real Application Scenario



Abstract
1. Introduction
2. Related Works
3. Open Issues and Goal of this Work
4. Experimental Evaluation
4.1. Nuclei Segmentation Methods
4.2. Datasets
4.3. Experimental Set-Up
5. Results
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CAD | computer-aided diagnosis |
| DSSP | decision support system for pathologists |
| LIS | laboratory information system |
| WSI | Whole Slide Image |
| H&E | hematoxylin & eosin |
| GBM | glioblastoma |
| LGG | lower grade glioma |
| HNSC | head and neck squamous cell carcinoma |
| LUSC | lung squamous cell carcinoma |
| DA | domain adaptation |
| UDA | unsupervised domain adaptation |
| CNN | convolutional neural network |
| RCNN | regional CNN |
| FCN | fully connected network |
| HED | holistically-nested edge detector |
| DIMAN | Deep Interval-Marker-Aware Network |
| TP | true positive |
| FN | false negative |
| FP | false positive |
| F1 | F1-score |
| P | precision |
| R | recall |
| ODI | object-level Dice index |
| OHD | object-level Hausdorff distance |
References
- Qi, X.; Xing, F.; Foran, D.J.; Yang, L. Robust segmentation of overlapping cells in histopathology specimens using parallel seed detection and repulsive level set. IEEE Trans. Biomed. Eng. 2012, 59, 754–765. [Google Scholar] [CrossRef] [PubMed]
- Xing, F.; Yang, L. Robust Nucleus/Cell Detection and Segmentation in Digital Pathology and Microscopy Images: A Comprehensive Review. IEEE Rev. Biomed. Eng. 2016, 9, 234–263. [Google Scholar] [CrossRef] [PubMed]
- Dey, P. Cancer Nucleus: Morphology and Beyond. Diagn. Cytopathol. 2010, 38, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Gurcan, M.N.; Boucheron, L.; Can, A.; Madabhushi, A.; Rajpoot, N.; Yener, B. Histopathological image analysis: A review. IEEE Rev. Biomed. Eng. 2009, 2, 147–171. [Google Scholar] [CrossRef]
- Nawaz, S.; Yuan, Y. Computational pathology: Exploring the spatial dimension of tumor ecology. Cancer Lett. 2015, 380, 296–303. [Google Scholar] [CrossRef]
- Chen, J.-M.; Qu, A.-P.; Wang, L.-W.; Yuan, J.-P.; Yang, F.; Xiang, Q.-M.; Maskey, N.; Yang, G.-F.; Liu, J.; Li, Y. New breast cancer prognostic factors identified by computer-aided image analysis of he stained histopathology images. Sci. Rep. 2015, 5, 10690. [Google Scholar] [CrossRef][Green Version]
- Gandomkar, Z.; Brennan, P.C.; Mello-Thoms, C. Computer-based image analysis in breast pathology. J. Pathol. Inform. 2016, 7, 43. [Google Scholar] [CrossRef]
- Veta, M.; Pluim, J.P.W.; van Diest, P.J.; Viergever, M.A. Breast cancer histopathology image analysis: A review. IEEE Trans. Biomed. Eng. 2014, 61, 1400–1411. [Google Scholar] [CrossRef]
- Veta, M.; Diest, P.J.V.; Willems, S.M.; Wang, H.; Madabhushi, A.; Cruz-Roa, A.; Gonzalez, F.; Larsen, A.B.; Vestergaard, J.S.; Dahl, A.B.; et al. Assessment of algorithms for mitosis detection in breast cancer histopathology images. Med. Image Anal. 2015, 20, 237–248. [Google Scholar] [CrossRef]
- Alturkistani, H.A.; Tashkandi, F.M.; Mohammedsaleh, Z.M. Histological stains: A literature review and case study. Glob. J. Health Sci. 2016, 8, 72–79. [Google Scholar] [CrossRef]
- Kapuscinski, J. DAPI: A DNA-Specific Fluorescent Probe. Biotech. Histochem. 1995, 70, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Titford, M. The long history of hematoxylin. Biotech. Histochem. 2005, 80, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Bándi, P.; Geessink, O.; Manson, Q.; van Dijk, M.; Balkenhol, M.; Hermsen, M.; Bejnordi, B.E.; Lee, B.; Paeng, K.; Zhong, A.; et al. From detection of individual metastases to classification of lymph node status at the patient level: The camelyon17 challenge. IEEE Trans. Med. Imaging 2018, 38, 550–560. [Google Scholar] [CrossRef]
- Titford, M.; Bowman, B. What May the Future Hold for Histotechnologists? Who Will Be Affected? What Will Drive the Change? Lab. Med. 2012, 43, 5–10. [Google Scholar] [CrossRef]
- Pantanowitz, L.; Szymas, J.; Yagi, Y.; Wilbur, D. Whole slide imaging for educational purposes. J. Pathol. Inform. 2012, 3, 46. [Google Scholar] [CrossRef]
- Stoler, M.H.; Ronnett, B.M.; Joste, N.E.; Hunt, W.C.; Cuzick, J.; Wheeler, C.M. The Interpretive Variability of Cervical Biopsies and Its Relationship to HPV Status. Am. J. Surg. Pathol. 2015, 39, 729–736. [Google Scholar] [CrossRef]
- Kowal, M.; Filipczuk, P.; Obuchowicz, A.; Korbicz, J.; Monczak, R. Computer-aided diagnosis of breast cancer based on fine needle biopsy microscopic images. Comput. Biol. Med. 2013, 43, 1563–1572. [Google Scholar] [CrossRef]
- Sirinukunwattana, K.; Raza, S.E.A.; Tsang, Y.-w.; Snead, D.R.J.; Cree, I.A.; Rajpoot, N.M. Locality Sensitive Deep Learning for Detection and Classification of Nuclei in Routine Colon Cancer Histology Images. IEEE Trans. Med. Imaging 2016, 35, 1196–1206. [Google Scholar] [CrossRef]
- Xing, F.; Xie, Y.; Yang, L. An automatic learning-based framework for robust nucleus segmentation. IEEE Trans. Med. Imaging 2016, 35, 550–566. [Google Scholar] [CrossRef]
- Gurcan, M.N.; Pan, T.; Shimada, H.; Saltz, J. Image analysis for neuroblastoma classification: Segmentation of cell nuclei. In Proceedings of the International Conference of the IEEE Engineering in Medicine and Biology Society, New York, NY, USA, 30 August–3 September 2006. [Google Scholar] [CrossRef]
- Zhang, C.; Sun, C.; Pham, T.D. Segmentation of clustered nuclei based on concave curve expansions. J. Microsc. 2013, 251, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Verma, R.; Sharma, S.; Bhargava, S.; Vahadane, A.; Sethi, A. A dataset and a technique for generalised nuclear segmentation for computational pathology. IEEE Trans. Med. Imaging 2017, 36, 1550–1560. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Qi, J.; Pan, L.; Wali, S. Integrating deep convolutional neural networks with marker-controlled watershed for overlapping nuclei segmentation in histopathology images. Neurocomputing 2020, 376, 166–179. [Google Scholar] [CrossRef]
- Al-Kofahi, Y.; Lassoued, W.; Lee, W.; Roysam, B. Improved automatic detection and segmentation of cell nuclei in histopathology images. IEEE Trans. Biomed. Eng. 2010, 57, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Janowczyk, A.; Madabhushi, A. Deep learning for digital pathology image analysis: A comprehensive tutorial with selected use cases. J. Pathol. Inform. 2016, 7, 29. [Google Scholar] [CrossRef]
- Xu, J.; Xiang, L.; Liu, Q.; Gilmore, H.; Wu, J.; Tang, J.; Madabhushi, A. Stacked sparse autoencoder (ssae) for nuclei detection on breast cancer histopathology images. IEEE Trans. Med. Imaging 2016, 35, 119–130. [Google Scholar] [CrossRef]
- Irshad, H.; Veillard, A.; Roux, L.; Racoceanu, D. Methods for Nuclei Detection, Segmentation, and Classification in Digital Histopathology: A Review—Current Status and Future Potential. IEEE Rev. Biomed. Eng. 2014, 7, 97–114. [Google Scholar] [CrossRef]
- Naylor, P.; Laé, M.; Reyal, F.; Walter, T. Nuclei segmentation in histopathology images using deep neural networks. In Proceedings of the IEEE 14th International Symposium on Biomedical Imaging, Melbourne, Australia, 18–21 April 2017; pp. 933–936. [Google Scholar]
- Hou, L.; Agarwal, A.; Samaras, D.; Kurc, T.M.; Gupta, R.R.; Saltz, J.H. Robust histopathology image analysis: To label or to synthesise? In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Long Beach, CA, USA, 16–20 June 2019; pp. 8533–8542. [Google Scholar]
- Liu, D.; Zhang, D.; Song, Y.; Zhang, F.; ODonnell, L.; Huang, H.; Chen, M.; Cai, W. Unsupervised Instance Segmentation in Microscopy Images via Panoptic Domain Adaptation and Task Re-Weighting. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Seattle, WA, USA, 16–18 June 2020; pp. 4242–4251. [Google Scholar]
- Yi, F.; Huang, J.; Yang, L.; Xie, Y.; Xiao, G. Automatic extraction of cell nuclei from H&E-stained histopathological images. J. Med. Imaging 2017, 4, 027502. [Google Scholar]
- Boyle, D.P.; McArt, D.G.; Irwin, G.; Wilhelm-Benartzi, C.S.; Lioe, T.F.; Sebastian, E.; McQuaid, S.; Hamilton, P.W.; James, J.A.; Mullan, P.B.; et al. The prognostic significance of the aberrant extremes of p53 immunophenotypes in breast cancer. Histopathology 2014, 65, 340–352. [Google Scholar] [CrossRef]
- Li, J.; Yang, S.; Huang, X.; Da, Q.; Yang, X.; Hu, Z.; Duan, Q.; Wang, C.; Li, H. Signet ring cell detection with a semi-supervised learning framework. In Proceedings of the International Conference on Information Processing in Medical Imaging, Hong Kong, China, 2–7 June 2019; pp. 842–854. [Google Scholar]
- Zhou, Y.; Dou, Q.; Chen, H.; Qin, J.; Heng, P.-A. Sfcn-opi: Detection and fine-grained classification of nuclei using sibling fcn with objectness prior interaction. In Proceedings of the Thirty-Second AAAI Conference on Artificial Intelligence, New Orleans, LA, USA, 2–7 February 2018. [Google Scholar]
- Qu, H.; Riedlinger, G.; Wu, P.; Huang, Q.; Yi, J.; De, S.; Metaxas, D. Joint segmentation and fine-grained classification of nuclei in histopathology images. In Proceedings of the International Symposium on Biomedical Imaging, Venice, Italy, 8–11 April 2019; pp. 900–904. [Google Scholar]
- Naylor, P.; Laé, M.; Reyal, F.; Walter, T. Segmentation of nuclei in histopathology images by deep regression of the distance map. IEEE Trans. Med. Imaging 2018, 38, 448–459. [Google Scholar] [CrossRef]
- Otsu, N. A threshold selection method from gray-level histograms. Automatica 1975, 11, 23–27. [Google Scholar] [CrossRef]
- Huang, D.-C.; Hung, K.-D.; Chan, Y.-K. A computer assisted method for leukocyte nucleus segmentation and recognition in blood smear images. J. Syst. Softw. 2012, 85, 2104–2118. [Google Scholar] [CrossRef]
- Nawandhar, A.A.; Yamujala, L.; Kumar, N. Image segmentation using thresholding for cell nuclei detection of colon tissue. In Proceedings of the International Conference on Advances in Computing, Communications and Informatics (ICACCI), Kochi, India, 10–13 August 2015. [Google Scholar] [CrossRef]
- Meyer, F.; Beucher, S. Morphological segmentation. J. Vis. Commun. Image Rep. 1990, 1, 21–46. [Google Scholar] [CrossRef]
- Chen, X.; Zhou, X.; Wong, S. Automated segmentation, classification, and tracking of cancer cell nuclei in time-lapse microscopy. IEEE Trans. Biomed. Eng. 2006, 53, 762–766. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, H.; Zhou, X. Nuclei Segmentation Using Marker-Controlled Watershed, Tracking Using Mean-Shift, and Kalman Filter in Time-Lapse Microscopy. IEEE Trans. Circuits Syst. 2006, 53, 2405–2414. [Google Scholar] [CrossRef]
- Fatakdawala, H.; Basavanhally, A.; Xu, J.; Bhanot, G.; Ganesan, S.; Feldman, M.; Tomaszewski, J.; Madabhushi, A. Expectation maximisation driven geodesic active contour with overlap resolution (EMaGACOR): Application to lymphocyte segmentationl on breast cancer histopathology. IEEE Trans. Biomed. Eng. 2010, 57, 1676–1689. [Google Scholar] [CrossRef]
- Yan, P.; Zhou, X.; Shah, M.; Wong, S. Automatic segmentation of high-throughput RNAi fluorescent cellular images. IEEE Trans. Inf. Technol. Biomed. 2008, 12, 109–117. [Google Scholar] [CrossRef]
- Kong, H.; Gurcan, M.; Belkacem-Boussaid, K. Partitioning histopathological images: An integrated framework for supervised color-texture segmentation and cell splitting. IEEE Trans. Med. Imaging 2011, 30, 1661–1677. [Google Scholar] [CrossRef]
- Plissiti, M.L.E.; Nikou, C. Overlapping cell nuclei segmentation using a spatially adaptive active physical model. IEEE Trans. Image Process. 2012, 21, 4568–4580. [Google Scholar] [CrossRef]
- Chen, H.; Qi, X.; Yu, L.; Dou, Q.; Qin, J.; Heng, P.-A. Dcan: Deep contour-aware networks for object instance segmentation from histology images. Med. Image Anal. 2017, 36, 135–146. [Google Scholar] [CrossRef]
- Zhu, J.-Y.; Park, T.; Isola, P.; Efros, A.A. Unpaired image-to-image translation using cycle-consistent adversarial networks. In Proceedings of the IEEE International Conference on Computer Vision, Venice, Italy, 22–29 October 2017. [Google Scholar]
- Mahmood, F.; Borders, D.; Chen, R.; McKay, G.N.; Salimian, K.J.; Baras, A.; Durr, N.J. Deep adversarial training for multi-organ nuclei segmentation in histopathology images. IEEE Trans. Med. Imaging 2019, 39, 3257–3267. [Google Scholar] [CrossRef]
- Ganin, Y.; Lempitsky, V. Unsupervised domain adaptation by backpropagation. In Proceedings of the International Conference on Machine Learning (ICML), Lille, France, 6–11 July 2015. [Google Scholar]
- Kim, T.; Jeong, M.; Kim, S.; Choi, S.; Kim, C. Diversify and match: A domain adaptive representation learning paradigm for object detection. In Proceedings of the Computer Vision and Pattern Recognition (CVPR), Long Beach, CA, USA, 16–20 June 2019; pp. 12456–12465. [Google Scholar]
- Pan, S.J.; Yang, Q. A survey on transfer learning. IEEE Trans. Knowl. Data Eng. 2009, 22, 1345–1359. [Google Scholar] [CrossRef]
- Tzeng, E.; Hoffman, J.; Saenko, K.; Darrell, T. Adversarial discriminative domain adaptation. In Proceedings of the Computer Vision and Pattern Recognition (CVPR), Honolulu, HI, USA, 21–26 July 2017; pp. 7167–7176. [Google Scholar]
- Chen, C.; Dou, Q.; Chen, H.; Qin, J.; Heng, P.-A. Synergistic image and feature adaptation: Towards cross-modality domain adaptation for medical image segmentation. In Proceedings of the Association for the Advancement of Artificial Intelligence (AAAI), Honolulu, HI, USA, 29–31 January 2019; pp. 865–872. [Google Scholar]
- Dou, Q.; Ouyang, C.; Chen, C.; Chen, H.; Heng, P.-A. Unsupervised cross-modality domain adaptation of convnets for biomedical image segmentations with adversarial loss. arXiv 2018, arXiv:1804.10916. [Google Scholar]
- Zhang, Y.; Miao, S.; Mansi, T.; Liao, R. Task driven generative modeling for unsupervised domain adaptation: Application to x-ray image segmentation. arXiv 2018, arXiv:1806.07201. [Google Scholar]
- Long, J.; Shelhamer, E.; Darrell, T. Fully convolutional networks for semantic segmentation. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Boston, MA, USA, 7–12 June 2015. [Google Scholar] [CrossRef]
- Xie, S.; Tu, Z. Holistically-nested edge detection. In Proceedings of the IEEE International Conference on Computer Vision (ICCV), Santiago, Chile, 7–13 December 2015. [Google Scholar] [CrossRef]
- Ronneberger, O.; Fischer, P.; Brox, T. U-net: Convolutional networks for biomedical image segmentation. In Medical Image Computing and Computer-Assisted Intervention; Lecture Notes in Computer Science; Springer: Cham, Switzerland, 2015; pp. 234–241. [Google Scholar] [CrossRef]
- He, K.; Gkioxari, G.; Dollár, P.; Girshick, R. Mask r-cnn. In Proceedings of the IEEE International Conference on Computer Vision, Venice, Italy, 22–29 October 2017; pp. 2961–2969. [Google Scholar]
- Xie, X.; Li, Y.; Zhang, M.; Shen, L. Robust segmentation of nucleus in histopathology images via Mask R-CNN. In Brainlesion: Glioma, Multiple Sclerosis, Stroke and Traumatic Brain Injuries; Lecture Notes in Computer Science; Springer: Cham, Switzerland, 2018; Volume 11383. [Google Scholar]
- Wilcoxon, F. Individual comparisons by ranking methods. Biometrics 1945, 1, 80–83. [Google Scholar] [CrossRef]
- Demšar, J. Statistical Comparisons of Classifiers over Multiple Data Sets. J. Mach. Learn. Res. 2006, 7, 1–30. [Google Scholar]





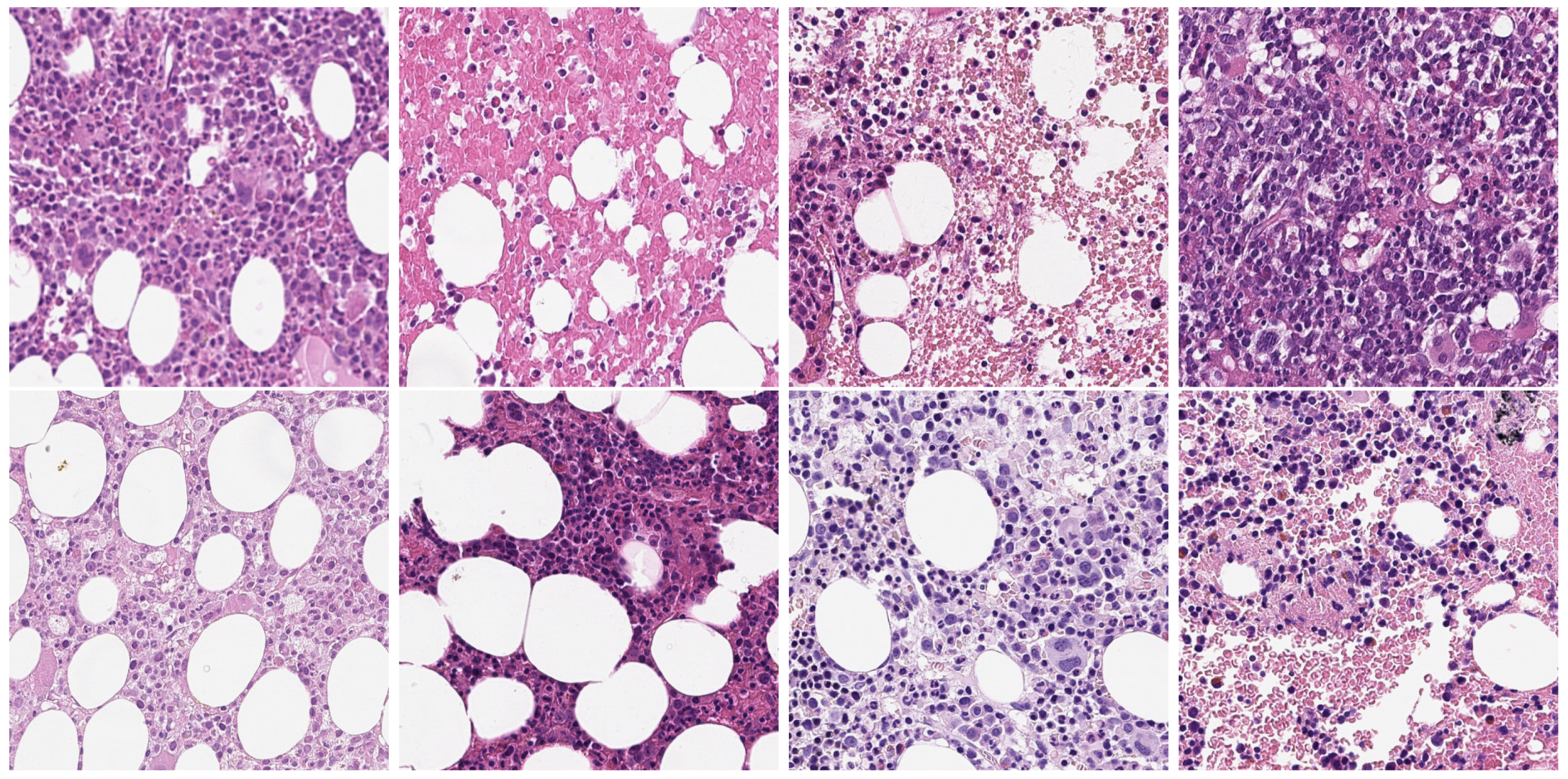{kind=link}
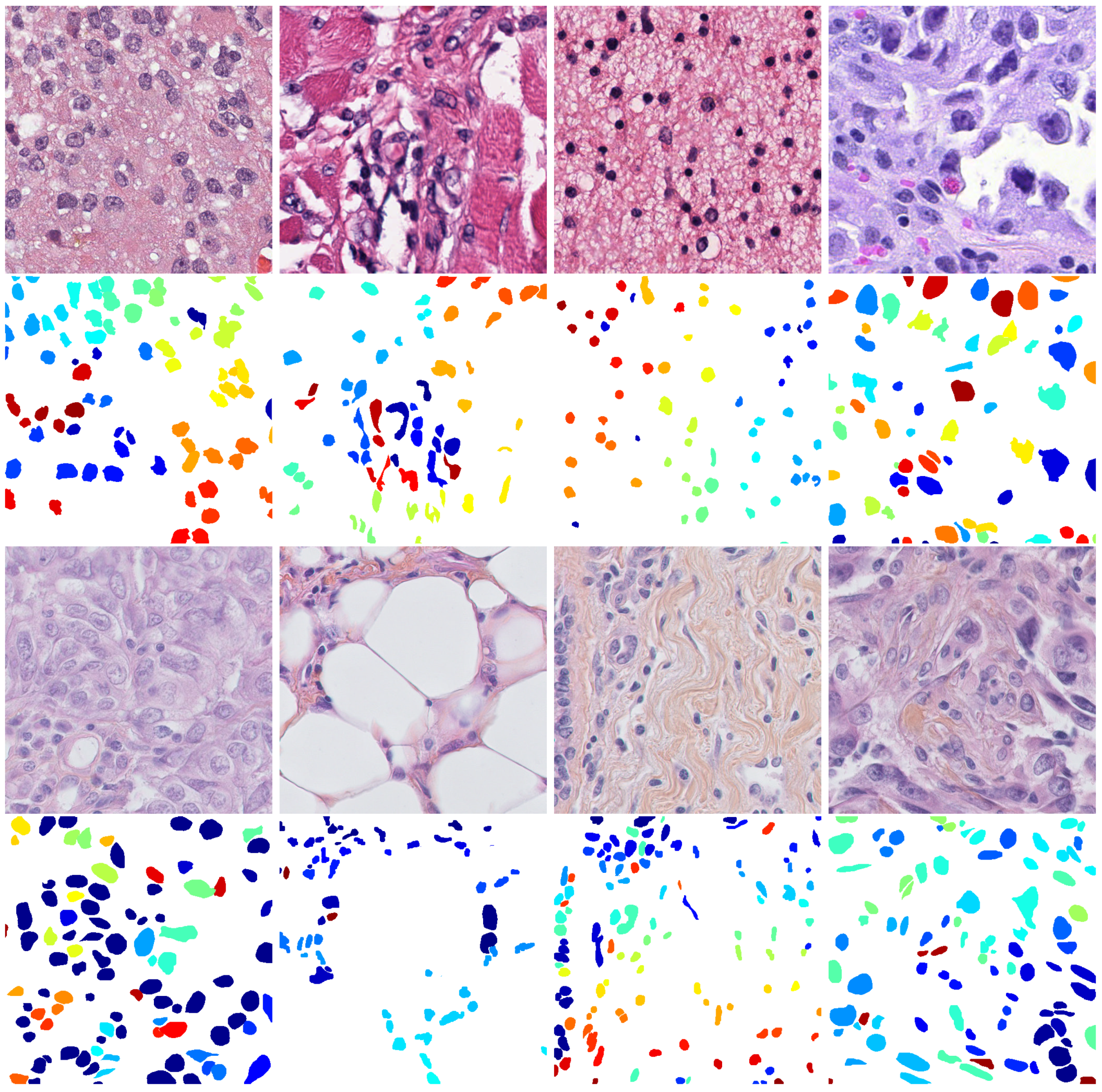{kind=link}
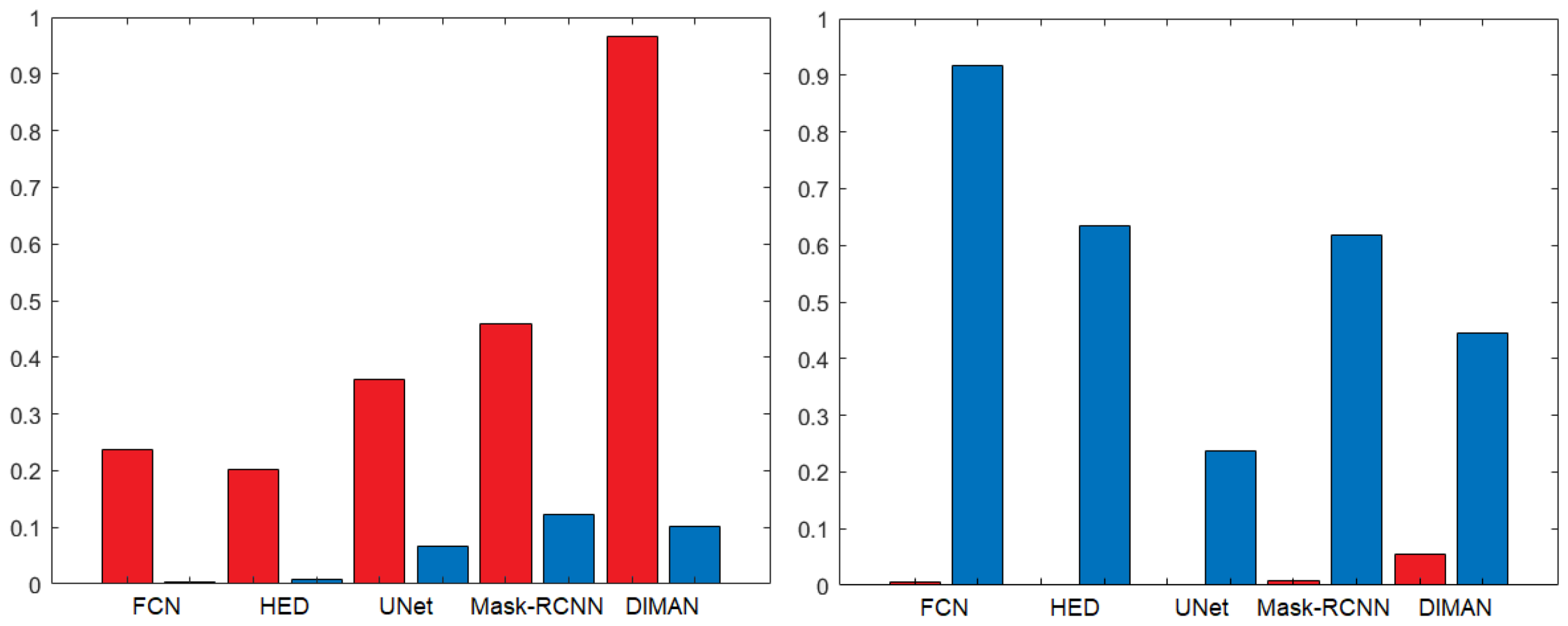{kind=link}
{kind=link}
| Method | Target | Source | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MIC17 | BNS | ||||||||||
| F1 | P | R | ODI | OHD | F1 | P | R | ODI | OHD | ||
| FCN | MIC17 | 0.74 | 0.76 | 0.73 | 0.69 | 15.60 | 0.58 | 0.49 | 0.73 | 0.63 | 12.90 |
| BNS | 0.52 | 0.60 | 0.46 | 0.54 | 23.16 | 0.73 | 0.73 | 0.74 | 0.69 | 14.75 | |
| HED | MIC17 | 0.79 | 0.85 | 0.74 | 0.71 | 16.03 | 0.73 | 0.70 | 0.77 | 0.70 | 15.30 |
| BNS | 0.52 | 0.70 | 0.42 | 0.55 | 26.40 | 0.77 | 0.83 | 0.73 | 0.71 | 17.13 | |
| UNet | MIC17 | 0.73 | 0.72 | 0.75 | 0.70 | 15.95 | 0.66 | 0.58 | 0.80 | 0.71 | 13.70 |
| BNS | 0.48 | 0.59 | 0.41 | 0.52 | 28.32 | 0.70 | 0.73 | 0.71 | 0.69 | 18.84 | |
| Mask R-CNN | MIC17 | 0.69 | 0.66 | 0.75 | 0.71 | 15.63 | 0.57 | 0.48 | 0.77 | 0.68 | 15.06 |
| BNS | 0.53 | 0.57 | 0.50 | 0.56 | 21.52 | 0.68 | 0.66 | 0.72 | 0.68 | 18.96 | |
| DIMAN | MIC17 | 0.71 | 0.69 | 0.77 | 0.72 | 16.91 | 0.67 | 0.60 | 0.82 | 0.74 | 14.30 |
| BNS | 0.61 | 0.66 | 0.60 | 0.62 | 19.56 | 0.73 | 0.73 | 0.76 | 0.72 | 16.75 | |
| Method | Target | Source | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MIC17 | BNS | ||||||||||
| F1 | P | R | ODI | OHD | F1 | P | R | ODI | OHD | ||
| FCN | GBM | 0.79 | 0.83 | 0.75 | 0.68 | 13.53 | 0.77 | 0.75 | 0.79 | 0.69 | 12.01 |
| HNSC | 0.76 | 0.78 | 0.76 | 0.71 | 13.96 | 0.54 | 0.42 | 0.77 | 0.64 | 12.45 | |
| LCC | 0.70 | 0.64 | 0.77 | 0.70 | 13.32 | 0.49 | 0.39 | 0.66 | 0.60 | 11.89 | |
| LUSC | 0.71 | 0.78 | 0.66 | 0.67 | 21.59 | 0.51 | 0.41 | 0.68 | 0.58 | 15.23 | |
| HED | GBM | 0.81 | 0.87 | 0.76 | 0.72 | 12.80 | 0.82 | 0.84 | 0.79 | 0.72 | 12.64 |
| HNSC | 0.80 | 0.87 | 0.75 | 0.70 | 18.23 | 0.71 | 0.66 | 0.79 | 0.71 | 15.89 | |
| LCC | 0.78 | 0.79 | 0.77 | 0.74 | 13.37 | 0.70 | 0.62 | 0.81 | 0.72 | 10.78 | |
| LUSC | 0.76 | 0.85 | 0.69 | 0.69 | 19.73 | 0.69 | 0.68 | 0.70 | 0.65 | 21.87 | |
| Mask R-CNN | GBM | 0.80 | 0.82 | 0.79 | 0.71 | 12.28 | 0.79 | 0.77 | 0.81 | 0.72 | 12.22 |
| HNSC | 0.69 | 0.67 | 0.77 | 0.69 | 16.90 | 0.48 | 0.38 | 0.81 | 0.68 | 15.93 | |
| LCC | 0.59 | 0.48 | 0.77 | 0.73 | 14.17 | 0.42 | 0.29 | 0.76 | 0.67 | 12.43 | |
| LUSC | 0.69 | 0.69 | 0.69 | 0.69 | 19.16 | 0.57 | 0.49 | 0.70 | 0.66 | 19.64 | |
| UNet | GBM | 0.79 | 0.80 | 0.79 | 0.71 | 11.80 | 0.81 | 0.80 | 0.82 | 0.73 | 11.13 |
| HNSC | 0.72 | 0.71 | 0.73 | 0.67 | 17.60 | 0.61 | 0.53 | 0.80 | 0.71 | 14.38 | |
| LCC | 0.71 | 0.65 | 0.79 | 0.74 | 13.20 | 0.57 | 0.43 | 0.84 | 0.71 | 10.88 | |
| LUSC | 0.69 | 0.70 | 0.68 | 0.67 | 21.20 | 0.64 | 0.58 | 0.73 | 0.68 | 18.40 | |
| DIMAN | GBM | 0.85 | 0.87 | 0.84 | 0.74 | 11.14 | 0.83 | 0.81 | 0.85 | 0.76 | 9.54 |
| HNSC | 0.67 | 0.64 | 0.76 | 0.69 | 18.93 | 0.64 | 0.56 | 0.85 | 0.75 | 13.29 | |
| LCC | 0.62 | 0.53 | 0.77 | 0.75 | 16.55 | 0.59 | 0.46 | 0.84 | 0.75 | 12.38 | |
| LUSC | 0.71 | 0.72 | 0.71 | 0.71 | 21.03 | 0.62 | 0.55 | 0.73 | 0.68 | 21.99 | |
| Data Subset | R | G | B | R Nuclei | B Nuclei | G Nuclei |
|---|---|---|---|---|---|---|
| MIC17 | 175 ± 17.9 | 117 ± 24.1 | 163 ± 21.3 | 103 ± 24.4 | 61 ± 21.3 | 118 ± 24.0 |
| BNS | 204 ± 12.8 | 185 ± 19.3 | 204 ± 10.6 | 157 ± 10.8 | 138 ± 11.7 | 178 ± 6.9 |
| GBM | 178 ± 16.8 | 132 ± 11.8 | 170 ± 8.5 | 102 ± 28.7 | 66 ± 24.1 | 118 ± 22.2 |
| HNSC | 177 ± 13.3 | 119 ± 13.5 | 164 ± 8.2 | 117 ± 26.0 | 72 ± 21.8 | 125 ± 11.1 |
| LCC | 173 ± 26.6 | 102 ± 26.9 | 149 ± 19.6 | 89 ± 11.5 | 43 ± 6.6 | 97 ± 12.0 |
| LUSC | 172 ± 17.1 | 116 ± 30.5 | 170 ± 31.3 | 102 ± 24.8 | 62 ± 21.1 | 130 ± 30.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Putzu, L.; Fumera, G. An Empirical Evaluation of Nuclei Segmentation from H&E Images in a Real Application Scenario. Appl. Sci. 2020, 10, 7982. https://doi.org/10.3390/app10227982
Putzu L, Fumera G. An Empirical Evaluation of Nuclei Segmentation from H&E Images in a Real Application Scenario. Applied Sciences. 2020; 10(22):7982. https://doi.org/10.3390/app10227982
Chicago/Turabian StylePutzu, Lorenzo, and Giorgio Fumera. 2020. "An Empirical Evaluation of Nuclei Segmentation from H&E Images in a Real Application Scenario" Applied Sciences 10, no. 22: 7982. https://doi.org/10.3390/app10227982
APA StylePutzu, L., & Fumera, G. (2020). An Empirical Evaluation of Nuclei Segmentation from H&E Images in a Real Application Scenario. Applied Sciences, 10(22), 7982. https://doi.org/10.3390/app10227982