Erythropoietin Protects Against Cognitive Impairment and Hippocampal Neurodegeneration in Diabetic Mice

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Protocol
2.3. Morris Water Maze
2.4. Histology
2.5. Data Analysis
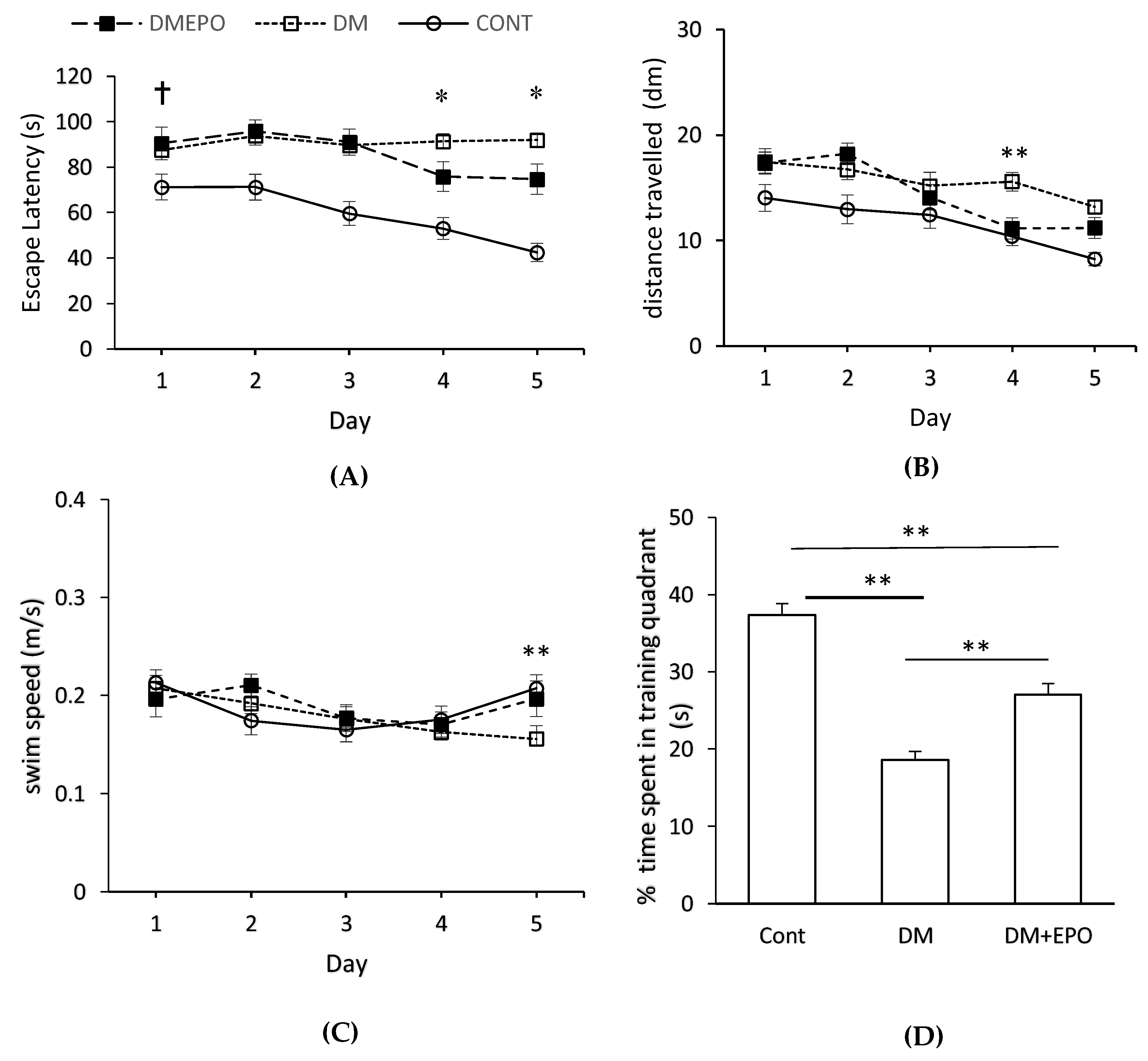
3. Results
3.1. Water Maze
3.1.1. Day 1
3.1.2. Within Group Comparisons
3.1.3. Between Group Comparisons
3.1.4. Probe Trial
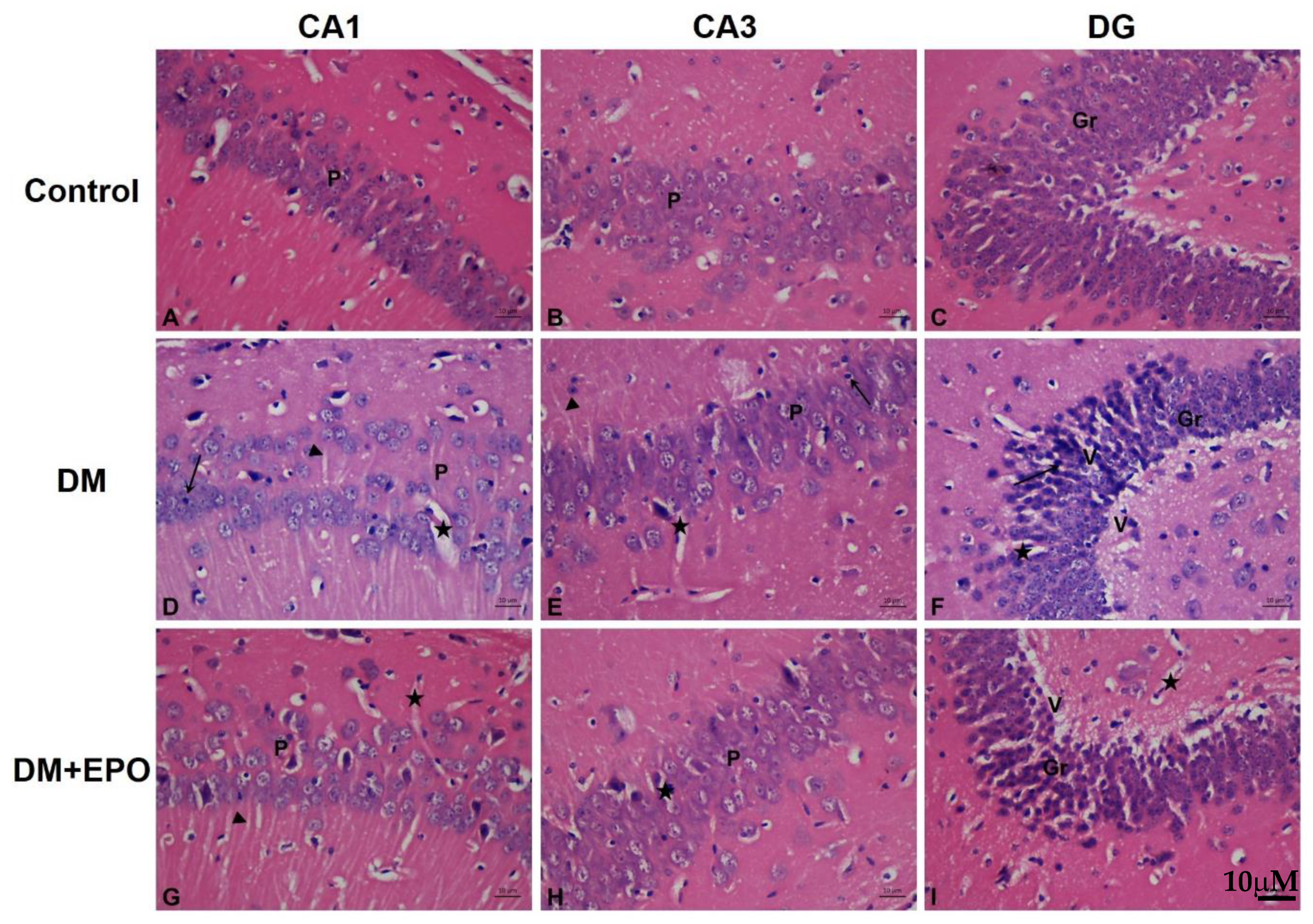
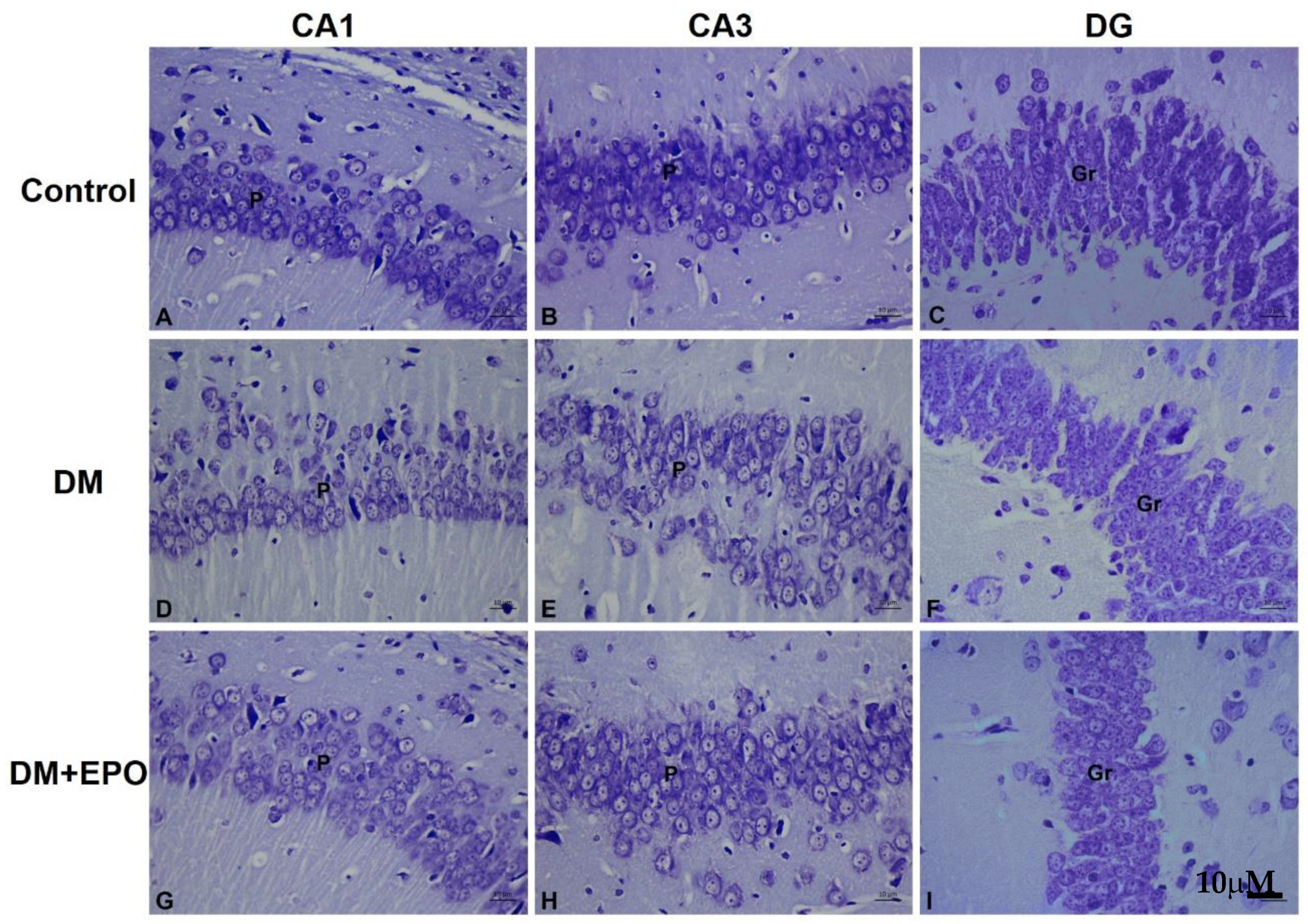
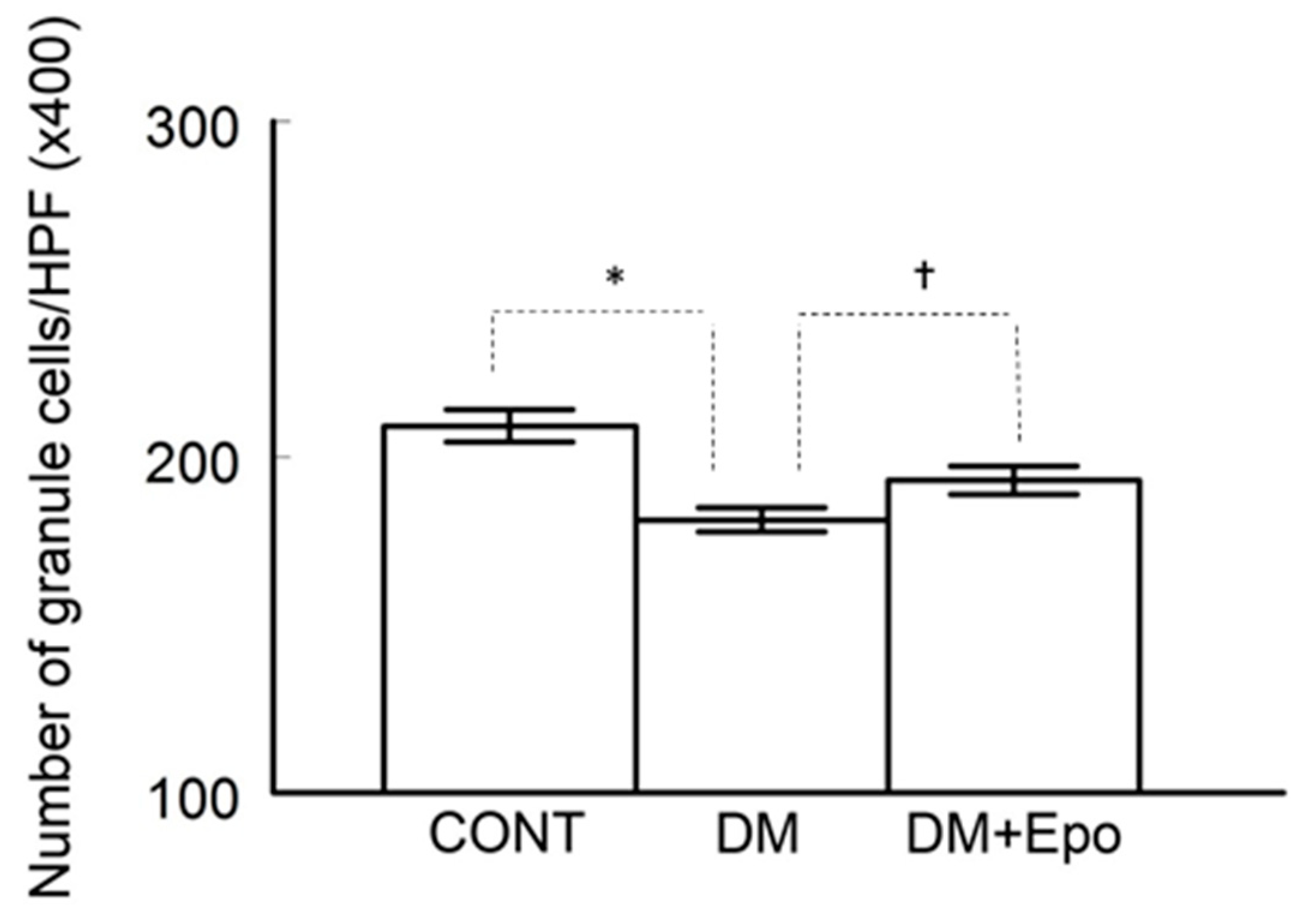
3.2. Histology of the Hippocampus
3.2.1. Hemotoxylin and Eosin Staining
3.2.2. Cresyl Violet Stain
3.2.3. Cell Count
3.3. Blood Glucose
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De la Monte, S.M. Type 3 diabetes is sporadic Alzheimers disease: Mini-review. Eur. Neuropsychopharmacol. 2014, 24, 1954–1960. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.; Tyagi, E.; Shukla, R.; Nath, C. A study of brain insulin receptors, AChE activity and oxidative stress in rat model of ICV STZ induced dementia. Neuropharmacology 2009, 56, 779–787. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S.M.; Tong, M. Brain metabolic dysfunction at the core of Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Biessels, G.J.; Urban, I.J.A.; Gispen, W.H. Hippocampal synaptic plasticity in streptozotocin-diabetic rats: Impairment of long-term potentiation and facilitation of long-term depression. Neuroscience 1999, 90, 737–745. [Google Scholar] [CrossRef]
- Magarinos, A.; McEwen, B. Experimental diabetes in rats causes hippocampal dendritic and synaptic reorganisation and increased glucocorticoid reactivity to stress. Proc. Natl. Acad. Sci. USA 2000, 97, 11056–11061. [Google Scholar] [CrossRef] [PubMed]
- Beauquis, J.; Saravia, F.; Coulaud, J.; Roig, P.; Dardenne, M.; Homo-Delarche, F.; De Nicola, A. Prominently decreased hippocampal neurogenesis in a spontaneous model of type 1 diabetes, the nonobese diabetic mouse. Exp. Neurol. 2008, 210, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.V.; Frier, B.M.; Strachan, M.W. The relationship between type 2 diabetes and cognitive dysfunction: Longitudinal studies and their methodological limitations. Eur. J. Pharmacol. 2004, 490, 169–175. [Google Scholar] [CrossRef]
- Biessels, G.J.; Staekenborg, S.; Brunner, E.; Brayne, C.; Scheltens, P. Risk of dementia in diabetes mellitus: A systematic review. Lancet Neurol. 2006, 5, 64–74. [Google Scholar] [CrossRef]
- Jelkmann, W. Regulation of erythropoietin production. J. Physiol. 2011, 589, 1251–1258. [Google Scholar] [CrossRef]
- Kumral, A.; Tuzun, F.; Oner, M.G.; Genc, S.; Duman, N.; Ozkan, H. Erythropoietin in neonatal brain protection: The past, the present and the future. Brain Dev. 2011, 33, 632–643. [Google Scholar] [CrossRef]
- Maiese, K.; Li, F.; Chong, Z.Z. New avenues of exploration for erythropoietin. JAMA 2005, 293, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, C.T.; Asavaritikrai, P.; Teng, R.; Jia, Y. Role of erythropoietin in the brain. Crit. Rev. Oncol. Hematol. 2007, 64, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Al Shaibani, T.; Ramakers, G. Erythropoietin decreases the excitatory neurotransmitter release probability and enhances synaptic plasticity in mice hippocampal slices. Brain Res. 2011, 1410, 33–37. [Google Scholar] [CrossRef]
- Sargin, D.; Friedrichs, H.; El-Kordi, A.; Ehrenreich, H. Erythropoietin as neuroprotective and neuroregenerative treatment strategy: Comprehensive overview of 12 years of preclinical and clinical research. Best Pract. Res. Clin. Anaesthesiol. 2010, 24, 573–594. [Google Scholar] [CrossRef] [PubMed]
- Sakanaka, M.; Wen, T.-C.; Matsuda, S.; Masuda, S.; Morishito, E.; Nagao, M.; Sasaki, R. In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc. Natl. Acad. Sci. USA 1998, 95, 4635–4640. [Google Scholar] [CrossRef] [PubMed]
- Jun, Y.; Xu, J.; Huang, Y.; Song, Y.; Zhen, J.; Ma, X.; Xu, J.; Xue, H.; Zhang, X.; Xing, X. Erythropoietin pre-treatment prevents cognitive impairments following status epilepticus in rats. Brain Res. 2009, 1282, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Shingo, T.; Sorokan, S.T.; Shimazaki, T.; Weiss, S. Erythropoietin regulates the in vitro and in vivo production of neuronal progenitors by mammalian forebrain neural stem cells. J. Neurosci. 2001, 21, 9733–9743. [Google Scholar] [CrossRef]
- Chong, Z.Z. Erythropoietin Is a Novel Vascular Protectant Through Activation of Akt1 and Mitochondrial Modulation of Cysteine Proteases. Circulation 2002, 106, 2973–2979. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Z.; Wang, Y.; Zhang, R.; Chopp, M. Treatment of stroke with erythropoietin enhances neurogenesis and angiogenesis and improves neurological function in rats. Stroke 2004, 35, 1732–1737. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Z.G.; Rhodes, K.; Renzi, M.; Zhang, R.L.; Kapke, A.; Lu, M.; Pool, C.; Heavner, G.; Chopp, M. Post-ischemic treatment with erythropoietin or carbamylated erythropoietin reduces infarction and improves neurological outcome in a rat model of focal cerebral ischemia. Br. J. Pharmacol. 2007, 151, 1377–1384. [Google Scholar] [CrossRef]
- Yu, T.; Li, L.; Chen, T.; Liu, Z.; Liu, H.; Li, Z. Erythropoietin attenuates advanced glycation endproducts-induced toxicity of Schwann cells in vitro. Neurochem. Res. 2015, 40, 698–712. [Google Scholar] [CrossRef]
- Subiros, N.; Del Barco, D.G.; Coro-Antich, R.M. Erythropoietin: Still on the neuroprotection road. Ther. Adv. Neurol. Disord. 2012, 5, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Maiese, K. Erythropoietin and diabetes mellitus. World J. Diabetes 2015, 6, 1259–1273. [Google Scholar] [CrossRef] [PubMed]
- Maiese, K. Novel applications of trophic factors, Wnt and WISP for neuronal repair and regeneration in metabolic disease. Neural Regen. Res. 2015, 10, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Silverberg, D.; Wexler, D.; Iaina, A.; Schwartz, D. The interaction between heart failure and other heart diseases, renal failure, and anemia. Semin. Nephrol. 2006, 26, 296–306. [Google Scholar] [CrossRef]
- Lewis, E.F.; Pfeffer, M.A.; Feng, A.; Uno, H.; McMurray, J.J.; Toto, R.; Gandra, S.R.; Solomon, S.D.; Moustafa, M.; Macdougall, I.C.; et al. Darbepoetin alfa impact on health status in diabetes patients with kidney disease: A randomized trial. Clin. J. Am. Soc. Nephrol. 2011, 6, 845–855. [Google Scholar] [CrossRef]
- Yu, T.; Li, L.; Bi, Y.; Liu, Z.; Liu, H.; Li, Z. Erythropoietin attenuates oxidative stress and apoptosis in Schwann cells isolated from streptozotocin-induced diabetic rats. J. Pharm. Pharmacol. 2014, 66, 1150–1160. [Google Scholar] [CrossRef]
- Wang, M.; Yan, W.; Liu, Y.; Hu, H.; Sun, Q.; Chen, X.; Zang, W.; Chen, L. Erythropoietin ameliorates diabetes-associated cognitive dysfunction in vitro and in vivo. Sci. Rep. 2017, 7, 2801. [Google Scholar] [CrossRef]
- Kristensen, P.L.; Pedersen-Bjergaard, U.; Kjaer, T.W.; Olsen, N.V.; Dela, F.; Holst, J.J.; Faber, J.; Tarnow, L.; Thorsteinsson, B. Influence of erythropoietin on cognitive performance during experimental hypoglycemia in patients with type 1 diabetes mellitus: A randomized cross-over trial. PLoS ONE 2013, 8, e59672. [Google Scholar] [CrossRef]
- Deeds, M.C.; Anderson, J.M.; Armstrong, A.S.; Gastineau, D.A.; Hiddinga, H.J.; Jahangir, A.; Eberhardt, N.L.; Kudva, Y.C. Single dose streptozotocin-induced diabetes: Considerations for study design in islet transplantation models. Lab. Anim. 2011, 45, 131–140. [Google Scholar] [CrossRef]
- Biessels, G.J.; Kamal, A.; Ramakers, G.M.; Urban, I.J.; Spruijt, B.M.; Erkelens, D.W.; Gispen, W.H. Place Learning and Hippocampal Synaptic Plasticity in Streptozotocin-Induced Diabetic Rats. Diabetes 1996, 45, 1259–1266. [Google Scholar] [CrossRef]
- Kamal, A.; Biessels, G.J.; Gispen, W.H.; Ramakers, G.M. Synaptic transmission changes in the pyramidal cells of the hippocampus in streptozotocin-induced diabetes mellitus in rats. Brain Res. 2006, 1073–1074, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Rajab, E.; Abdeen, Z.; Hassan, Z.; Alsaffar, Y.; Mandeel, M.; Al Shawaaf, F.; Al-Ansari, S.; Kamal, A. Cognitive performance and convulsion risk after experimentally-induced febrile-seizures in rat. Int. J. Dev. Neurosci. 2014, 34, 19–23. [Google Scholar] [CrossRef]
- Gallagher, M.; Burwell, R.; Burchinal, M. Severity of spatial learning impairment in aging: Development of a learning index for performance in the Morris water maze. Behav. Neurosci. 1993, 107, 618–626. [Google Scholar] [CrossRef]
- Lindner, M.D. Reliability, distribution, and validity of age-related cognitive deficits in the Morris water maze. Neurobiol. Learn. Mem. 1997, 68, 203–220. [Google Scholar] [CrossRef]
- El-Falougy, H.; Kubikova, E.; Benuska, J. The microscopical structure of the hippocampus in the rat. Bratisl. Lek. Listy 2008, 109, 106–110. [Google Scholar]
- Bancroft, J.D.; Gamble, M. Theory and Practice of Histological Techniques, 6th ed.; Churchill Livingstone: London, UK, 2008. [Google Scholar]
- Manschot, S.M.; Biessels, G.J.; Cameron, N.E.; Cotter, M.A.; Kamal, A.; Kappelle, L.J.; Gispen, W.H. Angiotensin converting enzyme inhibition partially prevents deficits in water maze performance, hippocampal synaptic plasticity and cerebral blood flow in streptozotocin-diabetic rats. Brain Res. 2003, 966, 274–282. [Google Scholar] [CrossRef]
- Soares, E.; Prediger, R.D.; Nunes, S.; Castro, A.A.; Viana, S.D.; Lemos, C.; De Souza, C.M.; Agostinho, P.; Cunha, R.A.; Carvalho, E.; et al. Spatial memory impairments in a prediabetic rat model. Neuroscience 2013, 250, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.N.; Younan, S.M.; Youssef, M.F.; Rashed, L.A.; Mohamady, I. A histological and functional study on hippocampal formation of normal and diabetic rats. F1000Resarch 2013, 2, 151. [Google Scholar] [CrossRef] [PubMed]
- Golembewski, E.K.; Wales, S.Q.; Aurelian, L.; Yarowsky, P.J. The HSV-2 protein ICP10PK prevents neuronal apoptosis and loss of function in an in vivo model of neurodegeneration associated with glutamate excitotoxicity. Exp. Neurol. 2007, 203, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, L.; Ling, S.; Zhang, X. Expression changes of growth-associated protein-43 (GAP-43) and mitogen-activated protein kinase phosphatase-1 (MKP-1) and in hippocampus of streptozotocin-induced diabetic cognitive impairment rats. Exp. Neurol. 2007, 206, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.J.; Tan, Y.F.; Yue, J.T.; Vranic, M.; Wojtowicz, J.M. Impairment of hippocampal neurogenesis in streptozotocin-treated diabetic rats. Acta Neurol. Scand. 2008, 117, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.G.; Zhang, W.; Grunberger, G.; Sima, A.A. Hippocampal neuronal apoptosis in type 1 diabetes. Brain Res. 2002, 946, 221–231. [Google Scholar] [CrossRef]
- Ahmadpour, S.; Sadeghi, Y.; Sheibanifar, M.H.H. Neuronal death in dentate gyrus and ca3 in diabetic rats: Effects of insulin and ascorbic acid. Hormozan J. Med. Sci. 2010, 13, 13–16. [Google Scholar]
- Jafari Anarkooli, I.; Sankian, M.; Ahmadpour, S.; Varasteh, A.R.; Haghir, H. Evaluation of Bcl-2 family gene expression and Caspase-3 activity in hippocampus STZ-induced diabetic rats. Exp. Diabetes Res. 2008, 2008, 638467. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, L.; Bennis, Y.; Velly, L.; Grandvuillemin, I.; Pisano, P.; Bruder, N.; Guillet, B. Erythropoietin protects newborn rat against sevoflurane-induced neurotoxicity. Paediatr. Anaesth. 2014, 24, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Ratilal, B.O.; Arroja, M.M.; Rocha, J.P.; Fernandes, A.M.; Barateiro, A.P.; Brites, D.M.; Pinto, R.M.; Sepodes, B.M.; Mota-Filipe, H.D. Neuroprotective effects of erythropoietin pretreatment in a rodent model of transient middle cerebral artery occlusion. J. Neurosurg. 2014, 121, 55–62. [Google Scholar] [CrossRef]
- Jang, W.; Park, J.; Shin, K.J.; Kim, J.S.; Kim, J.S.; Youn, J.; Cho, J.W.; Oh, E.; Ahn, J.Y.; Oh, K.W.; et al. Safety and efficacy of recombinant human erythropoietin treatment of non-motor symptoms in Parkinson’s disease. J. Neurol. Sci. 2014, 337, 47–54. [Google Scholar] [CrossRef]
- Wood, S.C.; Rao, T.D.; Frey, A.B. Multidose streptozotocin induction of diabetes in BALB/cBy mice induces a T cell proliferation defect in thymocytes which is reversible by interleukin-4. Cell. Immunol. 1999, 192, 1–12. [Google Scholar] [CrossRef]
- Fenjves, E.S.; Ochoa, M.S.; Cabrera, O.; Mendez, A.J.; Kenyon, N.S.; Inverardi, L.; Ricordi, C. Human, nonhuman primate, and rat pancreatic islets express erythropoietin receptors. Transplantation 2003, 75, 1356–1360. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, L.; Dey, S.; Alnaeeli, M.; Suresh, S.; Rogers, H.; Teng, R.; Noguchi, C. Erythropoietin action in stress response, tissue maintenance and metabolism. Int. J. Mol. Sci. 2014, 15, 10296–10333. [Google Scholar] [CrossRef] [PubMed]
- Katz, O.; Stuible, M.; Golishevski, N.; Lifshitz, L.; Tremblay, M.L.; Gassmann, M.; Mittelman, M.; Neumann, D. Erythropoietin treatment leads to reduced blood glucose levels and body mass: Insights from murine models. J. Endocrinol. 2010, 205, 87–95. [Google Scholar] [CrossRef]
- Chen, L.N.; Sun, Q.; Liu, S.Q.; Hu, H.; Lv, J.; Ji, W.J.; Wang, M.; Chen, M.X.; Zhou, J. Erythropoietin improves glucose metabolism and pancreatic beta-cell damage in experimental diabetic rats. Mol. Med. Rep. 2015, 12, 5391–5398. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.T.; Chu, K.; Park, J.E.; Jung, K.H.; Jeon, D.; Lim, J.Y.; Lee, S.K.; Kim, M.; Roh, J.K. Erythropoietin improves memory function with reducing endothelial dysfunction and amyloid-beta burden in Alzheimer’s disease models. J. Neurochem. 2012, 120, 115–124. [Google Scholar] [CrossRef]
- Belanger, A.; Lavoie, N.; Trudeau, F.; Massicotte, G.; Gagnon, S. Preserved LTP and water maze learning in hyperglycaemic-hyperinsulinemic ZDF rats. Physiol. Behav. 2004, 83, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Biessels, G.J.; Duis, S.E.; Gispen, W.H. Learning and hippocampal synaptic plasticity in streptozotocin-diabetic rats: Interaction of diabetes and ageing. Diabetologia 2000, 43, 500–506. [Google Scholar] [CrossRef]
- Wright, D.G.; Wright, E.C.; Narva, A.S.; Noguchi, C.T.; Eggers, P.W. Association of Erythropoietin Dose and Route of Administration with Clinical Outcomes for Patients on Hemodialysis in the United States. Clin. J. Am. Soc. Nephrol. 2015, 10, 1822–1830. [Google Scholar] [CrossRef]
- Leuner, B.; Gould, E.; Shors, T.J. Is there a link between adult neurogenesis and learning? Hippocampus 2006, 16, 216–224. [Google Scholar] [CrossRef]
- Snyder, J.S.; Soumier, A.; Brewer, M.; Pickel, J.; Cameron, H.A. Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature 2011, 476, 458–461. [Google Scholar] [CrossRef]
- Ho, N.; Sommers, M.S.; Lucki, I. Effects of diabetes on hippocampal neurogenesis: Links to cognition and depression. Neurosci. Biobehav. Rev. 2013, 37, 1346–1362. [Google Scholar] [CrossRef]
- Tsai, P.T.; Ohab, J.J.; Kertesz, N.; Groszer, M.; Matter, C.; Gao, J.; Liu, X.; Wu, H.; Carmichael, S.T. A critical role of erythropoietin receptor in neurogenesis and post-stroke recovery. J. Neurosci. 2006, 26, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chopp, M.; Mahmood, A.; Meng, Y.; Qu, C.; Xiong, Y. Impact of inhibition of erythropoietin treatment-mediated neurogenesis in the dentate gyrus of the hippocampus on restoration of spatial learning after traumatic brain injury. Exp. Neurol. 2012, 235, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Y.; Asavaritikrai, P.; Prchal, J.T.; Noguchi, C.T. Endogenous erythropoietin signaling is required for normal neural progenitor cell proliferation. J. Biol. Chem. 2007, 282, 25875–25883. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Fan, S.; Song, D.; Wang, Z.; Ma, S.; Li, S.; Li, X.; Xu, M.; Xu, M.; Wang, X. Long-term streptozotocin-induced diabetes in rats leads to severe damage of brain blood vessels and neurons via enhanced oxidative stress. Mol. Med. Rep. 2013, 7, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Appelros, P.; Samuelsson, M.; Lindell, D. Lacunar infarcts: Functional and cognitive outcomes at five years in relation to MRI findings. Cerebrovasc. Dis. 2005, 20, 34–40. [Google Scholar] [CrossRef]
- Prasad, S.; Sajja, R.K.; Naik, P.; Cucullo, L. Diabetes Mellitus and Blood-Brain Barrier Dysfunction: An Overview. J. Pharmacovigil. 2014, 2, 125. [Google Scholar] [CrossRef]
- Anagnostou, A.; Liu, Z.; Steiner, M.; Chin, K.; Lee, E.; Kessimian, N.; Noguchi, C.T. Erythropoietin receptor mRNA expression in human endothelial cells. Proc. Natl. Acad. Sci. USA 1994, 91, 3974–3978. [Google Scholar] [CrossRef]
- Wang, L.; Chopp, M.; Gregg, S.R.; Zhang, R.L.; Teng, H.; Jiang, A.; Feng, Y.; Zhang, Z.G. Neural progenitor cells treated with EPO induce angiogenesis through the production of VEGF. J. Cereb. Blood Flow Metab. 2008, 28, 1361–1368. [Google Scholar] [CrossRef]
- Ribatti, D.; Presta, M.; Vacca, A.; Ria, R.; Giuliani, R.; Dell’Era, P.; Nico, B.; Roncali, L.; Dammacco, F. Human erythropoietin induces a pro-angiogenic phenotype in cultured endothelial cells and stimulates neovascularization in vivo. Blood 1999, 93, 2627–2636. [Google Scholar]
- Chapillon, P.; Debouzie, A. BALB/c mice are not so bad in the Morris water maze. Behav. Brain Res. 2000, 117, 115–118. [Google Scholar] [CrossRef]
- Francis, D. Stress-induced disturbances in Morris water-maze performance: Interstrain variability. Physiol. Behav. 1995, 58, 57–65. [Google Scholar] [CrossRef]
- Upchurch, M.; Wehner, J.M. Differences between Inbred Strains of Mice in Morris Water Maze Performance. Behav. Genet. 1988, 18, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Alnaeeli, M.; Wang, L.; Piknova, B.; Rogers, H.; Li, X.; Noguchi, C.T. Erythropoietin in brain development and beyond. Anat. Res. Int. 2012, 2012, 953264. [Google Scholar] [CrossRef] [PubMed]
- Grasso, G.; Buemi, M.; Alafaci, C.; Sfacteria, A.; Passalacqua, M.; Sturiale, A.; Calapai, G.; De Vico, G.; Piedimonte, G.; Salpietro, F.M.; et al. Beneficial effects of systemic administration of recombinant human erythropoietin in rabbits subjected to subarachnoid hemorrhage. Proc. Natl. Acad. Sci. USA 2002, 99, 5627–5631. [Google Scholar] [CrossRef] [PubMed]
- Brines, M.L.; Ghezzi, P.; Keenan, S.; Agnello, D.; de Lanerolle, N.C.; Cerami, C.; Itri, L.M.; Cerami, A. Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc. Natl. Acad. Sci. USA 2000, 97, 10526–10531. [Google Scholar] [CrossRef] [PubMed]
- Artola, A.; Kamal, A.; Ramakers, G.M.; Biessels, G.J.; Gispen, W.H. Diabetes mellitus concomitantly facilitates the induction of long-term depression and inhibits that of long-term potentiation in hippocampus. Eur. J. Neurosci. 2005, 22, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Tsien, J.Z.; Huerta, P.T.; Tonegawa, S. The Essential Role of Hippocampal CA1 NMDA Receptor–Dependent Synaptic Plasticity in Spatial Memory. Cell 1996, 87, 1327–1338. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Blood Glucose Baseline (mmol/L) | Blood Glucose End (mmol/L) | Statistical Comparisons: Baseline vs. End |
|---|---|---|---|
| control | 8.6 ± 0.5 | 7.8 ± 0.4 | p = 0.1733, t = 1.4478 |
| Diabetic | 18.6 ± 0.7 ** | 31.6 ± 0.7 ** | p = 1.945 × 10−8, t = −13.0162 |
| Diabetic + EPO | 17.9 ± 0.6 ** | 30.1 ± 1.3 ** | p =1.5916 × 10−6, t = −8.6924 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Othman, M.A.M.; Rajab, E.; AlMubarak, A.; AlNaisar, M.; Bahzad, N.; Kamal, A. Erythropoietin Protects Against Cognitive Impairment and Hippocampal Neurodegeneration in Diabetic Mice. Behav. Sci. 2019, 9, 4. https://doi.org/10.3390/bs9010004
Othman MAM, Rajab E, AlMubarak A, AlNaisar M, Bahzad N, Kamal A. Erythropoietin Protects Against Cognitive Impairment and Hippocampal Neurodegeneration in Diabetic Mice. Behavioral Sciences. 2019; 9(1):4. https://doi.org/10.3390/bs9010004
Chicago/Turabian StyleOthman, Manal A. M., Ebrahim Rajab, Ahmed AlMubarak, Mohammed AlNaisar, Noora Bahzad, and Amer Kamal. 2019. "Erythropoietin Protects Against Cognitive Impairment and Hippocampal Neurodegeneration in Diabetic Mice" Behavioral Sciences 9, no. 1: 4. https://doi.org/10.3390/bs9010004
APA StyleOthman, M. A. M., Rajab, E., AlMubarak, A., AlNaisar, M., Bahzad, N., & Kamal, A. (2019). Erythropoietin Protects Against Cognitive Impairment and Hippocampal Neurodegeneration in Diabetic Mice. Behavioral Sciences, 9(1), 4. https://doi.org/10.3390/bs9010004