Cellular Redox Imbalance and Neurochemical Effect in Cognitive-Deficient Old Rats


 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal
2.2. Behavioral Test
2.3. Sedation and Sample Collection
2.4. Biochemical Analysis
2.4.1. Oxidative Stress Markers
Glutathione GSH Quantification
Malondialdehyde (MDA) Quantification
Assay for Calcium-Dependent Cytosolic Phospholipase A2 (PLA2) Activity
2.4.2. Amino Acid Analysis
Sample Preparation and Derivatization
Instrumentation and Chromatographic Conditions
2.5. Data Processing and Statistical Analysis
3. Results
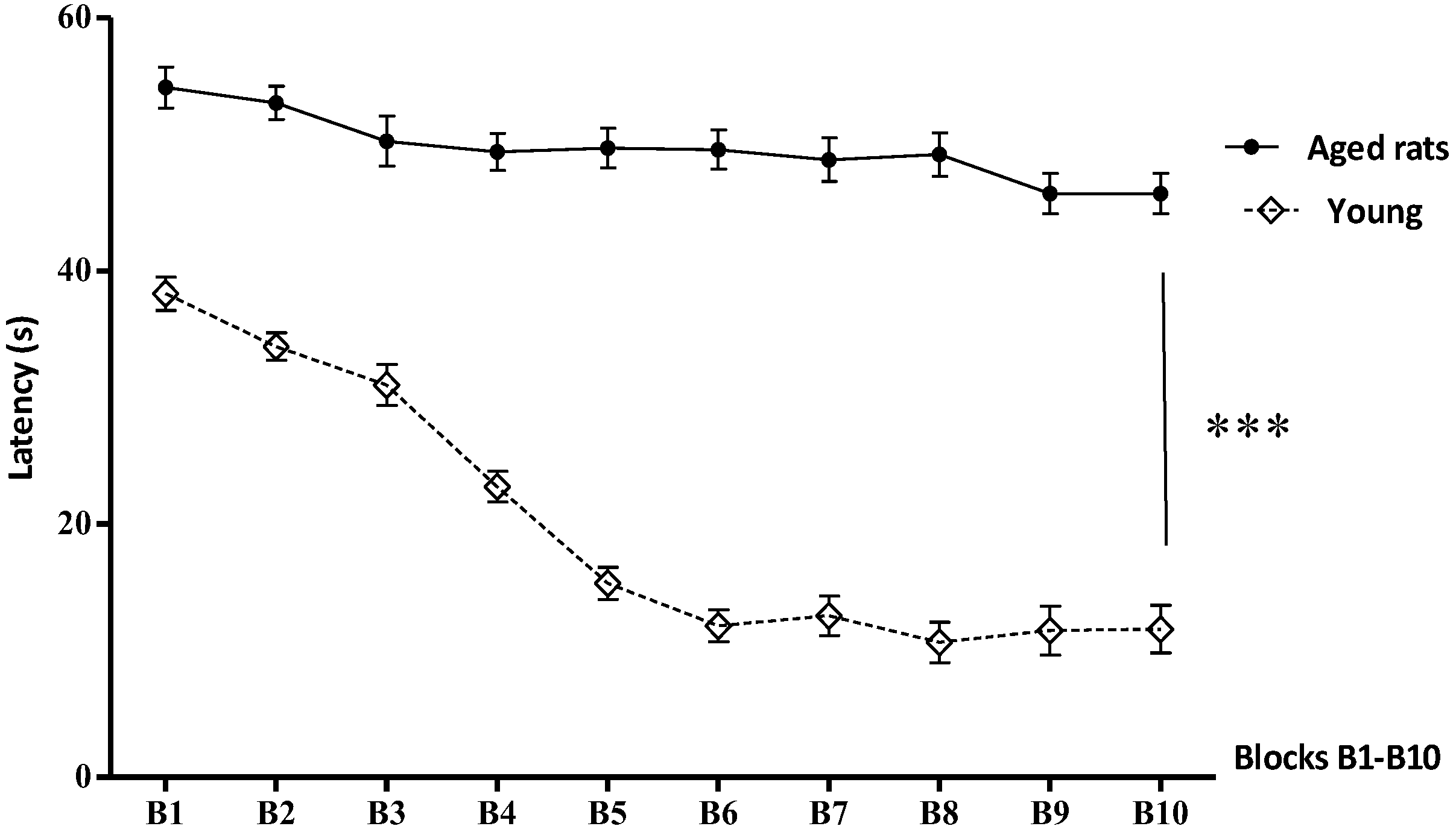
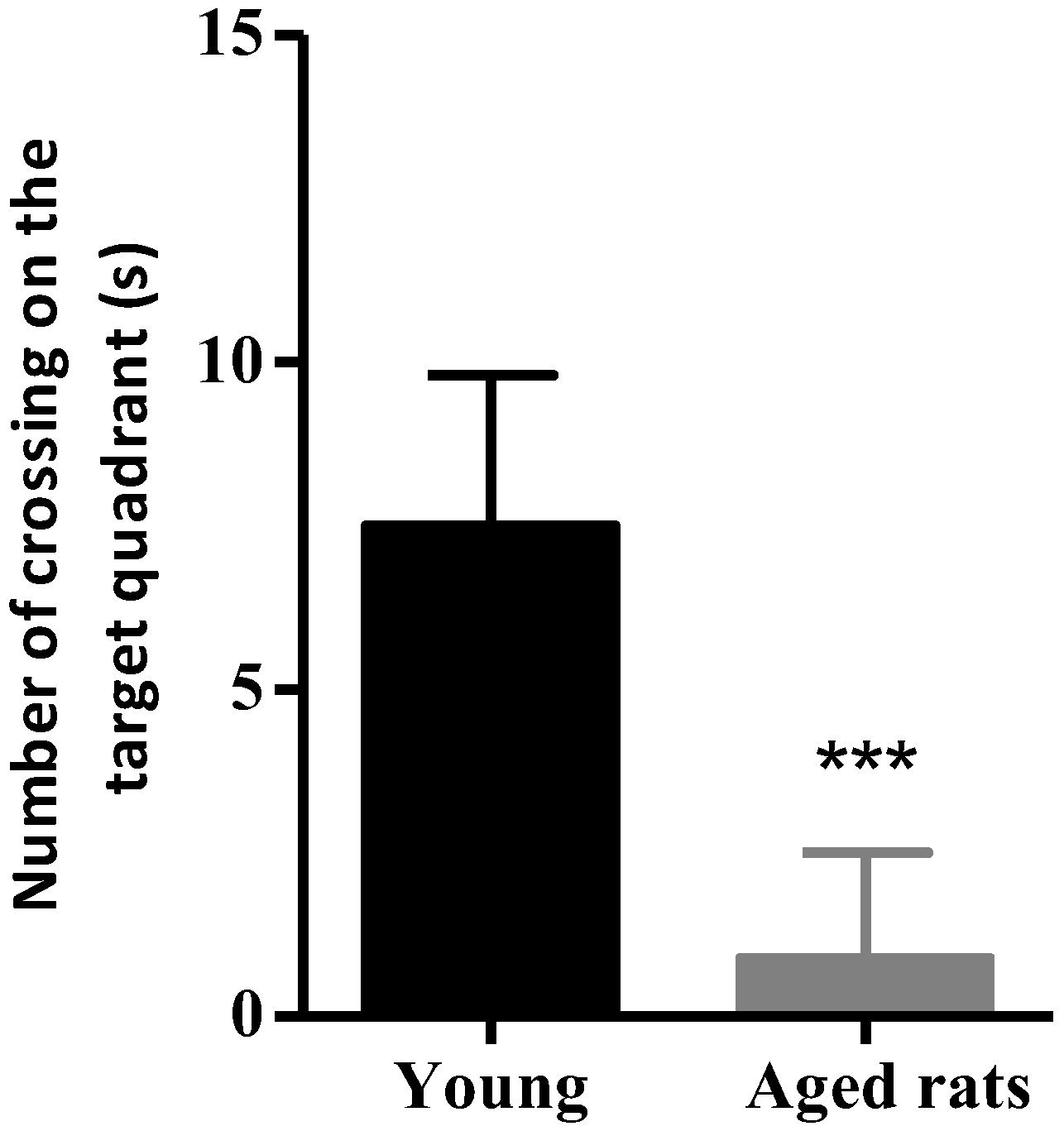
3.1. Behavioural Studies
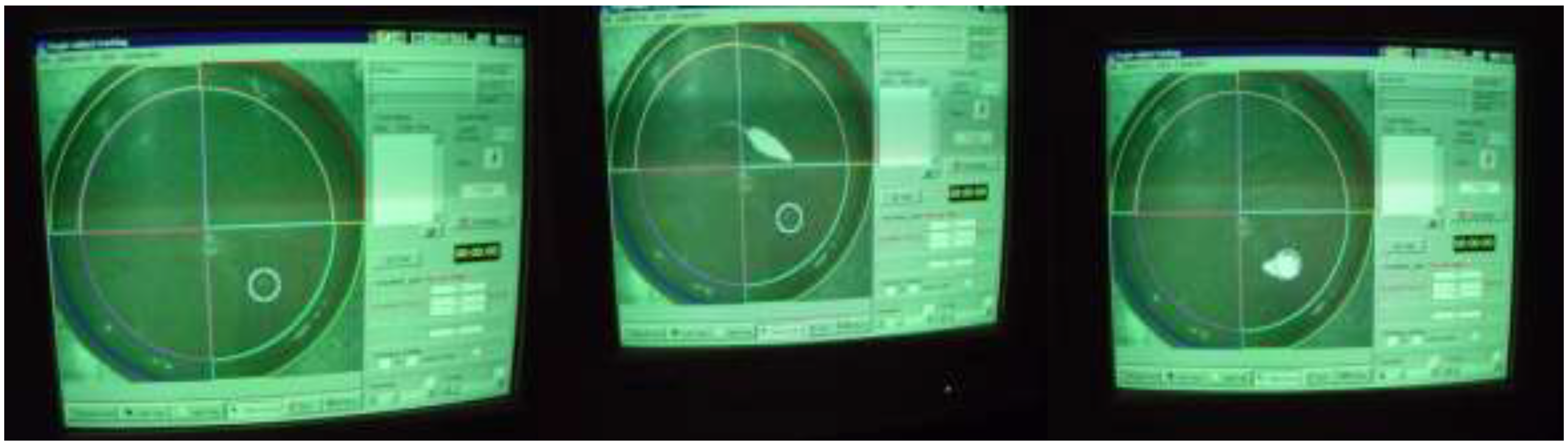
Morris Water Maze
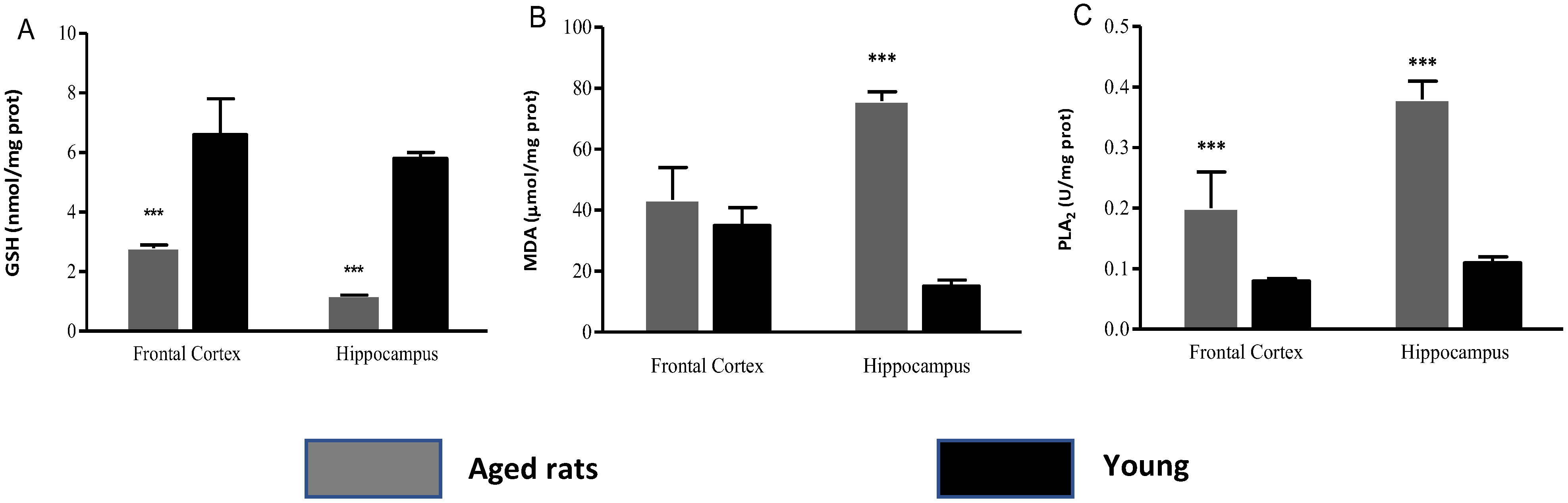
3.2. Biochemical Indicators
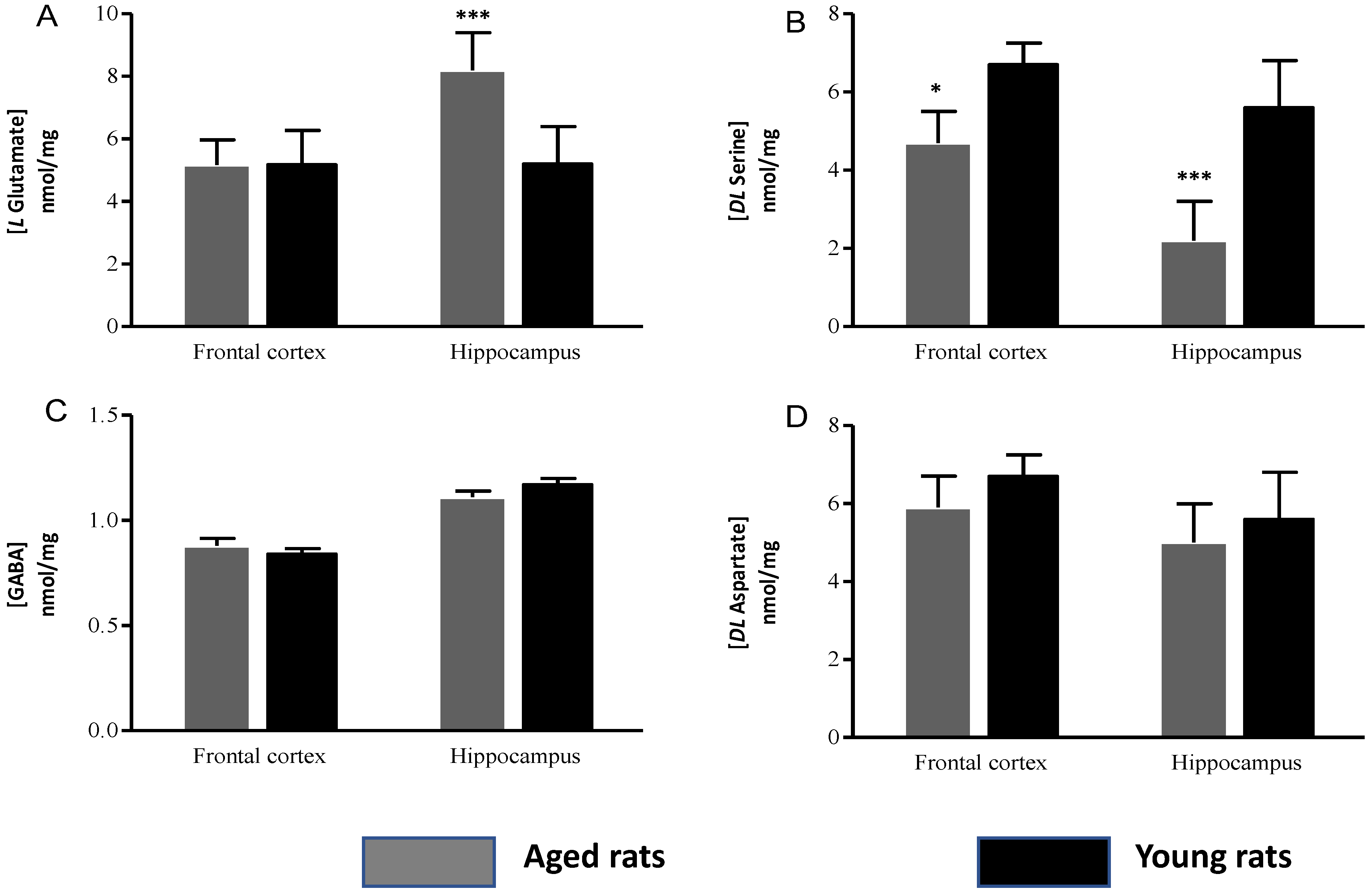
Amino Acid Concentrations
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, Y.; Li, P.; Feng, J.; Minghu, W. Dysfunction of NMDA receptors in Alzheimer’s disease. Neurol. Sci. 2016, 37, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Mari, M.; Morales, A.; Colell, A.; Garcia-Ruiz, C.; Fernandez-Checa, J. Mitochondrial Glutathione, a Key Survival Antioxidant. ARS 2009, 11, 2685–2700. [Google Scholar] [CrossRef] [PubMed]
- Bergado, J.A.; Almaguer, W.; Rojas, J.; Capdevila, V.; Frey, J.U. Spatial and emotional memory in aged rats: A behavioral-statistical analysis. Neuroscience 2011, 172, 256–269. [Google Scholar] [CrossRef] [PubMed]
- Krämer, T.; Grob, T.; Menzel, L.; Hirnet, T.; Griemert, E.; Radyushkin, K.; Thal, S.C.; Methner, A.; Schaefer, M.K.E. Dimethyl fumarate treatment after traumatic brain injury prevents depletion of antioxidative brain glutathione and confers neuroprotection. J. Neurochem. 2017, 143 (Suppl. 5), 523–533. [Google Scholar] [CrossRef] [PubMed]
- Gemelli, T.; de Andrade, R.B.; Rojas, D.B.; Zanatta, Â.; Schirmbeck, G.H.; Funchal, C.; Wajner, M.; Dutra-Filho, C.S.; Wannmacher, C.M.D. Chronic Exposure to β-Alanine Generates Oxidative Stress and Alters Energy Metabolism in Cerebral Cortex and Cerebellum of Wistar Rats. Mol. Neurobiol. 2018, 55 (Suppl. 6), 5101–5110. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Aguado, R.; Almaguer-Melian, W.; Diaz, C.M.; Lorigados, L.; Bergado, J. Behavioral and biochemical effects of glutathione depletion in the rat brain. Brain Res. Bull. 2001, 55 (Suppl. 3), 327–333. [Google Scholar] [CrossRef]
- Bonasera, S.; Arikkath, J.; Boska, M.D.; Chaudoin, T.R.; De Korver, N.W.; Goulding, E.H.; Traci, A.; Hoke, T.A.; Mojtahedzedah, V.; Reyelts, C.D.; et al. Age-related changes in cerebellar and hypothalamic function accompany non-microglial immune gene expression, altered synapse organization, and excitatory amino acid neurotransmission deficits. Aging 2016, 8, 2153–2164. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, V.; Chauhan, A. Abnormalities in membrane lipids, membrane-associated proteins, and signal transduction in Autism. In Austim Oxidative Stress, Inflammation and Immune Abnormalities; Chauhan, A., Ed.; CRC Press: Boca Raton, FL, USA, 2010; pp. 177–207. [Google Scholar]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Basselin, M.; Chang, L.; Chen, M.; Bell, J.M.; Rapoport, S. Chronic Administration of Valproic Acid Reduces Brain NMDA Signaling via Arachidonic Acid in Unanesthetized Rats. Neurochem. Res. 2008, 33, 2229–2240. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.Y.; Kannan, S.; Chen, Y.J.; Tan, F.C.; Ong, W.Y.; Go, M.L.; Verma, C.S.; Low, C.M.; Lam, Y. A New Generation of Arachidonic Acid Analogues as Potential Neurological Agent Targeting Cytosolic Phospholipase A2. Sci. Rep. 2017, 7, 13683. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Reddy, P.H. Role of glutamate and NMDA receptors in Alzheimer’s disease. J. Alzheimers Dis. 2017, 57 (Suppl. 4), 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Billard, J.M. D-serine signalling as a prominent determinant of neuronal-glial dialogue in the healthy and diseased brain. J. Cell. Mol. Med. 2008, 12, 1872–1884. [Google Scholar] [CrossRef] [PubMed]
- Sasabe, J.; Miyoshi, Y.; Suzuki, M.; Mita, M.; Konno, R.; Matsuoka, M.; Hamase, K.; Aiso, S. D-amino acid oxidase controls motoneuron degeneration through D-serine. Proc. Natl. Acad. Sci. USA 2012, 109, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Nuechterlein, K.H.; Subotnik, K.L.; Green, M.F.; Ventura, J.; Asarnow, R.F.; Gitlin, M.J.; Yee, C.M.; Gretchen-Doorly, D.; Mintz, J. Neurocognitive predictors of work outcome in recent-onset schizophrenia. Schizophr. Bull. 2011, 37, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Ishima, T.; Hashimoto, K. Supplementation with D-serine prevents the onset of cognitive deficits in adult offspring after maternal immune activation. Sci. Rep. 2016, 6, 37261. [Google Scholar] [CrossRef] [PubMed]
- Olferd, E.; Cross, B.; McWillian, D.; McWillian, A. Guidelines for the Use of Animal in Neuroscience Research; Canadian Council on Care (CCAC), Bradda Printing Services Inc.: Ottawa, ON, Canada, 1997; pp. 163–165. [Google Scholar]
- Van Praag, H.; Shubert, T.; Zhao, C.; Gage, F.H. Exercise enhances learning and hippocampal neurogenesis in aged mice. J. Neurosci. 2005, 25 (Suppl. 38), 8680–8685. [Google Scholar] [CrossRef] [PubMed]
- Almaguer-Melian, W.; Cruz-Aguado, R.; Riva, C.L.; Kendrick, K.M.; Frey, J.U.; Bergado, J. Effect of LTP-reinforcing paradigms on neurotransmitter release in the dentate gyrus of young and aged rats. Biochem. Biophys. Res. Commun. 2005, 327, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Tombaugh, G.C.; Rowe, W.B.; Chow, A.R.; Michael, T.H.; Rose, G.M. Theta-frequency synaptic potentiation in CA1 in vitro distinguishes cognitively impaired from unimpaired aged Fischer 344 rats. J. Neurosci. 2002, 22, 9932–9940. [Google Scholar] [CrossRef] [PubMed]
- Rowe, W.B.; Spreekmeester, E.; Meaney, M.J.; Quirion, R.; Rochford, J. Reactivity to novelty in cognitively-impaired and cognitively unimpaired aged rats and young rats. Neuroscience 1998, 83, 669–680. [Google Scholar] [CrossRef]
- Tietze, F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: Applications to mammalian blood and other tissues. Anal. Biochem. 1969, 27, 502–522. [Google Scholar] [CrossRef]
- Buege, J.A.; Aust, S.D. Microsomal lipid peroxidation. Methods Enzymol. 1978, 52, 302–310. [Google Scholar] [PubMed]
- Lucas, K.K.; Dennis, E.A. Distinguishing phospholipase A2 types in biological samples by employing group-specific assays in the presence of inhibitors. Prostag. Other Lipid Mediat. 2005, 77, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Shukitt-Hale, B.; Casadesus, G.; Cantuti-Castelvetri, I.; Joseph, J.A. Effect of age on object exploration, habituation, and response to spatial and nonspatial change. Behav. Neurosci. 2001, 115, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.; Wilson-Delfosse, A.L.; Mieyal, J.J. Dysregulation of Glutathione Homeostasis in Neurodegenerative Diseases. Nutrients 2012, 4, 1399–1440. [Google Scholar] [CrossRef] [PubMed]
- Allam, F.; Dao, T.; Gaurav, C.; Bohar, R.; Farzan, J. Grape Powder supplementation prevents oxidative stress-induced anxiety-like behaviour, memory impairment and high blood pressure in rats. J. Nutr. 2013, 8, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Lezcano, L.; Jimenez-Martin, J.; Díaz-Hung, M.L.; Alberti-Amador, E.; Wong-Guerra, M.; González-Fraguela, M.E.; Estupiñán-Díaz, B.; Serrano-Sánchez, T.; Francis-Turner, L.; Delgado-Ocaña, S.; et al. Motor dysfunction and alterations in glutathione concentration, cholinesterase activity, and BDNF expression in substantia nigra pars compacta in rats with pedunculopontine lesion. Neuroscience 2017, 348, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. ER calcium and Alzheimer’s disease: In a state of flux. Sci. Signal. 2010, 3, pe10. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.S.; Kellom, M.; Reese, E.A.; Rapoport, S.I.; Kim, H.-W. Dysregulated glutamate and dopamine transporters in postmortem frontal cortex from bipolar and schizophrenic patients. J. Affect. Disord. 2012, 136, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Chalimoniuk, M.; Stolecka, A.; Zieminska, E.; Stepien, A.; Langfort, J.; Strosznaider, J.B. Involvement of multiple protein kinases in cPLA2 phosphorylation, arachidonic acid release, and cell death in vivo and in vitro models of 1-methyl-4-phenylpyridinium-induced parkinsonism—The possible key role of PKG. J. Neurochem. 2009, 110, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Last, V.; Williams, A.; Werling, D. Inhibition of cytosolic phospholipase A2 prevents prion peptide-induced neuronal damage and co-localisation with beta iii tubulin. BMC Neurosci. 2012, 13, 106. [Google Scholar] [CrossRef] [PubMed]
- Beckhauser, T.F.; Francis-Oliveira, J.; De Pasquale, R. Reactive Oxygen Species: Physiological and Physiopathological Effects on Synaptic Plasticity. J. Exp. Neurosci. 2016, 10 (Suppl. 1), 23–48. [Google Scholar] [CrossRef] [PubMed]
- Serrano, F.; Chang, A.; Hernandez, C.; Pautler, R.G.; Sweatt, J.D.; Klann, E. NADPH oxidase mediates beta-amyloid peptide-induced activation of ERK in hippocampal organotypic cultures. Mol. Brain 2009, 2, 31. [Google Scholar] [CrossRef] [PubMed]
- Adibhatla, R.M.; Hatcher, J.F.; Dempsey, R.J. Phospholipase A2, hydroxyl radicals, and lipid peroxidation in transient cerebral ischemia. ARS 2003, 5 (Suppl. 5), 647–654. [Google Scholar] [CrossRef] [PubMed]
- Cocco, T.; Di Paola, M.; Papa, S.; Lorusso, M. Arachidonic acid interaction with the mitochondrial electron transport chain promotes reactive oxygen species generation. Free Radic. Biol. Med. 1999, 27 (Suppl. 1–2), 1–2). [Google Scholar] [CrossRef]
- Jiang, R.; Chen, S.; Shen, Y.; Wu, J.; Chen, S.; Wang, A.; Wu, S.; Zhao, X. Higher Levels of Lipoprotein Associated Phospholipase A2 is associated with Increased Prevalence of Cognitive Impairment: The APAC Study. Sci. Rep. 2016, 6, 33073. [Google Scholar] [CrossRef] [PubMed]
- Shelat, P.B.; Chalimoniuk, M.; Wang, J.H.; Strosznajder, J.B.; Lee, J.C.; Sun, A.Y.; Simonyi, A.; Sun, G.Y. Amyloid beta peptide and NMDA induce ROS from NADPH oxidase and AA release from cytosolic phospholipase A2 in cortical neurons. J. Neurochem. 2008, 106 (Suppl. 1), 45–55. [Google Scholar] [CrossRef] [PubMed]
- Snyder, E.M.; Nong, Y.; Almeida, C.G.; Paul, S.; Moran, T.; Choi, E.Y.; Nairn, A.C.; Salter, M.W.; Lombroso, P.J.; Gouras, G.K.; et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat. Neurosci. 2005, 8, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Calon, F.; Lim, G.P.; Morihara, T.; Yang, F.; Ubeda, O.; Salem, N., Jr.; Frautschy, S.A.; Cole, G.M. Dietary n-3 polyunsaturated fatty acid depletion activates caspases and decreases NMDA receptors in the brain of a transgenic mouse model of Alzheimer’s disease. Eur. J. Neurosci. 2005, 22, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Bergado, A.; Lucas, M.; Richter-Levin, G. Emotional tagging—A simple hypothesis in a complex reality. Prog. Neurobiol. 2011, 94 (Suppl. 1), 64–76. [Google Scholar] [CrossRef] [PubMed]
- Baxter, P.S.; Bell, K.F.S.; Hasel, P.; Kaindl, A.M.; Fricker, M.; Thomson, D.; Cregan, S.P.; Gillingwater, T.H.; Hardingham, G.H. Synaptic NMDA receptor activity is coupled to the transcriptional control of the glutathione system. Nat. Commun. 2015, 6, 6761. [Google Scholar] [CrossRef] [PubMed]
- Foster, C.F. Dissecting the age-related decline on spatial learning and memory tasks in rodent models: N-methyl-d-aspartate receptors and voltage-dependent Ca2+ channels in senescent synaptic plasticity. Prog. Neurobiol. 2012, 96 (Suppl. 3), 283–303. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, Y.; Yang, Z. Myocardial infarction induces cognitive impairment by increasing the production of hydrogen peroxide in adult rat hippocampus. Neurosci. Lett. 2014, 560, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.D.; Antigenic, A.; Gancsos, M.; Ichinose, M.; Corlett, P.R.; Krystal, J.H.; Wang, X.J. Linking microcircuit dysfunction to cognitive impairment: Effects of disinhibition associated with schizophrenia in a cortical working memory model. Cereb. Cortex 2014, 24, 859–872. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Suzuki, M.; Sasabe, J.; Takahashi, S.; Unekawa, M.; Mashima, K.; Iizumi, T.; Hamase, K.; Konno, R.; Aiso, S.; et al. Cellular origin and regulation of D- and L serine in in vitro and in vivo models of cerebral ischemia. J. Cereb. Blood Flow Metab. 2014, 34, 1928–1935. [Google Scholar] [CrossRef] [PubMed]
- Balu, D.T.; Coyle, J.T. The NMDA receptor ‘glycine modulatory site’ in schizophrenia: D-serine, glycine, and beyond. Curr. Opin. Pharmacol. 2015, 20, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Benneyworth, M.A.; Li, Y.; Basu, A.C.; Bolshakov, V.Y.; Coyle, J.T. Cell selective conditional null mutations of serine racemase demonstrate a predominate localization in cortical glutamatergic neurons. Cell. Mol. Neurobiol. 2012, 32, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Imanishi, N.; Mita, M.; Hamase, K.; Aiso, S.; Sasabe, J. Heterogeneity of D-Serine Distribution in the Human Central Nervous System. ASN Neuro 2017, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wolosker, H.; Balu, D.T.; Coyle, J.T. The Rise and Fall of the d-Serine-Mediated Gliotransmission Hypothesis. Trends Neurosc. 2016, 39, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Vanoni, M.A.; Cosma, A.; Mazzeo, D.; Mattevi, A.; Todone, F.; Curti, B. Limited proteolysis and X-ray crystallography reveal the origin of substrate specificity and of the rate-limiting product release during oxidation of D-amino acids catalyzed by mammalian D-amino acid oxidase. Biochemistry 1997, 36, 5624–5632. [Google Scholar] [CrossRef] [PubMed]
- Sasabe, J.; Chiba, T.; Yamada, M.; Okamoto, K.; Nishimoto, I.; Matsuoka, M.; Aiso, S. D-Serine is a key determinant of glutamate toxicity in amyotrophic lateral sclerosis. EMBO J. 2007, 26, 4149–4159. [Google Scholar] [CrossRef] [PubMed]
- Miladinovic, T.; Nashed, M.G.; Singh, G. Overview of Glutamatergic Dysregulation in Central Pathologies. Biomolecules 2015, 5, 3112–3141. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Qiao, H.; Wen, L.; Zhou, W.; Zhang, Y. D-serine enhances impaired long-term potentiation in CA1 subfield of hippocampal slices from aged senescence-accelerated mouse prone/8. Neurosci. Lett. 2005, 379, 7–12. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Blocks (B) | Aged Rats (Mean ± SD Mean) | Young (Mean ± SD Mean) |
|---|---|---|
| B1 | 54.5 ± 1.62 | 38.20 ± 1.32 |
| B2 | 53.30 ± 1.33 | 34.02 ± 1.087 |
| B3 | 50.26 ± 1.97 | 30.99 ± 1.61 |
| B4 | 49.41 ± 1.48 | 22.96 ±1.20 |
| B5 | 49.7 ± 1.56 | 15.30 ± 1.27 |
| B6 | 49..59 ± 1.54 | 11.96 ± 1.26 |
| B7 | 48.8 ± 1.70 | 12.75 ± 1.57 |
| B8 | 48.2 ± 1.73 | 10.66 ± 1.59 |
| B9 | 46.12 ± 1.60 | 11.58 ± 1.94 |
| B10 | 46.06 ± 1.7 | 11.70 ± 1.92 |
| Number of Crossings | 0.9 ± 1.6 | 7.5 ± 2.3 |
| Biochemical Indicators | Frontal Cortex Aged Rats | Frontal Cortex Young Rats | Hippocampus Aged Rats | Hippocampus Young Rats |
|---|---|---|---|---|
| GSH | 1.3 ± 0.1 | 5.45 ± 1.2 | 0.76 ± 0.22 | 5.15 ± 0.2 |
| MDA | 43.42 ± 10.6 | 34.93 ± 5.95 | 80.31 ± 3.01 | 15.84 ± 2.02 |
| PLA2 | 0.2 ± 0.06 | 0.08 ± 0.0 | 0.35 ± 0.03 | 0.11 ± 0.01 |
| L Glutamate | 5.29 ± 0.8 | 5.17 ± 1.10 | 10.24 ± 1.2 | 6.89 ± 1.2 |
| GABA | 0.88 ± 0.03 | 0.94 ± 0.02 | 1.11 ± 0.03 | 1.17 ±0.03 |
| DL Serine | 4.85 ± 0.8 | 6.07 ± 0.55 | 2.13 ± 1.02 | 5.52 ± 1.2 |
| DL Aspartate | 5.35 ± 0.72 | 5.4 ± 0.55 | 5.02 ± 1.03 | 5.16 ± 1.2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Fraguela, M.E.; Blanco-Lezcano, L.; Fernandez-Verdecia, C.I.; Serrano Sanchez, T.; Robinson Agramonte, M.D.l.A.; Cardellá Rosales, L.L. Cellular Redox Imbalance and Neurochemical Effect in Cognitive-Deficient Old Rats. Behav. Sci. 2018, 8, 93. https://doi.org/10.3390/bs8100093
González-Fraguela ME, Blanco-Lezcano L, Fernandez-Verdecia CI, Serrano Sanchez T, Robinson Agramonte MDlA, Cardellá Rosales LL. Cellular Redox Imbalance and Neurochemical Effect in Cognitive-Deficient Old Rats. Behavioral Sciences. 2018; 8(10):93. https://doi.org/10.3390/bs8100093
Chicago/Turabian StyleGonzález-Fraguela, Maria Elena, Lisette Blanco-Lezcano, Caridad Ivette Fernandez-Verdecia, Teresa Serrano Sanchez, Maria De los A. Robinson Agramonte, and Lidia Leonor Cardellá Rosales. 2018. "Cellular Redox Imbalance and Neurochemical Effect in Cognitive-Deficient Old Rats" Behavioral Sciences 8, no. 10: 93. https://doi.org/10.3390/bs8100093
APA StyleGonzález-Fraguela, M. E., Blanco-Lezcano, L., Fernandez-Verdecia, C. I., Serrano Sanchez, T., Robinson Agramonte, M. D. l. A., & Cardellá Rosales, L. L. (2018). Cellular Redox Imbalance and Neurochemical Effect in Cognitive-Deficient Old Rats. Behavioral Sciences, 8(10), 93. https://doi.org/10.3390/bs8100093