Colorectal Cancer: Genetic Abnormalities, Tumor Progression, Tumor Heterogeneity, Clonal Evolution and Tumor-Initiating Cells



Abstract
1. Introduction
2. Colorectal Carcinogenesis
2.1. Normal Intestinal Stem Cells
2.2. Oncogenic Transformation of Intestinal Stem Cells
2.3. Hereditary Syndromes Associated with Frequent Colorectal Cancer
2.4. The Serrated Pathway
3. Somatic Genetic Abnormalities in Colon Cancer
3.1. General Studies
3.2. PIK3CA Mutations
3.3. BRAF Mutations
3.4. KRAS Mutations
3.5. HER2 Mutations
3.6. Molecular Subtypes
3.7. Molecular Mechanisms of Resistance to Anti-EGFR Therapy
3.8. Molecular Abnormalities in Colitis-Associated Colorectal Cancer
3.9. Molecular Abnormalities Associated with the Serrated Pathway
3.10. Molecular Abnormalities in Synchronous Colorectal Cancer
3.11. Molecular Abnormalities Associated with Metastatic Disease
3.12. Intratumor Heterogeneity and Colorectal Cancer Evolution
3.13. Tumor Location
4. Gene Expression Studies
5. Animal Models of Colorectal Cancer
5.1. Colon Cancer Metastases
5.2. Early-Onset Colorectal Cancer
5.3. Genetic Abnormalities in Colorectal Neuroendocrine Carcinomas
5.4. Genetic Abnormalities in Small-Bowel Adenocarcinoma
5.5. Genetic Abnormalities in Colorectal Carcinomas with a Signet-Ring Cell Component
6. Colon Cancer Stem Cells
6.1. Membrane Markers
6.2. Functional Properties and Signaling Pathways
6.3. Transcription Factors and Colon Cancer Stem Cells
6.4. Colon Cancer Stem Cells and Chronic Inflammation
6.5. Cancer Stem Cells in Colorectal Neuroendocrine Cancer
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ACVR2A | Activin A Receptor type 2A |
| APC | Adenomatous Polyposis Coli |
| ARID1A | AT-Rich Interaction Domain 1A |
| ATM | Ataxia Telangiaectasia Mutated |
| ATOH1 | Atonal BHLH Transcritpion Factor 1 |
| bFGF | Basic fibroblast growth factor |
| BAX | Bcl-2-Associated X protein |
| BMP | Bone Morphogenetic Protein |
| BMPR1 | Bone Morphogenetic Protein Receptor 1 |
| BRAF | B-RAF |
| CASP8 | Caspase 8 |
| CDKN2A | Cyclin-Dependent Kinase |
| CDX2 | Caudal Homeobox 2 |
| CIN | Chromosomal Instability |
| CIMP | CpG Island Methylator Phenotype |
| CHEK2 | Checkpoint kinase 2 |
| CMS | Consensus Molecular Subtype |
| CNA | Copy Number Alteration |
| CRC | Colorectal Carcinoma |
| CtBP1 | C-terminal Binding Protein 1 |
| CTNNB1 | Catenin beta 1 |
| dARE | Depleted in AU-Rich Elements |
| EGFR | Epidermal Growth Factor Receptor |
| EIF3E | Eukaryotic Translation Initiation Factor Subunit 3E |
| EPCAM | Epithelial Cell Adhesion Molecule |
| ERB | Erythroblastic Leukemia Viral Oncogene Homolog |
| ERK | Extracellular Signal-Regulated Kinase |
| FBXW7 | F box and WD Repeat Domain Containing 7 |
| FAP | Familial Adenomatous Polyposis |
| GREM1 | Gremlin 1 |
| HLA | Human Leukocyte Antigen |
| HSP90 | Heat Shock Protein 90 |
| HLH | Helix-Loop-Helix |
| KRAS | Kristen Rat Sarcoma |
| LGR5 | Leucine-Rich Repeat-Containing G-Protein Coupled Receptor 5 |
| MAPK | Mitogen-Activated Protein Kinase |
| MEK | MAPK Extracellular Signal-Regulated Kinase |
| MGMT | Methyl Guanine DNA Methyltransferase |
| MLH1 | MUTL Homolog 1 |
| MMR | Mis-Match Repair |
| MSH-1 | MutS Homolog 1 |
| MSH-2 | MutS Homolog 2 |
| MSH-6 | MutS Homolog 6 |
| MSI | Microsatellite Instability |
| MSS | Microsatellite Stable |
| MUTYH | MUTY Homolog |
| NFkB | Nuclear Factor Kappa-Light |
| NRAS | Neuroblastoma Rat Sarcoma |
| NK | Natural Killer |
| NTHL1 | NTH Like DNA Glycosylase 1 |
| P16 | Protein 16 |
| PDGRFA | Platelet Derived Growth Factor Receptor Alpha |
| PGE2 | Prostglandin E2 |
| PI3K | Phosphatidyl-Inositol-3 Kinase |
| PI3KCA | Phosphatidyl-Inositol-3 Kinase Catalytic |
| PMS2 | PostMeiotic Segregation increased 2 |
| POLD1 | Polymerase δ1 |
| POLE | DNA Polymerase E subunit |
| PTEN | Phosphatase and Tensin Homolog |
| PTGS2 | Prostglandin-Endoperoxide Synthase 2 |
| PTPRK | Protein Tyrosine Phosphatase Receptor Type K |
| RFN43 | Ring Finger Protein 43 |
| RSPO2 | R-Spondin 2 |
| RSPO3 | R-Spondin 3 |
| SCID | Severe Immunodeficiency |
| SHH | Sonic HedgeHog |
| SMAD4 | Small Mother Against Decapentaplegic 4 |
| SMO | Smoothened, Frizzled Class Receptor |
| SOX2 | SRY-Box 2 |
| SOX9 | SRY-Box 9 |
| STK11 | Serine/Threonine Kinase 11 |
| TCGA | The Cancer Genoma Atlas |
| TCF7L2 | Transcription Factor 7 Like 2 |
| TGF-β | Transforming Growth Factor-Beta |
| TGFBR2 | Transforming Growth Factor Receptor 2 |
| TIAM1 | T Cell Lymphoma Invasion and Metastasis |
| VEGF | Vascular Endothelial Growth Factor |
| VEGFR | Vascular Endothelial Growth Factor Receptor |
| WNT | Wingless-Type MMTV Integration Site Signaling |
| YAP1 | Yes-Associate Protein 1 |
References
- Clevers, H. The intestinal crypt, a prototype stem cell compartment. Cell 2013, 154, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; van de Watering, M.; Clevers, H. The intestinal stem cell. Genes Dev. 2008, 22, 1856–1864. [Google Scholar] [CrossRef] [PubMed]
- Powell, A.E.; Wang, Y.; Li, Y.; Poulin, E.J.; Means, A.L.; Washington, M.K.; Higginbotham, J.N.; Juchheim, A.; Prasad, N.; Levy, S.E.; et al. The pan-ErbB negative regulator Lrig1 an intestinal stem cells marker that functions as a tumor suppressor. Cell 2012, 49, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Yui, S.; Nakamura, T.; Sato, T.; Nemoto, Y.; Mizutani, T.; Zheng, X.; Ichinose, S.; Nagaishi, T.; Okamoto, R.; Tsuchiya, K.; et al. Functional engraftment of colon epithelium expanded in vitro from a single adult Lgr5+ stem cell. Nat. Med. 2012, 18, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Louvard, D.; Sigaux, F.; Robine, S. The power of one. Sci. Transl. Med. 2012, 4, 130fs7. [Google Scholar] [CrossRef] [PubMed]
- Buczacki, S.; Zecchini, H.I.; Nicholson, A.; Russell, R.; Vermeulen, L.; Kemp, R.; Winton, D.J. Intestinal label-retaining cells are secretory precursors expressing LGR5. Nature 2013, 495, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. A unifying theory for the crypt. Nature 2013, 495, 53–54. [Google Scholar] [CrossRef] [PubMed]
- Gehart, H.; Clevers, H. Repairing organs: Lessons from intestine and liver. Trends Genet. 2015, 341, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, C.; Kljavin, N.M.; Ybarra, N.M.; de Sauvage, F.J. Lgr5+ stem cells are indispensable for radiation-induced intestinal regeneration. Cell Stem Cell 2014, 14, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Sipos, F.; Muzes, G. Injury-associated reacquiring of intestinal stem cell function. World J. Gastroenterol. 2015, 21, 2005–2010. [Google Scholar] [CrossRef] [PubMed]
- Van Es, J.H.; Sato, T.; van de Wetering, M.; Lyubimova, A.; Yee Nee, A.N.; Gregorieff, A.; Sasaki, N.; Zeinstra, L.; van den Born, M.; Korving, J.; et al. Dll1+ secretory progenitor cells revert to stem cells upon crypt damage. Nat. Cell Biol. 2012, 14, 1099–1104. [Google Scholar] [CrossRef] [PubMed]
- Tetteh, P.W.; Basak, O.; Farin, H.F.; Wiebrands, K.; Kretzschmar, K.; Begthel, H.; van den Born, M.; Korving, J.; de Sauvage, F.; van Es, J.H.; et al. Replacement of lost LGR5-positive stem cells through plasticity of their enterocyte-lineage daughters. Cell Stem Cell 2016, 18, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Farin, H.F.; vanEs, J.H.; Clevers, H.; Langer, R.; Karp, J.M. Niche-independent high-purity cultures of LGR5+ intestinal stem cells and their progeny. Nat. Methods 2014, 11, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Grun, D.; Lyubimova, A.; Kester, L.; Wiebrands, K.; Basak, O.; Sasaki, N.; Clevers, H.; van Oudenaarden, A. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature 2015, 525, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Colman, M.; Schewe, M.; Stigter, E.; Gerrits, J.; Pras-Raves, M.; Sacchetti, A.; Horrusweld, M.; Ooh, K.; Snippert, H.; Vrenoven-Duif, N.; et al. Interplay between metabolic identities in the intestinal crypt support stem cell function. Nature 2017, 543, 424–427. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Sachs, N.; Wiebrands, K.; Ellenbrock, S.; Fumagalli, A.; Lyubinova, A.; Begthel, A.; van den Born, M.; van Es, J.; Karthaus, W.; et al. Reg4+ deep secretory cells function as epithelial niche for Lgr5+ stem cells in colon. Proc. Natl. Acad. Sci. USA 2016, 113, E5399–E5407. [Google Scholar] [CrossRef] [PubMed]
- Barriga, F.M.; Montagni, E.; Mana, M.; Mendez-Lago, M.; Hernando-Mamblona, X.; Sevillano, M.; Guillaumet-Adkins, A.; Rodriguez-Esteban, G.; Buczocki, S.J.A.; Gut, M.; et al. Mex3a marks a slowly dividing subpopulation of Lgr5+ intestinal stem cells. Cell Stem Cell 2017, 20, 801–816. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Modeling development and disease with organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef] [PubMed]
- Gijerevski, N.; Sachs, N.; Manfrin, A.; Giger, S.; Bragina, M.E.; Ordóñez-Morán, P.; Clevers, H.; Lutolf, M.P. Designer matrices for intestinal stem cells and organoid culture. Nature 2016, 539, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Ritsma, L.; Ellenbrock, S.I.J.; Zomer, A.; Snippert, H.J.; de Sauvage, F.J.; Simons, B.D.; Clevers, H.; van Rheenen, J. Intestinal crypt homeostasis revealed at single-stem-cell level by in vivo live imaging. Nature 2014, 507, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Barry, E.R.; Morikawa, T.; Butler, B.; Shrestha, K.; de la Rosa, R.; Yan, K.S.; Fuchs, C.S.; Magness, S.T.; Smits, R.; Ogino, S.; et al. Restriction of intestinal stem cells expansion and the regenerative response by YAP. Nature 2013, 493, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.K.; Spit, M.; Jordens, I.; Low, T.Y.; Stange, D.E.; van de Wetering, M.; van Es, J.H.; Mohammed, S.; Heck, A.J.; Maurice, M.M.; et al. Tumor suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 2012, 488, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Schuijers, J.; Junker, J.P.; Mokry, M.; Hatzis, P.; Koo, B.K.; Sasselli, V.; van der Flier, L.G.; Cuppen, E.; van Oudenaarden, A.; Clevers, H. Ascl2 acts as an R-spondin responsive switch to control stemness in intestinal crypts. Cell Stem Cell 2015, 16, 158–170. [Google Scholar] [CrossRef] [PubMed]
- De Lau, W.; Peng, W.C.; Gros, P.; Clevers, H. The R-spondin (LGR5/RNF43 module): Regulator of WNT signal strength. Genes Dev. 2014, 28, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.M.; Calabrese, P.; Miller, A.; Muñoz, N.M.; Grady, W.M.; Shibata, D.; Liskay, R.M. Single cell lineage tracing reveals a role for TGFβR2 in intestinal stem cell dynamics and differentiation. Proc. Natl. Acad. Sci. USA 2016, 113, 12192–12197. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.S.; Jand, C.Y.; Cheng, J.; Zheng, G.X.Y.; Larkin, K.A.; Luca, V.C.; Chia, L.A.; Mah, A.T.; Tery, J.M.; Ootani, A. Non-equivalence of Wnt and R-spondin ligands during Lgr5+ intestinal stem-cell self-renewal. Nature 2017, 545, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Sangiorgi, E.; Capecchi, M.R. Bmi1 is expressed in vivo in intestinal stem cells. Nat. Genet. 2008, 40, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.S.; Chia, L.A.; Li, X.; Ootani, A.; Su, L.; Lee, J.Y.; Su, N.; Luo, Y.; Heilshorn, S.C.; Annieva, M.R.; et al. The intestinal stem cell markers Bmi1 and Lgr5 identify two functionally distinct populations. Proc. Natl. Acad. Sci. USA 2012, 109, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.S.; Gevaert, O.; Zheng, G.; Anchaug, B.; Probert, C.S.; Larkin, K.A.; Davies, P.S.; Cehng, Z.; Raddis, J.S.; Han, A.; et al. Intestinal enteroendocrine lineage cells possess homeostatic and injury-inducible stem cell activity. Cell Stem Cell 2017, 21, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, U.; Saxena, M.; O’Neill, N.; Saadatpur, A.; Yvan, G.C.; Herbert, Z.; Murata, K.; Shidvasani, R. Dynamic reorganization of chromatin accessibility signatures during differentiation of secretory precursors into Lgr5+ intestinal stem cells. Cell Stem Cell 2017, 21, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, M.; Li, L.; Lengner, C.J. Hierarchy and plasticity in the intetsinal cell compartment. Trends Cell Biol. 2017, 27, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Snipper, H.J.; van der Flier, L.; Sato, T.; van Es, J.H.; van den Born, M.; Kroon-Veenboer, C.; Barker, N.; Klein, A.M.; van Rheenen, J.; Simons, B.D.; et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 2010, 143, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Garcia, C.; Klein, A.M.; Simons, B.D.; Winton, D.J. Intestinal stem cell replacement follows a pattern of neutral drift. Science 2010, 330, 822–825. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; Morrissey, E.; van der Heijden, M.; Nicholson, A.M.; Sottoriva, A.; Buczacki, S.; Kemp, R.; Tavaré, S.; Winton, D.J. Defining stem cell dynamics in models of intestinal tumor initiation. Science 2013, 342, 995–998. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.C.; Van Den Berg, D.; Kang, H.; Hsieh, C.L.; Lieber, M.R. Large chromosome deletions, duplications, and gene conversion events accumulate with age in normal human colon crypts. Aging Cell 2013, 12, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Sieudeja, K.; Nassari, S.; Gervais, L.; Skorski, P.; Lameiras, S.; Stolfa, D.; Zande, M.; Bernard, V.; Frio, T.R.; Bardin, A.J. Frequent somatic mutation in adult intestinal stem cells drives neoplasia and genetic mosaicism during aging. Cell Stem Cell 2015, 27, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.M.; Cereser, B.; Melton, S.; Fletcher, A.G.; Rodriguez-Justo, M.; Tadrous, P.J.; Humphries, A.; Elia, G.; McDonald, S.A.; Wright, N.A.; et al. Quantification of crypt and stem cell evolution in the normal and neoplastic human colon. Cell Rep. 2014, 8, 940–947. [Google Scholar] [CrossRef] [PubMed]
- Jung, P.; Sommer, C.; Barriga, F.M.; Buczacki, S.J.; Hernonda Momblona, X.; Duran-Frigola, M.; Aloy, P.; Slebach, M.; Winton, D.J.; Batlle, E.; et al. Isolation of human colon stem cells using surface expression of PTK7. Cell Rep. 2015, 5, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Phelps, R.A.; Chidester, S.; Dehghanizadeh, S.; Phelps, J.; Sandoval, I.T.; Rai, K.; Broadbent, T.; Sarkar, S.; Burt, R.W.; Jones, D.A. A two-step model for colon adenoma initiation and progression caused by APC loss. Cell 2009, 137, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Dow, L.E.; O’Rourke, K.P.; Simon, J.; Tschaharganeh, D.F.; van Es, J.H.; Clevers, H.; Lowe, S.W. Apc restoration promotes cellular differentiation and reestablishes crypt homeostasis in colorectal cancer. Cell 2015, 161, 1539–1552. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Yamada, Y.; Semi, K.; Yagi, M.; Tanaka, A.; Itakura, F.; Aoki, K.; Kunisada, T.; Woltjen, K.; Haga, H.; et al. Cellular context-dependent consequences of Apc mutations of gene regulation and cellular behavior. Proc. Natl. Acad. Sci. USA 2017, 114, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Chen, W.; Parmigiani, G.; Diehl, F.; Beerenwinkel, N.; Antal, T.; Trauls, A.; Nowak, M.A.; Siegel, C.; Velculescu, V.E.; et al. Comparative lesion sequencing provides insights into tumor evolution. Proc. Natl. Acad. Sci. USA 2008, 105, 4283–4288. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, S.I.; Sotiriou, S.K.; Pateras, I.S.; Santoni, F.; Sougioultzis, S.; Edgren, H.; Almusa, H.; Robyr, D.; Guipponi, M.; Saarela, J.; et al. A single-nucleotide substitution mutate phenotype revealed by exome sequencing of human colon adenomas. Cancer Res. 2012, 72, 6279–6289. [Google Scholar] [CrossRef] [PubMed]
- Schwitalla, S.; Ziegler, P.; Horst, D.; Becker, V.; Kerle, I.; Begus-Nahrmann, Y.; Lechel, A.; Rudolph, K.L.; Langer, R.; Slotta-Huspenina, J.; et al. Loss of p53 in enterocytes generates an inflammatory microenvironment enabling invasion and lymph node metastasis of carcinogen-induced colorectal tumors. Cancer Cell 2013, 23, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Leystra, A.; Deming, D.; Zahm, C.; Farhoud, M.; Olson, T.J.; Hadac, J.N.; Nettekoven, L.A.; Albrecht, D.M.; Clipson, L.; Sullivan, R.; et al. Mice expressing activated PI3K rapidly develop advanced colon cancer. Cancer Res. 2012, 72, 2931–2936. [Google Scholar] [CrossRef] [PubMed]
- Deming, D.; Leystra, A.; Nettekoven, L.; Sievers, C.; Miller, D.; Middlebrooks, M.; Clipson, L.; Albrecht, D.; Bacher, J.; Washington, M.K.; et al. PI3KCA and APC mutations are synergistic in the development of intestinal cancers. Oncogene 2013, 33, 2245–2254. [Google Scholar] [CrossRef] [PubMed]
- Onuma, K.; Ochiai, M.; Orihashi, K.; Takahashi, M.; Imai, T.; Nakagama, H.; Hippo, Y. Genetic reconstitution of tumorigenesis in primary intestinal cells. Proc. Natl. Acad. Sci. USA 2013, 110, 11121–11132. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.; Gao, R.; Navin, N. Tumor evolution: Linear, branching, neutral or punctuated? Biochim. Biophys. Acta 2017, 1867, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Sievers, C.K.; Grady, W.; Halberg, R.; Rickardt, P. New insights into the earliest stages of colorectal tumorigenesis. Exp. Rev. Gastroenterol. Hepatol. 2017, 11, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuis, M.; Vasen, H. Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): A review of the literature. Crit. Rev. Oncol. Hematol. 2007, 61, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Christie, M.; Jorissen, R.N.; Mouradov, D.; Sakthianandeswaren, A.; Li, S.; Day, F.; Tsui, C.; Lipton, L.; Desai, J.; Jones, I.T.; et al. Different APC genotypes in proximal and distal sporadic colorectal cancers suggest distinct WNT/β-catenin signaling thresholds for tumorigenesis. Oncogene 2013, 32, 4675–4682. [Google Scholar] [CrossRef] [PubMed]
- Fodde, R.; Kuipers, J.; Rosenberg, C.; Smits, R.; Kielman, M.; Gaspar, C.; van Es, J.H.; Breukel, C.; Wiegant, J.; Giles, R.H.; et al. Mutations in the APC tumor suppressor gene cause chromosomal instability. Nat. Cell Biol. 2001, 3, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, M.; Furuya, T.; Shiraki, A.; Sato, T.; Oga, A.; Sasaki, K. The relationship of DNA ploidy to chromosomal instability in primary human colorectal cancers. Cancer Res. 1999, 59, 5283–5285. [Google Scholar] [PubMed]
- Hautey, W.; McIlhatton, M.; Ebede, K.; Kenedy, B.; Haucioglu, B.; Zhang, J.; Brock, G.; Honeg, K.; Groda, J. Mutational mechanisms that activate Wnt signaling and predict outcomes on colorectal cancer patients. Cancer Res. 2017, 78, 617–630. [Google Scholar]
- Walther, A.; Johnstone, E.; Swanton, C.; Midgley, R.; Tomlinson, I.; Kerr, D. Genetic prognostic and predictive markers in colorectal cancer. Nat. Rev. Cancer 2009, 9, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Schepers, A.G.; Snipper, H.J.; Stange, D.E.; van den Born, M.; van Es, J.H.; van de Wetering, M.; Clevers, H. Lineage tracing reveals Lgr5+ stem cell activity in the mouse intestinal adenomas. Science 2012, 337, 730–735. [Google Scholar] [CrossRef] [PubMed]
- Boman, B.M.; Fields, J.Z.; Bohnham-Carter, O.; Runquist, O.A. Computer modelling implicates stem cell overproduction in colon cancer initiation. Cancer Res. 2001, 61, 8408–8411. [Google Scholar] [PubMed]
- Boman, B.M.; Huang, E. Human colon cancer stem cells: A new paradigm in gastrointestinal oncology. J. Clin. Oncol. 2008, 26, 2828–2838. [Google Scholar] [CrossRef] [PubMed]
- Boman, B.M.; Wicha, M.S.; Fields, J.Z.; Runquist, O.A. Symmetric division of human cancer stem cells—A key mechanism in tumor growth that should be targeted in future therapeutic approaches. Clin. Pharmacol. Ther. 2007, 81, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Boman, B.M.; Walters, R.; Fields, J.Z.; Kovatich, A.J.; Zhang, T.; Isenberg, G.A.; Goldstein, S.D.; Palazzo, J.P. Colonic crypt changes during adenoma development in familial adenomatous polyposis: Immunohistochemical evidence for expansion of the crypt base cell population. Am. J. Pathol. 2004, 165, 1489–1498. [Google Scholar] [CrossRef]
- Boman, B.M.; Fields, J.Z.; Cavanaugh, K.L.; Guetter, A.; Runquist, O.A. How dysregulated colonic crypt dynamics cause stem cell overpopulation and initiate colon cancer. Cancer Res. 2008, 6, 3304–3313. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.S.; Wu, H.Y.; Yu, H.P.; Zhou, Q.; Zhang, Y.F.; Huang, Q. Expression of Lgr5 in human colorectal carcinogenesis and its potential correlation with β-catenin. Int. J. Colorectal Dis. 2010, 25, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Uchida, H.; Yamazaki, K.; Fukuma, M.; Yamada, T.; Hayashida, T.; Hasegawa, H.; Kitajima, M.; Kitagawa, Y.; Sakamoto, M. Overexpression of leucin-rich repeat-containing G protein-coupled receptor 5 in colorectal cancer. Cancer Sci. 2010, 10, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.M.; Graham, T.A.; Elin, G.; Wright, N.A.; Rodriguez-Justo, M. Characterization of LGR5 stem cells in colorectal adenomas and in carcinomas. Sci. Rep. 2015, 5, 8654. [Google Scholar] [CrossRef] [PubMed]
- Jang, B.G.; Kim, H.S.; Kim, K.J.; Rhee, Y.Y.; Kim, W.H.; Kang, G.H. Distribution of intestinal stem cell markers in colorectal precancerous lesions. Histopathology 2016, 68, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.; Xu, G.; Zhu, L.; Pang, Z.; Zheng, W.; Li, Z.; Yuan, A. Temporal and spatial changes of cells positive for stem-like markers in different compartments and stages of human colorectal adenoma-carcinoma sequence. Oncotarget 2017, 8, 45311–45322. [Google Scholar] [CrossRef] [PubMed]
- Shih, I.M.; Wang, T.L.; Traverso, G.; Romans, K.; Hamilton, S.R.; Ben-Sasson, S.; Kinzler, K.W.; Vogelstein, B. Top-down morphogenesis of colorectal tumors. Proc. Natl. Acad. Sci. USA 2001, 18, 2640–2645. [Google Scholar] [CrossRef] [PubMed]
- Preston, S.L.; Wong, W.M.; Chan, A.; Poulsom, R.; Jeffery, R.; Goodlad, R.A.; Mandir, N.; Elia, G.; Novelli, M.; Bodmer, W.F.; et al. Botom-up histogenesis of colorectal adenomas: Origin in the monocryptal adenoma and initial expansion by crypt fission. Cancer Res. 2003, 63, 3819–3825. [Google Scholar] [PubMed]
- Langlands, A.; Almet, A.; Appleton, P.; Newton, I.P.; Osborne, J.M.; Näthke, I.S. Paneth cell-rich regions separated by a cluster of LGR5+ cells initiate crypt fission in the intestinal stem cell niche. PLoS Biol. 2016, 14, e1002491. [Google Scholar] [CrossRef] [PubMed]
- Al-Karusi, M.R.; Smartt, H.J.; Greenhough, A.; Collard, T.J.; Emery, E.D.; Williams, A.C.; Paraskeva, C. LGR5 promotes survival in human colorectal adenoma cells and is upregulated by PGE2: Implications for targeting adenoma stem cells with NSAIDs. Carcinogenesis 2013, 34, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Moreau, H.; Vasen, H.F.; Hes, F.J. MUTYH-associated polyposis (MAP). Crit. Rev. Oncol. Hematol. 2011, 79, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Palles, C.; Cazier, J.P.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Salguero, I.; et al. Germline mutations in the proof-reading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013, 45, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Freeman-Mills, L.; Rayner, E.; Glaire, M.; Briggs, S.; Vermeulen, L.; Fessler, E.; Medema, J.P.; Boot, A.; Morrem, H.; et al. Somatic POLE proofreading domain mutation, immune response, and prognosis in colorectal cancer: A retrospective, pooled biomarker study. Lancet Gastroneterol. Hepatol. 2106, 1, 2007–2016. [Google Scholar] [CrossRef]
- Weren, R.D.; Ligtenberg, M.J.; Kets, C.M.; de Voer, R.M.; Verwiel, E.T.; Spruijt, L.; van Zelst-Stams, W.A.; Jongmans, M.C.; Gilissen, C.; Hehir-Kwa, J.Y.; et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat. Genet. 2015, 47, 668–671. [Google Scholar] [CrossRef] [PubMed]
- Jasperson, K.W.; Tuohy, T.M.; Neklason, D.W.; Burt, R.W. Hereditary and familial colon cancer. Gastroenterology 2010, 138, 2044–2058. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, E.; Leedham, S.; Lewis, A.; Segditsas, S.; Becker, M.; Cuadrado, P.R.; Davis, H.; Kaur, K.; Heinimann, K.; Howarth, K.; et al. Hereditary mixed polyposis syndrome is caused by a 40-Kb upstream duplication that leads to increased and ectopic expression of the BMP antagonist GREM1. Nat. Genet. 2012, 44, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Davis, H.; Irshad, S.; Bausai, M.; Rafferty, H.; Boitsova, T.; Bardella, C.; Jaeger, E.; Lewis, A.; Freeman-Mills, L.; Giner, F.C.; et al. Aberrant epithelial GREM1 expression initiates colonic tumorigenesis from cells outside the stem cell niche. Nat. Med. 2015, 21, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Yurgelun, M.B.; Kulke, M.H.; Fuchs, C.S.; Allen, B.A.; Uno, H.; Hornick, J.L.; Ukaegbu, C.I.; Brais, L.K.; McNamara, P.G.; Mayer, R.J.; et al. Cancer susceptibility gene mutations in individuals with colorectal cancer. J. Clin. Oncol. 2017, 35, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, R.; Frankel, W.L.; Swanson, B.; Zhao, W.; Yilmaz, A.; Miller, K.; Bacher, J.; Bigley, C.; Nelsen, L.; Goodfellow, P.J.; et al. Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer. JAMA Oncol. 2017, 3, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Carragher, L.A.; Snell, K.R.; Giblett, S.M.; Aldridge, V.S.; Patel, B.; Cook, S.J.; Winton, D.J.; Marais, R.; Pritchsrd, C.A. V600EBraf induces gastrointestinal crypt senescence and promotes tumour progression through enhanced CpG methylation of p16INK4a. EMBO Mol. Med. 2010, 2, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Riemer, P.; Sreekumar, A.; Reinke, S.; Rad, R.; Schafer, R.; Sers, C.; Blaker, H.; Hermann, B.G.; Morkel, M. Transgenic expression of oncogenic BRAF induces loss of stem cells in the mouse intestine, which is antagonized by β-catenin activity. Oncogene 2015, 34, 3164–3175. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, N.; Feng, Y.; Stolfi, C.; Kurosu, Y.; Green, M.; Lin, J.; Green, M.E.; Sentani, K.; Yasui, W.; McMahon, M.; et al. BRAFV600E cooperates with CDX2 inactivation to promote serrated colorectal tumorigenesis. eLife 2017, 6, e20331. [Google Scholar] [CrossRef] [PubMed]
- Landau, M.S.; Kuan, S.F.; Chiosea, S.; Pai, R.K. BRAF-mutated microsatellite stable colorectal carcinoma: An aggressive adenocarcinoma with reduced CDX2 and increased cytokeratin 7 immunohistochemical expression. Hum. Pathol. 2014, 45, 1704–1712. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.M.; Lee, T.H.; Cho, N.Y.; Kang, G.H. Loss of CDX2 expression is associated with poor prognosis in colorectal cancer patients. World J. Gastroenterol. 2015, 21, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Dalerba, P.; Sahoo, D.; Paik, S.; Guo, X.; Yothers, G.; Song, N.; Wilcox-Fogel, N.; Forgò, E.; Rajendran, P.S.; Miranda, S.P.; et al. CDX2 as a prognostic biomarker in stage II and stage III colon cancer. N. Engl. J. Med. 2016, 374, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Tong, K.; Pellon-Cardenas, O.; Sirihorachai, V.R.; Warder, B.N.; Kothari, O.A.; Perakatt, A.O.; Fokas, E.E.; Fullem, R.L.; Zhou, A.; Thackray, J.K.; et al. Degree of tissue differentiation dictates susceptibility to BRAF-driven colorectal cancer. Cell Rep. 2017, 21, 3833–3845. [Google Scholar] [CrossRef] [PubMed]
- Rad, R.; Cadinanos, J.; Rad, L.; Varela, I.; Strong, A.; Kriegl, L.; Constantino-Casas, F.; Eser, S.; Hieber, M.; Seidler, B.; et al. A genetic progression model of BrafV600E-induced intestinal tumorigenesis reveals targets for therapeutic intervention. Cancer Cell 2013, 24, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Drew, D.A.; Devers, T.J.; O’Brien, M.J.; Horelik, N.A.; Levine, J.; Rosenberg, D.W. HD chromoendoscopy coupled with DNA mass spectrometry profiling identifies somatic mutations in microdissected human proximal aberrant crypt foci. Mol. Cancer Res. 2014, 12, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, D.W.; Yang, S.; Plaeu, D.C.; Greenspan, E.J.; Stevens, R.G.; Rajan, T.T.; Heinen, C.D.; Levine, J.; Zhou, Y.; O’Brien, M.J. Mutations in BRAF and KRAS differentially distinguish serrated versus non-serrated hyperplastic aberrant crypt foci in humans. Cancer Res. 2007, 67, 3551–3554. [Google Scholar] [CrossRef] [PubMed]
- Drew, D.A.; Mo, A.; Grady, J.J.; Stevens, R.J.; Levine, J.; Brenner, B.M.; Anderson, J.C.; Foroahar, F.; O’Brien, M.J.; Devrs, T.J.; et al. Proximal aberrant crypt foci associate with synchronous neoplasia and are primed for neoplastic progression. Mol. Cancer Res. 2018, 16, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Goel, A. Molecular classification and correlates in colorectal cancers. J. Mol. Diagn. 2008, 10, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Walther, A.; Houlston, R.; Toulinson, I. Association between chromosomal instability and prognosis in colorectal cancer: A meta-analysis. Gut 2008, 57, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Jass, J.R. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology 2007, 50, 113–130. [Google Scholar] [CrossRef] [PubMed]
- Zlobec, I.; Bihl, M.P.; Foerster, A.; Rufle, A.; Terracciano, L.; Lugli, A. Stratification and prognostic relevance of Jass’s molecular classification of colorectal cancer. Front. Oncol. 2012, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, M.; Morikawa, T.; Kuchiba, A.; Imamura, Y.; Qian, Z.R.; Nishihara, R.; Liao, X.; Waldron, L.; Hoshida, Y.; Huttenhower, C.; et al. Assessment of colorectal cancer molecular features along bowel subsites challenges the conception of distinct dichotomy of proximal versus distal colerectum. Gut 2012, 61, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R.T. Molecular genetics of colorectal cancer. Annu. Rev. Pathol. 2011, 6, 479–507. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Zhang, B.; Wang, J.; Wang, X.; Zhu, J.; Liu, Q.; Shi, Z.; Chambers, M.C.; Zimmerman, L.J.; Shaddox, K.F.; Kim, S.; et al. Proteogenomic characterization of human colon and rectal cancer. Nature 2014, 513, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Seshagiri, S.; Stawiski, E.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-spondin fusions in colon cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Bass, A.J.; Lawrence, M.S.; Brace, L.E.; Ramos, A.H.; Drier, Y.; Cibulskis, K.; Sougnez, C.; Voet, D.; Saksena, G.; Sivachenko, A.; et al. Genomic sequencing of colorectal adenocarcinomas identifies a recurrent VTI1A-TCF7L2 fusion. Nat. Genet. 2011, 10, 964–970. [Google Scholar] [CrossRef] [PubMed]
- Kloosterman, W.P.; Coebergh vasn den Braak, R.R.J.; Pieterse, M.; van Roosmalen, M.J.; Sieuwerts, A.M.; Stangl, C.; Brunekreef, R.; Lalmahomed, Z.S.; Oofts, S.S.; van Galen, A.; et al. A systematic analysis of oncogenic gene fusions in primary colon cancer. Colon Cancer 2017, 77, 3814–3822. [Google Scholar] [CrossRef] [PubMed]
- Iacopetta, B.J.; Welch, J.; Soong, R.; House, A.K.; Zhou, X.P.; Hamelin, R. Mutation of the transforming growth factor-β type II receptor gene in right-sided colorectal cancer: Relationship to clinicopathological features and genetic alterations. J. Pathol. 1998, 184, 390–395. [Google Scholar] [CrossRef]
- Fleming, N.; Jorissen, R.; Mouradov, D.; Christie, M.; Sakthianandeswaren, A.; Palmieri, M.; Day, F.; Li, S.; Tsui, C.; Lipton, L.; et al. SMAD2, SMAD3 and SMAD4 mutations in colorectal cancer. Cancer Res. 2013, 73, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Fessler, E.; Drost, J.; Van Hoof, S.R.; Linnekamp, J.F.; Wang, X.; Jansen, M.; De Sousa, E.; Melo, F.; Prasetyanti, P.R.; IJspeert, J.E.; et al. TGFβ signaling directs serrated adenomas to the mesenchymal colorectal subtype. EMBO Mol. Med. 2016, 8, 745–760. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Nishihara, R.; Chen, Y.; Li, W.; Shi, Y.; Masugi, Y.; Hamada, T.; Kosumi, K.; Liu, L.; da Silva, A.; et al. Aspirin exerts high anti-cancer activity in PI3KCA-mutant colon cancer cells. Oncotarget 2017, 8, 87379–87389. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Lochhead, P.; Nishihara, R.; Morikawa, T.; Kuchiba, A.; Yamauchi, M.; Imamura, Y.; Rong Qian, Z.; Baba, Y.; Shima, K.; et al. Aspirin use, tumor PI3KCA mutation, and colorectal cancer survival. N. Engl. J. Med. 2012, 367, 1596–1606. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Phipps, A.; Burnett-Hartman, A.N.; Adams, S.V.; Hardikar, S.; Cohen, S.A.; Kocarnika, J.M.; Ahnen, D.J.; Lindor, N.M.; Baron, J.A.; et al. Timing of aspirin and other nonsteroidal anti-inflammatory drug use among patients with colorectal cancer in relation to tumor markers and survival. J. Clin. Oncol. 2017, 35, 2806–2813. [Google Scholar] [CrossRef] [PubMed]
- Hamada, T.; Cao, Y.; Qian, Z.R.; Masugi, Y.; Nowak, J.A.; Yang, Y.; Song, M.; Mima, K.; Kasumi, K.; Liu, L.; et al. Aspirin use and colorectal cancer survival according to tumor CD274 (Programmed cell death 1 Ligand 1) expression status. J. Clin. Oncol. 2017, 35, 1836–1844. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Morikawa, T.; Lochhead, P.; Imamura, Y.; Kuchiba, A.; Yamauchi, M.; Nosho, K.; Qian, Z.R.; Nishihara, R.; Meyerhardt, J.A.; et al. Prognostic role of PI3KCA mutations in colorectal cancer: Cohort study and literature review. Clin. Cancer Res. 2012, 18, 2257–2268. [Google Scholar] [CrossRef] [PubMed]
- Haley, L.; Tseng, L.H.; Zheng, G.; Dudley, J.; Anderson, D.A.; Azad, N.S.; Gocke, C.D.; Eshleman, J.R.; Lin, M.T. Performance characteristics of next-generation sequencing in clinical mutation detection of colorectal cancers. Mod. Pathol. 2015, 28, 1390–1399. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Han, X.; Wang, J.; Wang, S.; Yang, H.; Lu, S.H.; Shi, Y. Prognostic impact of mutation profiling in patients with stage II and III colon cancer. Sci. Rep. 2016, 6, 24310. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.A.; Turner, E.H.; Beightol, M.B.; Jacobson, A.; Gooley, T.A.; Salipante, S.; Haraldsdottir, S.; Smith, C.; Scroggins, S.; Tait, J.; et al. Frequent PIK3CA mutations in colorectal and endometrial cancer with double somatic mismatch repair mutations. Gastroeterology 2016, 151, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Saffae Ardekani, G.S.; Jafornejad, S.M.; Tau, L.; Saeedi, A.; Li, G. The prognostic value of BRAF mutation in colorectal cancer and melanoma: A systematic review and meta-analysis. PLoS ONE 2012, 7, e47054. [Google Scholar] [CrossRef] [PubMed]
- Kayhanian, H.; Goode, E.; Sclafani, F.; Ang, J.E.; Gerlinger, M.; Gonzalez de Castro, D.; Shepherd, S.; Peckitt, C.; Rao, S.; Watkins, D.; et al. Treatment and survival outcome of BRAF-mutated metastatic colorectal cancer: A retrospective matched case-control study. Clin. Colorectal Cancer 2017, 17, e69–e76. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; Lin, Y.L.; Liau, J.Y.; Tsai, J.H.; Tseng, L.H.; Lin, L.I.; Liang, J.T.; Lin, B.R.; Hung, J.S.; Chang, Y.L.; et al. BRAF mutation may have different prognostic implications in early- and late-stage colorectal cancer. Med. Oncol. 2016, 33, 39. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, M.; Lochhead, P.; Morikawa, T.; Huttenhower, C.; Chan, A.T.; Giovannucci, E.; Fuchs, C.; Ogino, S. Colorectal cancxer: A tale of two sides or continuum? Gut 2012, 61, 794–797. [Google Scholar] [CrossRef] [PubMed]
- Prallahad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF (V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in multiple nonmelanoma cancer with BRAF V600 mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; McDonough, S.L.; Lenz, H.J.; Magliocco, A.M.; Atteya, C.E.; Diaz, L.A. Randomized trial of irinotecan and cetuximab with or without vemurafenib in BRAF-mutant metastatic colorectal cancer (SWOG S1406). J. Clin. Oncol. 2017, 35, 520. [Google Scholar] [CrossRef]
- Yaeger, R.; Yao, Z.; Hyman, D.; Hcehtman, J.F.; Vakiani, E.; Zhao, H.Y.; Su, W.; Wang, L.; Joelson, A.; Cercek, A.; et al. Mechanisms of acquired resistance to BRAF V600E inhibition in colon cancers converge on BRAF dimerization and are sensitive to inhibition. Cancer Res. 2017, 77, 6513–6523. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Yaeger, R.; Rodrik-Outmezguine, V.; Tao, A.; Torres, N.; Chang, M.; Drosten, M.; Zhao, H.; Cecchi, F.; Hembrough, T.; et al. Tumors with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 2017, 548, 234–238. [Google Scholar] [PubMed]
- Weisenberger, D.J.; Seigmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.M.; Deasi, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicenter, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Andre, T.; Lonardi, S.; Yeung, K.; Wong, M.; Morse, M.; McDermott, R.S.; Hill, A.G. Combination of nivolumab (nivo) + ipilimumab (ipi) in the treatment of patients (pts) with deficient DNA mismatch repair (dMMR)/high microsatellite instability (MSI-H) metastatic colorectal cancer (mCRC): CheckMate 142 study. J. Clin. Oncol. 2017, 35, 3531. [Google Scholar]
- Jones, J.C.; Renfro, L.A.; Al-Shamsi, H.O.; Schrock, A.B.; Rankin, A.; Zhang, B.Y.; Kasi, P.M.; Voss, J.S.; Leal, A.D.; Sun, J.; et al. Non-V600 BRAF mutations define a clinically distinct molecular subtype of metastatic colorectal cancer. J. Clin. Oncol. 2017, 35, 2624–2630. [Google Scholar] [CrossRef] [PubMed]
- Imamura, Y.; Morikawa, T.; Liao, X.; Lochhead, P.; Kuchiba, A.; Yamauchi, M.; Qian, Z.R.; Nishihara, R.; Meyerhardt, J.A.; Haigis, K.M.; et al. Specific mutations in KRAS codon 12 and 13, and patient prognosis in 1075 BRAF wild-type colorectal cancers. Clin. Cancer Res. 2012, 18, 4753–4763. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Geninger, P.; Rhodes, D.; Koopman, L.; Violette, S.; Bardeesy, N.; Settleman, J. A gene expression signature associated with “K-Ras-addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell 2009, 15, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Sweeney, M.F.; Yu, M.; Burger, A.; Greninger, P.; Benes, C.; Haber, D.A.; Settleman, J. Tak1 inhibition promotes apoptosis in KRAS-dependent colon cancers. Cancer Cell 2012, 148, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Rimbert, J.; Tachon, G.; Junca, A.; Villalva, C.; Tapon, L.K.; Tougeron, D. Association between clinicopathological characteristics and RAS mutation in colorectal cancern. Mod. Pathol. 2017, 31, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Haigis, K.M.; Kendall, K.R.; Wang, Y.; Cheung, A.; Haigic, M.C.; Glickman, J.M.; Niwa-Kawakita, M.; Sweet-Cordero, A.; Sebolt-Leopold, J.; Shannon, K.M.; et al. Differential effects of oncogenic KRAS and NRAS on proliferation, differentiation and tumor progression in the colon. Nat. Genet. 2008, 40, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Boutin, A.T.; Liao, W.T.; Wang, M.; Hwang, S.S.; Karpinets, T.V.; cheung, H.; Chu, G.C.; Jiang, S.; Hu, J.; Chang, K.; et al. Oncogenic Kras drives invasion and maintains metastases in colorectal cancer. Genes Dev. 2017, 31, 370–382. [Google Scholar] [CrossRef] [PubMed]
- Kavuri, S.; Jain, N.; Galimi, F.; Cottino, F.; Leto, S.M.; Migliardi, G.; Searleman, A.C.; Shen, W.; Monsey, J.; Trusolino, L.; et al. HER2 activating mutations are targets for colorectal cancer treatment. Cancer Discov. 2015, 5, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-targeted therapy with trastuzumab and lapatinib in tretament-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): A proof-of-concept, multicentre, open-label phase 2 trial. Lancet Oncol. 2016, 17, 738–746. [Google Scholar] [CrossRef]
- Sanz-Pamplona, R.; Lopez-Doriga, A.; Parè-Brunet, L.; Lázaro, K.; Bellido, F.; Alonso, M.H.; Aussó, S.; Guinó, E.; Beltrán, S.; Castro-Giner, F.; et al. Exome sequencing reveals AMER1 as a frequently mutated gene in colorectal cancer. Cancer Res. 2015, 21, 4709–4718. [Google Scholar] [CrossRef] [PubMed]
- Mur, P.; Aiza, G.; Sanz-Pamplona, R.; González, S.; Navarro, M.; Moreno, V.; Capellá, G.; Valle, L. AMER1 is a frequently mutated gene in colorectal cancer—Letter. Clin. Cancer Res. 2015, 21, 4985. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Phipps, A.I.; Limburg, P.J.; Baron, J.A.; Burnett-Hartman, A.N.; Weisenberger, D.J.; Laird, P.W.; Sinicrope, F.A.; Rosty, C.; Buchanan, D.D.; Potter, J.D.; et al. Association between molecular subtypes of colorectal cancer and patient survival. Gastroenterology 2015, 148, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Sinicrope, F.A.; Shi, Q.; Smyrk, T.C.; Thibodeau, S.N.; Dienstmann, R.; Guinney, J.; Bot, B.M.; Tejpar, S.; Delorenzi, M.; Goldberg, R.M.; et al. Molecular markers identify subtypes of stage III colon cancer associated with patient outcomes. Gastroenterology 2015, 148, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Sinicrope, F.A.; Mahoney, M.R.; Smyrk, T.C.; Thibodeau, S.N.; Warren, R.S.; Bertagmoli, M.M.; Nelson, G.D.; Goldeberg, R.M.; Sargent, D.J.; Alberts, S.R. Prognostic impact of deficient DNA mismatch repair in patients with stage III colon cancer from a randomized trial of FOLFOX-based adjuvant chemotherapy. J. Clin. Oncol. 2013, 31, 3664–3672. [Google Scholar] [CrossRef] [PubMed]
- Zaanan, A.; Shi, Q.; Taieb, J.; Alberts, S.R.; Meyers, J.P.; Smyrk, T.C.; Julie, C.; Zawadi, A.; Tabernero, J.; Mini, E.; et al. Role of seficient DNA mismatch repair status in patients with stage III colon cancer treated with FOLFOX adjuvant chemotherapy: A pooled analysis from two randomized clinical trials. JAMA Oncol. 2017, 4, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Taieb, J.; Le Malicot, K.; Shi, Q.; Penault Lorca, F.; Bouché, O.; Tabernero, J.; Mini, E.; Goldberg, R.M.; Folprecht, G.; Luc Van Laethem, J.; et al. Prognostic value of BRAF and KRAS mutations in MSI and MSS stage III colon cancer. J. Natl. Cancer Inst. 2016, 109, djw272. [Google Scholar] [CrossRef] [PubMed]
- Sinicrope, F.A.; Shi, Q.; Allegra, C.J.; Smyrk, T.C.; Tibodeau, S.N.; Goldberg, P.M.; Meyers, J.P.; Pogue-Geile, K.L.; Yothers, G.; Sargent, D.J.; et al. Association of DNA mismatch repair and mutations in BRAF and KRAS with survival after recurrence in stage III colon cancers: A secondary analysis of 2 randomized clinical trials. JAMA Oncol. 2017, 3, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Marchionni, L.; Novak, M.; Parmigiani, G.; Vogelstein, B. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Sawai, H.; Weber, T.K.; Rodriguez-Bigas, M.A.; Perucho, M. Somatic frameshift mutations in DNA mismatch repair and proapoptosis genes in hereditary nonpolyposis colorectal cancer. Cancer Res. 1998, 58, 997–1003. [Google Scholar] [PubMed]
- Yamaguchi, T.; Iijima, T.; Mori, T.; Takahashi, K.; Matsumoto, H.; Miyamoto, H.; Hishima, T.; Miyaki, M. Accumulation profile of frameshift mutations during development and progression of colorectal cancer from patients with hereditary nonpolyposis colorectal cancer. Dis. Colon Rectum 2006, 49, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, M.; Pinto, C.; Peixoto, A.; Veiga, I.; Lopes, P.; Henrique, R.; Baldaia, H.; Carneiro, F.; Seruca, R.; Tomlinson, I.; et al. Target gene mutational pattern in Lynch syndrome colorectal carcinomas according to tumour location and germline mutation. Br. J. Cancer 2015, 113, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Binder, H.; Hopp, L.; Schweiger, M.R.; Hoffmann, S.; Juhling, F.; Kerick, M.; Timmermann, B.; Siebert, S.; Grimm, C.; Nersisyan, L.; et al. Genomic and transcriptomic heterogeneity of colorectal tumors arising in Lynch syndrome. J. Pathol. 2017, 243, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Sekine, S.; Mori, T.; Ogawa, R.; Tanaka, M.; Yoshida, H.; Taniguchi, H.; Nakajima, T.; Sugano, K.; Yoshida, T.; Kato, M.; et al. Mismatch repair deficiency commonly precedes adenoma formation in Lynch syndrome-associated colorectal tumorigenesis. Mod. Pathol. 2017, 30, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Sahane, N.; Magnoli, F.; Bernasconi, B.; Tibiletti, M.G.; Romualdi, C.; Pedroni, M.; Pronz de Leon, M.; Magnani, G.; Reggiani-Bonetti, L.; Bertario, L.; et al. Aberrant DNA methylation profiles of inherited and sporadic colorectal cancer. Clin. Epigenet. 2015, 7, 131. [Google Scholar] [CrossRef] [PubMed]
- Bertotti, A.; Sassi, F. Molecular pathways: Sensitivity and resistance to anti-EGFR antibodies. Clin. Cancer Res. 2015, 21, 3377–3383. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Misale, S.; Bozic, I.; Tong, J.; Peraza-Penton, A.; Lallo, A.; Baldi, F.; Lin, K.H.; Truini, M.; Trusolino, L.; Bertotti, A.; et al. Vertical suppression of the EGFR pathway prevents onset of resistance in colorectal cancers. Nat. Commun. 2015, 6, 8305. [Google Scholar] [CrossRef] [PubMed]
- Bertotti, A.; Papp, E.; Jones, S.; Adleff, V.; Anagnostou, V.; Lupo, B.; Sausen, M.; Phallen, J.; Hruban, C.A.; Tokheim, C.; et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature 2015, 526, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Beaugerie, L.; Itzkowitz, S.H. Cancers complicating inflammatory bowel disease. N. Engl. J. Med. 2015, 372, 1441–1452. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.P.; Amstad, P.; Raja, K.; Ambs, S.; Nagashima, M.; Bennett, W.P.; Shields, P.G.; Ham, A.J.; Swenberg, J.A.; Marrogi, A.J.; et al. Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: A cancerprone chronic inflammatory disease. Cancer Res. 2000, 60, 3333–3337. [Google Scholar] [PubMed]
- Leedham, S.J.; Graham, T.A.; Oukrif, D.; McDonald, S.A.; Rodriguez-Justo, M.; Harrison, R.F.; Shepherd, N.A.; Novelli, M.R.; Jankowski, J.A.; Wright, N.A. Clonality, founder mutations, and field cancerization in human ulcerative colitis-associated neoplasia. Gastroenterology 2009, 136, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Kern, S.E.; Redston, M.; Seymour, A.B.; Caldas, C.; Powell, S.M.; Kornacki, S.; Kinzler, K.W. Molecular genetic profiles of colitis-associated neoplasms. Gastroenterology 1994, 107, 420–428. [Google Scholar] [CrossRef]
- Radish, A.; Hamilton, S.R. Genetic alterations in sporadic and Crohn’s-associated adenocarcinomas of the small intestine. Gastroenterology 1997, 113, 127–135. [Google Scholar]
- Hoque, A.T.; Hahn, S.A.; Schutte, M.; Kerm, S.E. DPC4 gene mutation in colitis associated neoplasia. Gut 1997, 40, 120–122. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Galandiuk, S.; Rodriguez-Jiusto, M.; Jeffrey, M.; Nicholson, A.M.; Cheng, Y.; Oukrif, D.; Elia, G.; Leedham, S.J.; McDonald, S.A.; Wright, N.A.; et al. Field cancerization in the intestinal epithelium of patients with Crohn’s ileocolitis. Gastroenterology 2012, 142, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Robles, A.; Traverso, G.; Zhang, M.; Roberts, N.J.; Khan, M.A.; Joseph, C.; Lauwers, G.Y.; Selaru, F.M.; Popoli, M.; Pittman, M.E.; et al. Whole-exome sequencing analyses of inflammatory bowel disease-associated colorectal cancers. Gastroenterology 2016, 150, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Shah, M.; Miller, V.; Kelsen, J.R.; Wang, K.; Heins, Z.J.; Ross, J.S.; He, Y.; Sanford, E.; Yantiss, R.K.; et al. Genomic alterations observed in colitis-associated cancers are distinct from those found in sporadic colorectal cancers and vary by type of inflammatory bowel disease. Gastroenterology 2016, 151, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Der Kraak, L.; Gros, P.; Beauchemin, N. Colitis-associated colon cancer: Is it in your genes? World J. Gastroenterol. 2015, 21, 11688–11699. [Google Scholar] [CrossRef] [PubMed]
- Ullman, T.A.; Itzkowitz, S.H. Intestinal inflammation and cancer. Gastroenterology 2011, 140, 1801–1818. [Google Scholar] [CrossRef] [PubMed]
- Cooks, T.; Pateras, I.S.; Tarcic, O.; Solomon, H.; Schetter, A.J.; Wilder, S.; Lozano, G.; Pikarsky, E.; Forshew, T.; Rosenfeld, N.; et al. Mutant p53 prolongs NF-kB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell 2013, 23, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Nathanson, J.W.; Yadron, N.E.; Farnan, J.; Kinnear, S.; Harrt, J.; Rubin, D.T. p53 mutations are associated with dysplasia and progression of dysplasia and progression of dysplasia in patients with Crohn’s disease. Dig. Dis. Sci. 2008, 53, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Aust, D.E.; Terdiman, J.P.; Willenbucher, R.F.; Chang, C.G.; Molinaro-Clark, A.; Baretton, G.B.; Loehrs, U.; Waldman, F.M. The APC/β-catenin pathway in ulcerative colitis-related colorectal carcinomas: A mutational analysis. Cancer 2002, 94, 1421–1427. [Google Scholar] [CrossRef] [PubMed]
- Rapozo, D.C.M.; Grinmann, A.B.; Carvalho, A.T.P.; de Souza, H.S.; Soares-Lima, S.C.; de Almeida Simão, T.; de Paiva, D.; Abby, F.; Albano, R.M.; Pinto, L.F. Analysis of mutations in TP53, APC, K-ras, and DCC genes in the non-dysplastiuc mucosa of patients with inflammatory bowel disease. Int. J. Colorectal Dis. 2009, 24, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Saraggi, D.; Fassan, M.; Mescoli, C.; Scarpa, M.; Valeri, N.; Michielan, A.; D’Incà, R.; Rugge, M. The molecular lanscape of colitis-associated carcinogenesis. Dig. Liver Dis. 2017, 49, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Keller, R.; Foerster, E.C.; Kohler, A.; Floer, B.; Winde, G.; Terpe, H.J.; Domschke, W. Diagnostic value of DNA image cytometry in ulcerative colitis. Dig. Dis. Sci. 2001, 46, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Befrits, R.; Hammarberg, C.; Rubio, C.; Jaramilo, E.; Tribukait, B. DNA aneuploidy and histologic dysplasia in long-standing ulcerative colitis. A 10-year follow-up study. Dis. Colon Rectum 1994, 37, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Rabinovitch, P.S.; Crispin, D.A.; Emond, M.J.; Koprowicz, K.M.; Bronner, M.P.; Brentnall, T.A. DNA fingerprinting abnormalities can distinguish ulcerative colitis patients with dysplasia and cancer from those who are dysplasia/cancer-free. Am. J. Pathol. 2003, 162, 665–672. [Google Scholar] [CrossRef]
- Schulmann, K.; Mori, Y.; Croog, V.; Yin, J.; Olaru, A.; Sterian, A.; Sato, F.; Wang, S.; Xu, Y.; Deacu, E.; et al. Molecular phenotype of inflammatory bowel disease-associated neoplasms with microsatellite instability. Gastroenterology 2005, 129, 74–85. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Svrcek, M.; El-Bchiri, J.; Chalastanis, A.; Capel, E.; Dumont, S.; Buhard, O.; Oliveira, C.; Seruca, R.; Bossard, C.; Mosnier, J.F.; et al. Inflammatory bowel disease-associated colorectal cancers showing microsatellite instability. J. Clin. Oncol. 2007, 25, 4231–4238. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Walker, N.I.; Leggett, B.A.; Whitehall, V.L.; Bettington, M.L.; Rosty, C. Sessile serrated adenomas with dysplasia: Morphological patterns and correlations with MLH1 immunohistochemistry. Mod. Pathol. 2017, 30, 1728–1738. [Google Scholar] [CrossRef] [PubMed]
- Bettington, M.L.; Walker, N.; Rosty, C.; Brown, I.S.; Clouston, A.D.; McKeone, D.M.; Pearson, S.A.; Klein, K.; Leggett, B.A.; Whitehall, V.L. A clinicopathological and molecular analysis of 200 traditional serrated adenomas. Mod. Pathol. 2015, 28, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.H.; Liau, J.Y.; Lin, Y.L.; Lin, L.I.; Cheng, Y.C.; Cheng, M.L.; Jeng, Y.M. Traditional serrated adenoma has two pathways of neoplastic progression that are distinct from the sessile serrated pathway of serrated pathway of colorectal carcinogenesis. Mod. Pathol. 2014, 27, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Bettington, M.; Walker, N.; Rosty, C.; Brown, I.; Clouston, A.; McKrone, D.; Pearson, S.A.; Leggeri, B.; Whitehall, V. Clinicopathological and molecular features of sessile serrated adenomas with dysplasia or carcinoma. Gut 2017, 66, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Fennell, L.J.; Jamieson, S.; McKeone, D.; Corish, T.; Rohdmann, M.; Furner, T.; Bettington, M.; Liu, C.; Kawamata, F.; Bond, C.; et al. MLH1-93 G/a polymorphism is associated with MLH1 promoter methylation and protein loss in dysplatic sessile serrated adenomas with BRAFV600E mutation. BMC Cancer 2018, 18, 35. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, G.S.; Musrap, N.; Saraon, P.; Treacy, A.; Scheffer, D.F.; Kirsch, R.; Riddel, R.; Diamandis, E.P. Bone morphogenetic protein antagonist gremlin-1 regulates colon cancer progression. Biol. Chem. 2015, 396, 163–183. [Google Scholar] [CrossRef] [PubMed]
- Leedham, S.J.; Chetty, R. Wnt disruption in colorectal polyps—The traditional serrated adenoma enters the fray. J. Pathol. 2016, 239, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Giannakis, M.; Hodis, E.; Mu, X.J.; Yamauchi, M.; Rosenbluh, J.; Cibulskis, K.; Saksena, G.; Lawrence, M.S.; Qian, Z.R.; Nishihara, R.; et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat. Genet. 2014, 46, 1264–1266. [Google Scholar] [CrossRef] [PubMed]
- Bond, C.E.; McKeone, D.; Kalimutho, M.; Bettington, M.L.; Pearson, S.A.; Dumenil, T.D.; Wockner, L.F.; Burge, M.; Leggett, B.A.; Whitehall, V.L. RNF43 and ZNRF3 are commonly altered in serrated pathway colorectal tumorigenesis. Oncotarget 2016, 7, 70589–70600. [Google Scholar] [CrossRef] [PubMed]
- Sekine, S.; Yamashita, S.; Tanabe, T.; Hashimoto, T.; Yoshida, H.; Taniguchi, H.; Kojima, M.; Shinmura, K.; Saito, Y.; Hiraoka, N.; et al. Frequent PTPRK-RSPO3 fusions and RNF43 mutations in colorectal traditional serrated adenoma. J. Pathol. 2016, 239, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.H.; Liau, J.Y.; Yuan, C.T.; Lin, Y.L.; Tseng, L.H.; Cheng, M.L.; Jeng, Y.M. RNF43 is an early and specific mutated gene in the serrated pathway, with increased frequency in traditional serrated adenoma and its associated malignancy. Am. J. Surg. Pathol. 2016, 40, 1352–1359. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Lai, J.; Ho, S.L.; Leung, W.K.; Law, W.L.; Lee, J.F.Y.; Chan, A.K.W.; Tsui, W.Y.; Chan, A.S.Y.; Lee, B.C.H.; et al. RNF43 germline and somatic mutation in serrated neoplasia pathway and its association with BRAF mutation. Gut 2017, 66, 1645–1656. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Yamashita, S.; Yoshida, H.; Taniguchi, H.; Ushijima, T.; Yamada, T.; Saito, Y.; Ochiai, A.; Sekine, S.; Hiraoka, N. WNT pathway gene mutations are associated with the presence of dysplasia in colorectal sessile serrated adenoma/polyps. Am. J. Surg. Pathol. 2017, 41, 1188–1197. [Google Scholar] [CrossRef] [PubMed]
- Shinmura, K.; Kahyo, T.; Kato, H.; Igarashi, H.; Matsuura, S.; Nakamura, S.; Kurachi, K.; Nakamura, T.; Ogawa, H.; Funai, K.; et al. RSPO fusion transcripts in colorectal cancer in Japanese population. Mol. Biol. Rep. 2014, 41, 5375–5384. [Google Scholar] [CrossRef] [PubMed]
- Storm, E.; Durinck, S.; De Sousa e Melo, F.; Tremayne, J.; Kljavin, N.; Tan, C.; Ye, X.; Chiu, C.; Pham, T.; Hongo, J.A.; et al. Targeting PTPRK-RSPO3 colon tumours promotes differentiation and loss of stem-cell function. Nature 2016, 529, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Kanth, P.; Bronner, M.P.; Boucher, K.; Burt, R.; Neckloson, D.; Hagedorn, C.; Delker, D. Gene signature in sessile serrated polyps identifies colon cancer subtype. Cancer Prev. Res. 2016, 9, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Rhee, Y.Y.; Bae, J.M.; Cho, N.Y.; Kang, G.H. Loss of CDX2/CK20 expression is associated with poorly differentiated carcinoma, the CpG island methylator phenotype, and adverse prognosis in microsatellite-unstable colorectal cancer. Am. J. Surg. Pathol. 2013, 37, 1532–1541. [Google Scholar] [CrossRef] [PubMed]
- Konishi, K.; Shen, L.; Jelinek, J.; Watanabe, Y.; Ahmed, S.; Kaneko, K.; Kogo, M.; Takano, T.; Imawari, M.; Hamilton, S.R.; et al. Concordant DNA methylation in synchronous colorectal carcinomas. Cancer Prev. Res. 2009, 2, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Nosho, K.; Kure, S.; Irahara, N.; Shima, K.; Baba, Y.; Spiegelman, D.; Meyerhardt, J.A.; Giovannucci, E.L.; Fuchs, C.S.; Ogino, S. A prospective cohort study shows unique epigenetic, genetic, and prognostic features of synchronous colorectal cancers. Gastroenterology 2009, 137, 1609–1620. [Google Scholar] [CrossRef] [PubMed]
- Malesci, A.; Basso, G.; Bianchi, P.; Fini, L.; Grizzi, F.; Celesti, G.; Di Caro, G.; Delconte, G.; Dattola, F.; Repici, A. Molecular heterogeneity and prognostic implications of synchronous advanced colorectal neoplasia. Br. J. Cancer 2014, 110, 1228–1235. [Google Scholar] [CrossRef] [PubMed]
- Arriba, M.; Sanchez, R.; Rueda, D.; Gómez, L.; García, J.L.; Rodríguez, Y.; Pajares, J.A.; Pérez, J.; Urioste, M.; Sarmiento, R.G.; et al. Toward a molecular classification of synchronous colorectal cancer: Clinical and molecular characterization. Clin. Colorectal Cancer 2017, 16, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, K.; Yao, T.; Konomoto, T.; Hayashi, K.; Fujishima, M.; Tsuneyoshi, M. Discordance of P53 mutations of synchronous colorectal carcinomas. Mod. Pathol. 2000, 13, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Pezeshkpour, G.; Phan, R.T. KRAS mutation status impacts diagnosis and treatment decision in a patient with two colon tumours: A case report. J. Clin. Pathol. 2015, 68, 83–85. [Google Scholar] [CrossRef] [PubMed]
- De Macedo, M.P.; de Melo, F.M.; Ribeiro, J.; de Mello, C.A.L.; Ferreira de Souza Begnami, M.D.; Soares, F.A.; Carraro, D.M.; Werneck da Cunha, I. RAS mutations vary between lesions in synchronous primary colorectal cancer: Testing only one lesion is not sufficient to guide anti-EGFR treatment decisions. Oncoscience 2015, 2, 125–130. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jesinghaus, M.; Pfarr, N.; Kloor, M.; Endris, V.; Tavernar, L.; Muckenhuber, A.; von Knebel Doeberitz, M.; Penzel, R.; Weichert, W.; Stenzinger, A. Genetic heterogeneity in synchronous colorectal cancers impacts genotyping approaches and therapeutic strategies. Genes Chromosomes Cancer 2016, 55, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Cereda, M.; Gambardella, G.; Benedetti, L.; Iannelli, F.; Patel, D.; Basso, G.; Guerra, R.F.; Mourikis, T.P.; Puccio, I.; Sinha, S.; et al. Patients with genetically heterogeneous synchronous colorectal cancer carry rare damaging germline mutations in immune-related genes. Nat. Commun. 2016, 7, 12072. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Chatila, W.; Lipsyc, M.; Hechtman, J.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.; et al. Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell 2018, 33, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.P.; Kim, J.E.; Hong, Y.S.; Ahn, S.M.; Chun, S.M.; Hong, S.M.; Jang, S.J.; Yu, C.S.; Kim, J.C.; Kim, T.W. Paired primary and metastatic tumor analysis of somatic mutations in synchronous and metachronous colorectal cancer. Cancer Res. Treat. 2017, 49, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Fujiyoshi, K.; Yamamoto, G.; Takahashi, A.; Arai, Y.; Yamada, M.; Kakuta, M.; Yamaguchi, K.; Akagi, Y.; Nishimura, Y.; Sakamoto, H.; et al. High concordance rate of KRAS/BRAF mutations and MSI-H between primary and corresponding metastases. Oncol. Rep. 2017, 37, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Jesinghaus, M.; Wolf, T.; Pfarr, N.; Muckenhuber, A.; Ahadova, A.; Warth, A.; Goeppert, B.; Sers, C.; Kloor, M.; Endris, V.; et al. Distinctive spatiotemporal stability of somatic mutations in metastasized microsatellite-stable colorectal cancer. Am. J. Surg. Pathol. 2015, 39, 1140–1147. [Google Scholar] [CrossRef] [PubMed]
- Jeantet, M.; Tougeron, D.; Tachon, G.; Cortes, U.; Archambaut, C.; Fromont, G.; Karayan-Tapon, L. High intra- and inter-tumoral heterogeneity of RAS mutations in colorectal cancer. Int. J. Mol. 2016, 17, 2015. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.M.; Jung, S.H.; An, C.H.; Lee, S.H.; Baek, I.P.; Kim, M.S.; Park, S.W.; Rhee, J.K.; Lee, S.H.; Chung, Y.J. Subclonal genomic architectures of primary and metastatic colorectal cancer based on intratumoral genetic heterogeneity. Clin. Cancer Res. 2015, 21, 4461–4472. [Google Scholar] [CrossRef] [PubMed]
- Sveen, A.; Loes, I.M.; Alagaratnam, S.; Nilsen, G.; Høland, M.; Lingjærde, O.C.; Sorbye, H.; Berg, K.C.; Horn, A.; Angelsen, J.H.; et al. Intra-patient inter-metastatic genetic heterogeneity in colorectal cancer as a key determinant of survival after curative liver resection. PLoS Genet. 2016, 12, e1006225. [Google Scholar] [CrossRef] [PubMed]
- Uchi, R.; Takahashi, Y.; Niida, A.; Shimamura, T.; Hirata, H.; Sugimachi, K.; Sawada, G.; Iwaya, T.; Kurashige, J.; Shinden, Y.; et al. Integrated multiregional analysis proposing a new model of colorectal cancer evolution. PLoS Genet. 2016, 12, e1005778. [Google Scholar] [CrossRef] [PubMed]
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D.; et al. A Big Bang model of human colorectal tumor growth. Nat. Genet. 2015, 47, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Salomon, M.P.; Sottoriva, A.; Zhao, J.; Toy, M.; Press, M.F.; Curtis, C.; Marjoram, P.; Siegmund, K.; Shibata, D. Many private mutations originate from the first few divisions of a human colorectal adenoma. J. Pathol. 2015, 237, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Sievers, C.K.; Zou, L.; Pickhardt, P.J.; Matkowskyj, K.A.; Albrecht, D.M.; Clipson, L.; Bacher, J.W.; Pooler, B.D.; Moawad, F.J.; Cash, B.D.; et al. Subclonal diversity arises early even in small colorectal tumours and contributes to differential growth fates. Gut 2017, 66, 2131–2140. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.J.; Werner, B.; Barnes, C.; Graham, T.A.; Sottoriva, A. Identification of neutral evolution across cancer types. Nat. Genet. 2016, 48, 238–244. [Google Scholar] [CrossRef] [PubMed]
- McFarland, C.D.; Korolev, K.; Kryukov, G.; Sunyaev, S.R.; Mirny, L.A. Impact of deleterious passenger mutations on cancer progression. Proc. Natl. Acad. Sci. USA 2013, 110, 2910–2913. [Google Scholar] [CrossRef] [PubMed]
- Mc Farland, C.D.; Mirny, L.A.; Korolev, K.S. Tug-of-war between driver and passenger mutations in cancer and other adaptive processes. Proc. Natl. Acad. Sci. USA 2014, 111, 15138–15143. [Google Scholar] [CrossRef] [PubMed]
- Humphries, A.; Cereser, B.; Gay, L.J.; Miller, D.S.; Das, B.; Gutteridge, A.; Elia, G.; Nye, E.; Jeffery, R.; Poulsom, R.; et al. Lineage tracing reveals multipotent stem cells maintain human adenomas and the pattern of clonal expansion in tumor evolution. Proc. Natl. Acad. Sci. USA 2013, 110, E2490–E2499. [Google Scholar] [CrossRef] [PubMed]
- Sottoriva, A.; Barnes, C.P.; Graham, T.A. Catch my drift? Making sense of genomic intra-tumor heterogeneity. Biochim. Biophys. Acta 2017, 1867, 95–100. [Google Scholar] [PubMed]
- Thliveris, A.; Schwefel, B.; Clipson, L.; Plesh, L.; Zahm, C.D.; Leystra, A.; Washington, M.K.; Sullivan, R.; Deming, D.A.; Newton, M.A.; et al. Transformation of epithelila cells through recruitment leads to polyclonal intestinal tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 11523–11528. [Google Scholar] [CrossRef] [PubMed]
- Thirlwell, C.; Will, O.C.; Domingo, E.; Graham, T.A.; McDonald, S.A.; Oukrif, D.; Jeffrey, R.; Gorman, M.; Rodriguez-Justo, M.; Ching-Aleong, J.; et al. Clonal assessment and clonal ordering of individual neoplastic crypts shows polyclonality of colorectal adenomas. Gastroenterology 2010, 138, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Borras, E.; San Lucas, A.; Chang, K.; Zhou, R.; Masand, G.; Fowler, J.; Mork, M.E.; You, Y.N.; Taggart, M.W.; McAllister, F.; et al. Genomic landscape of colorectal mucosa and adenomas. Cancer Prev. Res. 2016, 9, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Gausachs, M.; Borras, E.; Chang, K.; Gonzalez, S.; Azuara, D.; Amador, A.D.; Lopez-Doriga, A.; San Lucas, A.; San Juan, X.; Paules, M.; et al. Mutational heterogeneity in APC and KRAS arises at crypt level and leads to polyclonality in early colorectal tumorigenesis. Clin. Cancer Res. 2017, 23, 5936–5947. [Google Scholar] [CrossRef] [PubMed]
- Moon, B.S.; Jeong, W.J.; Park, J.; Kim, T.I.; Min do, S.; Choi, K.Y. Role of oncogenic K-Ras in cancer stem cell activation by aberrant Wnt/β-catenin signaling. J. Natl. Cancer Inst. 2014, 106, djt373. [Google Scholar] [CrossRef] [PubMed]
- Snippert, H.J.; Schepers, H.G.; van Es, J.H.; Simons, B.D.; Clevers, H. Biased competition between Lgr5 intestinal stem cells driven by oncogenic mutation induces clonal expansion. EMBO Rep. 2014, 15, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.H.; Raju, G.; Huff, C.; Ye, Y.; Gu, J.; Chen, J.S.; Hildebrandt, M.; Liang, H.; Menter, D.G.; Morris, J.; et al. The somatic mutation landscape of premalignant colorectal adenoma. Gut 2017. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Jung, S.H.; Kim, T.M.; Rhee, J.K.; Park, H.C.; Kim, M.S.; Chang, H.A.; Lee, S.H.; Chung, H.J. Whole-exome sequencing identified mutational profiles of high-grade colon adenomas. Oncoatrget 2017, 8, 6579–6588. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Lee, J.; Gong, J.R.; Cho, K.H. Percolation transition of cooperative mutational effects in colorectal tumorigenesis. Nat. Commun. 2017, 8, 1270. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Carduso, A.; Moran, S.; Musulen, E.; Moutinho, C.; Manzano, J.L.; Martinez-Balibrea, E.; Tierno, M.; Elez, E.; Landolfi, S.; Lorden, P.; et al. Epigenetic heterogeneity with colorectal tumors predicts shorter relapse-free and overall survival times for patients with locoregional cancer. Gastroenterology 2016, 151, 961–972. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; Rachiglio, A.M.; Lambiase, M.; Martinelli, E.; Fenizia, F.; Esposito, C.; Roma, C.; Troiani, T.; Rizzi, D.; Tatangelo, F.; et al. Heterogeneity of KRAS, NRAS, BRAF and PIK3CA mutations in metastatic colorectal cancer and potential effects on therapy in the CAPRI GOIM trial. Ann. Oncol. 2015, 26, 1710–1714. [Google Scholar] [CrossRef] [PubMed]
- Van Custem, E.; Lenz, H.J.; Kohne, C.H.; Heinemann, V.; Tejpar, S.; Melezinek, I.; Beier, F.; Stroh, C.; Rougier, P.; van Krieken, J.H.; et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J. Clin. Oncol. 2015, 33, 692–700. [Google Scholar]
- Laurent-Puig, P.; Pekin, D.; Normand, C.; Kotsopoulos, S.K.; Nizard, P.; Perez-Toralla, K.; Rowell, R.; Olson, J.; Srinivasan, P.; Le Corre, D.; et al. Clinical relevance of KRAS-muated subclones detected with picodroplet digital PCR in advanced colorectal cancer treated with anti-EGFR therapy. Clin. Cancer Res. 2015, 21, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Adua, D.; Di Fabio, F.; Ercolani, G.; Fiorentino, M.; Gruppioni, E.; Altimari, A.; Rojas Limpe, F.L.; Normanno, N.; Pinna, A.D.; Pinto, C. Heterogeneity in the colorectal primary tumor and the synchronous resected liver metastases prior to and after treatment with an anti-EGFR monoclonal antibody. Mol. Clin. Oncol. 2017, 7, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Arandottir, S.S.; Jeppesen, M.; Lamy, P.; Bramsen, J.B.; Nordentoft, I.; Knudsen, M.; Vang, S.; Madsen, M.R.; Thastrup, O.; Thastrup, J.; et al. Characterization of genetic intratumor heterogeneity in colorectal cancer and matching patient-derived spheroid cultures. Mol. Oncol. 2018, 12, 132–147. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Menter, D.G.; Kopetz, S. Right versus left colon cancer biology: Integrating the consensus molecular subtypes. J. Natl. Compr. Cancer Netw. 2017, 15, 411–419. [Google Scholar] [CrossRef]
- Stintzing, S.; Tejpar, S.; Gibbs, P.; Thiebach, L.; Lenz, H.J. Understanding the role of primary tumor localization in colorectal cancer treatment and outcomes. Eur. J. Cancer 2017, 84, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Pen, Q.; Lin, Q.; Chang, T.; Zou, L.; Xing, P.; Shen, P.; Zhu, Y. Identification of genomic expression differences between rightr-sided and left-sided colon cancer based on bioinformatics. Onco Targets Ther. 2018, 11, 609–618. [Google Scholar]
- Sinicrope, F.A.; Mahoney, M.R.; Yoon, H.H.; Smyrk, T.C.; Thibodeau, S.N.; Goldberg, R.M.; Nelson, G.D.; Sergent, D.J.; Alberts, S.R. Alliance for Clinical Trials in Oncology. Analysis of macular markers by anatomic tumor site in stage III colon carcinomas from adjuvant chemotherapy trial NCCTG NO147 (alliance). Clin. Cancer Res. 2015, 21, 5294–5304. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.T.; Jen-Kou, L.; Lin, C.H.; Yang, S.H.; Lin, C.C.; Wang, H.S.; Chen, W.S.; Lin, T.C.; Jiang, J.K.; Chang, S.C. Mutations in the RAS and PI3K pathways are associated with metastatic location in colorectal cancers. J. Surg. Oncol. 2015, 111, 905–910. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.; Weinberg, B.; Xiu, J.; El-Deiry, W.; Hwang, J.; Gatalica, Z.; Philip, P.; Shields, A.; Lenz, H.J.; Marshall, J.L. Comparative molecular analyses of left-sided, right-sided, and rectal cancers. Oncotarget 2017, 8, 86356–86368. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, F.; Tomasello, G.; Borgonovo, K.; Ghidini, M.; Turati, L.; Dallera, P.; Passalacqua, R.; Sgroi, G.; Barni, S. Prognostic survival associated with left-sided colon cancer: A systematic review and meta-analysis. JAMA Oncol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Arnold, D.; Lueza, B.; Douillard, J.Y.; Peeters, M.; Lenz, H.J.; Venook, A.; Heinemann, V.; Van Cutsem, E.; Pignon, J.P.; Tabernero, J.; et al. Prognostic and predictive value of primary tumour side in patients with RAS wild-type metastatic colorectal cancer treated with chemotherapy and EGFR directed antibodies in six randomized trials. Ann. Oncol. 2017, 28, 1713–1729. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, S.Y.; Lee, J.S.; Hong, Y.S.; Kim, J.E.; Kim, K.P.; Kim, J.; Jang, S.J.; Yoon, Y.K.; Kim, T.W. Primary tumor location predicts poor clinical outcome with cetuximab in RAS wild-type metastatic colorectal cancer. BMC Gastroenterol. 2017, 17, 121. [Google Scholar] [CrossRef] [PubMed]
- Taieb, J.; Kourie, H.R.; Emile, J.F.; Le Malicot, K.; Balogoun, R.; Tabernero, J.; Mini, E.; Folprecht, G.; Van Laethem, J.L.; Mulot, C.; et al. Association of prognostic value of primary tumor location in stage III colon cancer with RAS and BRAF mutational status. JAMA Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Loree, J.M.; Pereira, A.A.; Lam, M.; Willauer, A.N.; Raghav, K.; Dasari, A.; Morris, V.K.; Advani, S.M.; Menter, D.G.; Eng, C.; et al. Classifying colorectal cancer by tumor location rather than sidedness highlights a continuum in mutation profiles and consensus molecular subtypes. Clin. Cancer Res. 2018, 24, 1062–1072. [Google Scholar] [CrossRef] [PubMed]
- Eschrich, S.; Yang, I.; Bloom, G.; Kwong, K.Y.; Boulware, D.; Cantor, A.; Coppola, D.; Kruhøffer, M.; Aaltonen, L.; Orntoft, T.F.; et al. Molecular staging for survival prediction of colorectal cancer patients. J. Clin. Oncol. 2005, 23, 3526–3535. [Google Scholar] [CrossRef] [PubMed]
- Salazar, R.; Roepman, P.; Capella, G.; Moreno, V.; Simon, I.; Dreezen, C.; Lopez-Doriga, A.; Santos, C.; Marijnen, C.; Westerga, J.; et al. Gene expression signature to improve prognosis prediction of stage II and III colorectal cancer. J. Clin. Oncol. 2011, 29, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Pamplona, R.; Cordero, D.; Berenguer, A.; Lejbkowicz, F.; Rennert, H.; Salazar, R.; Biondo, S.; Sanjuan, X.; Pujana, M.A.; Rozek, L.; et al. Gene expression differences between colon and rectum tumors. Clin. Cancer Res. 2011, 17, 7303–7312. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Simon, I.; Moreno, V.; Roepman, P.; Tabernero, J.; Snel, M.; van’t Veer, L.; Salazar, R.; Bernards, R.; Capella, G. A combined oncogenic pathway signature of BRAF, KRAS and PI3KCA mutation improves colorectal cancer classification and cetuximab treatment prediction. Gut 2013, 62, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Perez-Villamil, B.; Romera-Lopez, A.; Hernandez-Prieto, S.; Lopez-Campos, G.; Calles, A.; Lopez-Asenjo, J.A.; Sanz-Ortega, J.; Fernandez-Perez, C.; Sastre, J.; Alfonso, R.; et al. Colon cancer molecular subtypes identified by expression profiling and associated to stroma mucinous type and differen clinical behavior. BMC. Cancer 2012, 12, 260. [Google Scholar] [CrossRef] [PubMed]
- Sadanandam, A.; Lyssiotis, C.A.; Homicsko, K.; Collisson, E.A.; Gibb, W.J.; Wullschleger, S.; Ostos, L.C.; Lannon, W.A.; Grotzinger, C.; Del Rio, M.; et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat. Med. 2013, 19, 619–625. [Google Scholar] [CrossRef] [PubMed]
- De Sousa E Melo, F.; Wang, X.; Jansen, M.; Fessler, E.; Trinh, A.; de Rooij, L.P.; de Jong, J.H.; de Boer, O.J.; van Leersum, R.; Bijlsma, M.F.; et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat. Med. 2013, 19, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Sadanandam, A.; Wang, X.; Sousa E Melo, F.; Gray, J.W.; Vermeulen, L.; Hanahan, D.; Medema, J.P. Reconciliation of classification systems defining molecular subtypes of colorectal cancer. Cell Cycle 2014, 13, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reynies, A.; Schilker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Barras, D.; Missaglia, E.; Wirapati, P.; Sieber, O.; Jorissen, R.; Love, C.; Molloy, P.; Jones, I.; McLaughlin, S.; Gibbs, P.; et al. BRAF V600E mutant colorectal cancer subtypes based on gene expression. Cancer Res. 2017, 23, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Dunne, P.D.; O’Reilly, P.; Coleman, H.G.; Gray, R.T.; Longley, D.B.; Johnston, P.G.; Salto-Tellez, M.; Lawler, M.; McArt, D.G. Startified analysis reveals chemokine-like factor (CKLF) as a potential prognostic marker in the MSI-immune consensus molecular subtype CMS1 of colorectal cancer. Oncotarget 2017, 7, 36632–36644. [Google Scholar]
- Lal, N.; White, B.; Goussous, G.; Pickles, O.; Mason, M.; Beggs, A.; Taniere, P.; Willcox, B.; Guinney, J.; Middleton, G. KRAS mutation and consensus molecular subtypes 2 and 3 are independently associated with reduced immune infiltration and reactivity in colorectal cancer. Clin. Cancer Res. 2018, 24, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Trinh, A.; Trumpi, K.; De Sousa Melo, F.; Wang, X.; de Jong, J.; Fessler, E.; Kuppen, P.; Reimers, M.; Swets, M.; Koopman, M.; et al. Practical and robust identification of molecular subtypes in colorectal cancer by immunohistochemistry. Clin. Cancer Res. 2016, 23, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Sveen, A.; Bruun, J.; Eide, P.W.; Eilersten, I.A.; Ramirez, L.; Murumagi, A.; Arjama, M.A.; Danielsen, S.A.; Kryeziu, K.; Elez, E.; et al. Colorectal cancer consensus molecular subtypes translated to preclinical models uncover potentially targetable cancer-cell dependencies. Clin. Cancer Res. 2018, 24, 794–806. [Google Scholar] [CrossRef] [PubMed]
- Linnekamp, J.F.; Hoof, S.R.V.; Prasetyanti, P.R.; Kandimalla, R.; Buikhuisen, J.Y.; Fessler, E.; Ramesh, P.; Lee, K.; Bochove, C.G.W.; de Jong, J.H.; et al. Consensus molecular subtypes of colorectal cancer are recapitulated in vitro and in vivo models. Cell Death Differ. 2018, 25, 616–633. [Google Scholar] [CrossRef] [PubMed]
- Isella, C.; Terrasi, A.; Bellomo, S.E.; Petti, C.; Galatola, G.; Muratore, A.; Mellano, A.; Senetta, R.; Cassenti, A.; Sonetto, C.; et al. Stromal contribution to the colorectal cancer transcriptome. Nat. Genet. 2015, 47, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Calon, A.; lonardo, E.; Berenguer-llergo, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.; Byrom, D.; et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet. 2015, 47, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Hartmaier, R.; Albacker, L.; Chmielecki, J.; Bailey, M.; He, J.; Goldberg, M.; Ramkisson, S.; Suh, J.; Elvin, J.; Chiacchia, S.; et al. High-throughput genomic profiling of adult solid tumors reveals novel insights into cancer pathogenesis. Cancer Res. 2017, 77, 2464–2475. [Google Scholar] [CrossRef] [PubMed]
- Dunne, P.D.; McArt, D.; Bradley, C.A.; O’Reilly, P.G.; Barret, H.L.; Cummins, R.; O’Grady, T.; Arthur, K.; Loughrey, M.B.; Allen, W.L.; et al. Challenging the cancer molecular stratification dogma: Intratumors heterogeneity undermines consensus molecular subtypes and potential diagnostic value in colorectal cancer. Clin. Cancer Res. 2016, 22, 4095–4104. [Google Scholar] [CrossRef] [PubMed]
- Dunne, P.D.; Alderdice, M.; O’Reilly, P.; Roody, A.C.; McCorry, A.M.B.; Richman, S.; Maughan, T.; McDade, S.S.; Johnston, P.G.; Longley, D.B.; et al. Cancer-cell intrinsic gene expression signatures overcome intratumoral heterogeneity bias in colorectal cancer patient classification. Nat. Commun. 2017, 8, 15657. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Courtois, E.T.; Sengupta, D.; Tan, Y.; Chen, K.H.; Goh, J.J.; Kong, S.L.; Chua, C.; Hon, L.K.; Tan, W.S.; et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 2017, 49, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Bramsen, J.B.; Rasmussen, M.H.; Ongen, H.; Mattesen, T.B.; Orntoft, M.B.W.; Arnadottir, S.S.; Sandoval, J.; Laguna, T.; Vang, S.; Oster, B.; et al. Molecular-subtype-specific biomarkers improve prediction of prognosis in colorectal cancer. Cell Rep. 2017, 19, 1268–1280. [Google Scholar] [CrossRef] [PubMed]
- Eide, P.W.; Bruun, J.; Lothe, R.A.; Sveen, A. CMScaller: An R package for consensus molecular subtyping of colorectal cancer pre-clinical models. Sci. Rep. 2017, 7, 16618. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Zhu, X.; Yan, T.; Yu, C.; Shen, C.; Hu, Y.; Hong, J.; Chen, H.; Fang, J.Y. Recurrence-associated gene signature optimizes recurrence-free survival prediction of colorectal cancer. Mol. Oncol. 2017, 11, 1544–1560. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Toyota, M.; Kondo, Y.; Lin, E.; Zhang, L.; Guo, Y.; Hernandez, N.S.; Chen, X.; Ahmed, S.; Konishi, K.; et al. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 18564–18569. [Google Scholar] [CrossRef] [PubMed]
- Purcell, R.V.; Visnovska, M.; Biggs, P.J.; Schmeier, S.; Frizelle, F.A. Distinct gut microbiome patterns associate with consensus molecular subtypes of colorectal cancer. Sci. Rep. 2017, 7, 11590. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.; Segditsas, S.; Deherugade, M.; Pollard, P.; Jeffery, R.; Nye, E.; Lockstone, H.; Davis, H.; Clark, S.; Stamp, G.; et al. Severe polyposis in Apc (1322T) mice is associated with submaximal Wnt signaling and increased expression of the stem cell marker Lgr5. Gut 2010, 59, 1680–1686. [Google Scholar] [CrossRef] [PubMed]
- Shibata, H.; Toyama, K.; Shioyka, H.; Ito, M.; Hirota, M.; Hasegawa, S.; Matsumoto, H.; Takano, H.; Akiyama, T.; Toyoshima, K.; et al. Rapid colorectal adenoma formation initiated by conditional targeting of the Apc gene. Science 1997, 278, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Samson, O.J.; Meniel, V.S.; Muncan, V.; Phesse, T.J.; Wilkins, J.A.; Reed, K.R.; Vass, J.K.; Athineos, D.; Clevers, H.; Clarke, A.R. Myc deletion rescues Apc deficiency in the small intestine. Nature 2007, 446, 676–679. [Google Scholar]
- Ashton, G.H.; Morton, J.P.; Myant, K.; Phesse, T.J.; Ridgway, R.A.; Marsh, V.; Wilkins, J.A.; Athineos, D.; Muncan, V.; Kemp, R.; et al. Focal adhesion kinase is required for intestinal regeneration and tumorigenesis downstream of Wnt/c-Myc signaling. Dev. Cell 2010, 19, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Jackstadt, R.; Samson, O.J. Mouse models of intestinal cancer. J. Pathol. 2016, 238, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Tian, A.; Benchabane, H.; Wang, Z.; Zimmerman, C.; Xin, N.; Perochon, J.; Kalna, G.; Sansom, O.J.; Cheng, C.; Cordero, J.B.; et al. Intestinal stem cell overproliferation resulting from inactivation of the APC tumor suppressor requires the transcription cofactors Earthbound and Erect wing. PLoS Genet. 2017, 13, e1006870. [Google Scholar] [CrossRef] [PubMed]
- Sansom, O.J.; Meniel, V.; Wilkins, J.A.; Cole, A.M.; Oien, K.A.; Marsh, V.; Jamieson, T.J.; Guerra, C.; Ashton, G.H.; Barbacid, M.; et al. Loss of APC allows phenotypic manifestation of the transforming properties on endogenous K-Ras oncogene in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 14122–14127. [Google Scholar] [CrossRef] [PubMed]
- Janssen, K.P.; Alberici, P.; Fsihi, H.; Gaspar, C.; Breukel, C.; Franken, P.; Rosty, C.; Abal, M.; El Marjou, F.; Smits, R.; et al. APC and oncogenic KRAS are synergistic in enhancing WNT signaling in intestinal tumor formation and progression. Gastroenterology 2006, 131, 1096–1099. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Bommer, G.T.; Zhao, J.; Green, M.; Sands, E.; Zhai, Y.; Brown, K.; Burberry, A.; Cho, K.R.; Fearon, E.R. Mutant KRas promotes hyperplasia and altered differentiation in colon epithelium but does not expand the presumptive crypt stem cell pool. Gastroenterology 2011, 141, 1003–1013. [Google Scholar] [CrossRef] [PubMed]
- Fearon, A.R.; Wicha, M.S. KRAS and cancer stem cells in APC-mutant colorectal cancer. J. Natl. Cancer Inst. 2014, 106, djt 444. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hung, K.E.; Maricevich, M.A.; Richard, L.G.; Chen, W.; Richardson, M.; Kunin, A.; Bronson, R.; Mahmood, U.; Kucherlapati, R. Development of mouse model for sporadic and metastatic colon tumors and its use in assessing drug treatment. Proc. Natl. Acad. Sci. USA 2010, 107, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Muno, N.M.; Upton, M.; Rojas, A.; Washington, M.K.; Lin, L.; Chytil, A.; Sozmen, E.G.; Madison, B.B.; Pozzi, A.; Moon, R.T.; et al. Transforming growth factor beta receptor type II inactivation induces the malignant transformation of intestinal neoplasms initiated by Apc mutation. Cancer Res. 2006, 66, 9837–9844. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Kametani, K.; Hashida, H.; Matsunaga, A.; Miyoshi, H.; Hosogi, H.; Aoki, M.; Oshima, M.; Hattori, M.; Takabayashi, A.; et al. SMAD4-deficient intestinal tumors recruit CCR1+ myeloid cells that promote invasion. Nat. Genet. 2007, 39, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Clarke, A.R.; Cummings, M.C.; Harrison, D.J. Interaction between murine germline mutations in p53 and APC predisposes to pancreatic neoplasia but not to increased intestinal malignancy. Oncogene 1995, 11, 1913–1920. [Google Scholar] [PubMed]
- Muller, P.A.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Sakai, E.; Echizen, K.; Yamada, Y.; Oshima, H.; Han, T.S.; Ohki, R.; Fujii, S.; Ochiai, A.; Robine, S.; et al. Intestinal cancer progression by mutant p53 through the acquisition of invasiveness associated with complex glandular formation. Oncogene 2017, 36, 5885–5896. [Google Scholar] [CrossRef] [PubMed]
- Tettew, P.W.; Kretzschmar, K.; Begthel, H.; van den Born, M.; Korving, J.; Morsink, F.; Farin, H.; van Es, J.; Offerhaus, J.; Clevers, H. Generation of an inducible colon-specific Cre enzyme mouse line for colon cancer research. Proc. Natl. Acad. Sci. USA 2016, 113, 11859–11864. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Johnson, R.; Desmet, M.; Snyder, P.W.; Fleet, J.C. Generation of a transgenic mouse for colorectal cancer research with intestinal cre expression limited to the large intestine. Mol. Cancer Res. 2010, 8, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Vries, R.G.; Snippert, H.G.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Stange, D.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- Van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Nadauld, L.; Ootani, A.; Corney, D.C.; Pai, R.K.; Gevaert, O.; Cantrell, M.A.; Rack, P.G.; Neal, J.T.; Chan, C.W.; et al. Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat. Med. 2014, 20, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.; van Jaarsveld, R.; Ponsioen, B.; Zimberlin, C.; van Boxtel, R.; Buijs, A.; Sachs, N.; Overmeer, R.M.; Offerhaus, G.J.; Begthel, H.; et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 2015, 521, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Matano, M.; Date, S.; Shimokawa, M.; Takano, A.; Fujii, M.; Ohta, Y.; Watanabe, T.; Kanai, T.; Sato, T. Modeling of human intestinal organoids. Nat. Med. 2015, 21, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Fujii, M.; Shimokawa, M.; Date, S.; Takano, A.; Matano, M.; Nauki, K.; Ohta, Y.; Tashimitsu, K.; Nakazato, Y.; Kawasaki, K.; et al. A colorectal tumor orgasnoid library demonstrates progressive loss of niche factor requirements during tumorigenesis. Cell Stem Cell 2016, 18, 827–838. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, K.; Loizou, E.; Livshits, G.; Scahtoff, E.M.; Baslan, T.; Manchado, E.; Simon, J.; Romesser, P.; Leach, B.; Han, T.; et al. Transplantation of engineered organoids enables rapid generation of metastatic mouse models of colorectal cancer. Nat. Biotechnol. 2017, 35, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, A.; Drost, J.; Suikerbuik, S.; van Boxtel, R.; de Ligt, J.; Offerhaus, G.J.; Begthel, H.; Beerling, E.; Tan, E.H.; Sansom, O.J.; et al. Genetic dissection of colorectal cancer progression by orthoptic transplantation of engineered cancer organoids. Proc. Natl. Acad. Sci. USA 2017, 114, E2357–E2364. [Google Scholar] [CrossRef] [PubMed]
- Roper, J.; Tammela, T.; Akkad, A.; Almeqdadi, M.; Santos, S.B.; Jacks, T.; Ylmaz, O.H. Colonoscopy-based colorectal cancer modeling in mice with CRISPR-Cas9 genome editing and organoid transplantation. Nat. Protoc. 2018, 13, 217–234. [Google Scholar] [CrossRef] [PubMed]
- Roper, J.; Tammela, T.; Cetinbas, N.M.; Akkad, A.; Ropghanian, A.; Rickelt, S.; Almeqdadi, M.; Wu, K.; Oberli, M.; Sanchez-Rivera, F.J.; et al. In vivo genome editing and organoid transplantation models of colorectal cancer and metastasis. Nat. Biotechnol. 2017, 35, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Sakai, E.; Nakayama, M.; Oshima, H.; Kouyama, Y.; Niida, A.; Fujii, S.; Ochiai, A.; Nakayama, K.I.; Mimori, K.; Suzuki, Y.; et al. Combined mutation of Apc, Kras and Tgfbr2 effectively drives metastasis of intestinal cancer. Cancer Res. 2017. [Google Scholar] [CrossRef]
- Drost, J.; van Boxtel, R.; Blokzjl, F.; Mizutani, T.; Sasaki, N.; Sasselli, V.; de Ligt, J.; Bahjati, S.; Grolleman, J.E.; van Wezel, T.; et al. Use of CRISPR-modified human stem cell organoids to study the origin of mutational signatures in cancer. Science 2017, 358, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Weren, R.D.; Ligtenberg, M.J.; Geurts van kessel, A.; de Voer, R.M.; Hoogerbrugge, N.; Kuiper, R.P. NTHL1 and MUTYH polyposis syndromes: Two sides of the same coin? J. Pathol. 2018, 244, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Brauer, C.; Hao, Z.; Elia, A.J.; Fortin, J.M.; Nechanitzky, R.; Brauer, P.M.; Sheng, Y.; Mana, M.D.; Chio, I.I.C.; Haight, J.; et al. Mule regulates the intestinal stem cell niche via the Wnt pathway and targets EphB3 for proteasomal and lysosomal degradation. Cell Stem Cell 2016, 19, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Brauer, C.; Khatun, R.; Elia, A.J.; Thu, K.L.; Ramachandran, P.; Baniasadi, S.P.; Hao, Z.; Jones, L.D.; Haight, J.; Sheng, Y.; et al. E3 ubiquitin ligase Mule targets β-catenin under conditions of hyperactive Wnt signaling. Proc. Natl. Acad. Sci. USA 2017, 114, E1148–E1157. [Google Scholar] [CrossRef] [PubMed]
- Myant, K.B.; Cammareri, P.; Hodder, M.C.; Wills, J.; Von Kriegsheim, A.; Gyorffy, B.; Rashid, M.; Polo, S.; Masopero, E.; Vaughan, L.; et al. HUWE1 is a critical colonic tumour suppressor gene that prevents MYC signaling, DNA damage accumulation and tumour initiation. EMBO Mol. Med. 2017, 9, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Babaei-Jadidi, R.; Li, N.; Saadedin, A.; Spencer-Dene, B.; Jandke, A.; Muhammad, B.; Ibrahim, E.E.; Muraleedharan, R.; Abunizadah, M.; Davis, H.; et al. FBXW7 infuences murine intestinal homeostasis and cancer, targeting NOTCH, JUN, and DEK for degradation. J. Exp. Med. 2011, 208, 295–312. [Google Scholar] [CrossRef] [PubMed]
- Khan, O.M.; Carvalho, J.; Spencer-Dene, B.; Mitter, R.; Frith, D.; Snijders, A.P.; Wood, S.A.; Behrens, A. The ubiquitinase USP9X regulates FBW7 stability and suppresses colorectal cancer. J. Clin. Investig. 2018, 128, 1326–1337. [Google Scholar] [CrossRef] [PubMed]
- Takada, M.; Zhang, W.; Suzuki, A.; Kuroda, T.S.; Yu, Z.; Inuzuka, H.; Gao, D.; Wan, L.; Zhuang, M.; Hu, L.; et al. FBW7 loss promotes chromosomal instability and tumorigenesis via Cyclin E1/CDK2-mediated phosphorylation of CENP-A. Cancer Res. 2017, 77, 4881–4893. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Tan, S.; Zou, F.; Yu, J.; Zhang, L. FBW7 mutations mediate resistance of colorectal cancer to targeted therapies by blocking Mcl-1 degradation. Oncogene 2017, 36, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Mathur, R.; Han Alver, B.; San Roman, A.; Wilson, B.G.; Wang, X.; Agoston, A.T.; Park, P.J.; Shivdasani, R.A.; Roberts, C.W. ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nat. Genet. 2017, 49, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Crespo, M.; Vilar, E.; Tsai, S.Y.; Chang, K.; Amin, S.; Srinivasan, T.; Zhang, T.; Pipalia, N.H.; Chen, H.J.; Whiterspoon, M.; et al. Colonic organoids derived from human induced pluripotent stem cells for modeling colorectal cancer and drug testing. Nat. Med. 2017, 23, 878–884. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.J.; Ewald, A.J. Acollective route to metastasis: Seeding by tumor cell clusters. Science 2016, 352, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Baldus, S.E.; Schaefer, K.L.; Engers, R.; Hartleb, D.; Stoecklein, N.H.; Gabbert, H.E. Prevalence and heterogeneity of KRAS, BRAF and PIK3CA mutations in primary colorectal adenocarcinomas and their corresponding metastases. Clin. Cancer Res. 2010, 16, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Brannon, A.R.; Vakiani, E.; Sylvester, B.E.; Scott, S.N.; McDermott, G.; Shah, R.H.; Kania, K.; Viale, A.; Oschwald, D.M.; Vacic, V.; et al. Comparative sequencing analysis reveals high genomic concordance between matched and metastatic colorectal cancer lesions. Genome Biol. 2014, 15, 454. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Ng, S.B.; Chua, C.; Leow, W.Q.; Chng, J.; Liu, S.Y.; Ramnarayaanan, K.; Gan, A.; Ho, D.L.; Ten, R.; et al. Multiregion ultra-deep sequencing reveals early intermixing and variable levels of intratumoral heterogeneity in colorectal cancer. Mol. Oncol. 2017, 11, 124–139. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Sun, W.; Zhou, Y.; Chen, F.; Chen, H.; Xia, D.; Xu, E.; Lai, M.; Wu, Y.; Zhang, H. Mutations of key driver genes in colorectal cancer progression and metastasis. Cancer Metastasis Rev. 2018, 37, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Sebagh, M.; Allard, M.A.; Bosselut, N.; Dao, M.; Vibert, E.; Lewin, M.; Lemoine, A.; Cherqui, D.; Adam, R.; Sa Cunha, A. Evidence of intermetastatic heterogeneity for pathological response and genetic mutations with colorectal liver metastases following preoperative chemotherapy. Oncotarget 2016, 7, 21591–21600. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ulinz, P.J.; Greenson, J.K.; Wu, R.; Fearon, E.R.; Hardima, K.M. Lymph node metastases in colon acncer are polyclonal. Clin. Cancer Res. 2017, clincanres.1425.2017. [Google Scholar] [CrossRef]
- Wei, Q.; Ye, Z.; Zhong, X.; Li, L.; Wang, C.; Myers, R.E.; Palazzo, J.P.; Fortuna, D.; Yan, A.; Waldman, S.A.; et al. Multiregion whole-exome sequencing of matched primary and metastatic tumors revealed genomic heterogeneity and suggests polyclonal seeding in colorectal cancer metastasis. Ann. Oncol. 2017, 28, 2135–2141. [Google Scholar] [CrossRef] [PubMed]
- Naxerova, K.; Reiter, J.G.; Brachtel, E.; Lennezz, J.; vsn de Wetering, M.; Rowan, A.; Cai, T.; Clevers, H.; Swanton, C.; Nowak, M.A.; et al. Origins of lymphatic and distant metastases in human colorectal cancer. Science 2017, 357, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Stigliano, V.; Sanchez-Mete, L.; Martayau, A.; Anti, M. Early-onset colorectal cancer: A sporadic or inherited disease? World J. Gastroenterol. 2014, 20, 12420–12430. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Pearlman, R.; Beightol, M.; Zhao, W.; Jones, D.; Frankel, W.L.; Goodfellow, P.J.; Yilmaz, A.; Miller, R.; Bacher, J.; et al. Assessmant of human sequencing as a replacement for Lynch syndrome screening and current molecular tests for patients with colorectal cancer. JAMA Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- De Voer, R.M.; Hahn, M.N.; Mesenkamp, A.R.; Hoischen, A.; Gilissen, C.; Henkes, A.; Spruijt, L.; van Zelst-Stams, W.A.; Kets, C.M.; Verwiel, E.T.; et al. Deleterious germline BLM mutations and the risk for early-onset colorectal cancer. Sci. Rep. 2015, 5, 14060. [Google Scholar] [CrossRef] [PubMed]
- De Voer, R.M.; Hahn, M.N.; Weren, R.D.; Mesenkamp, A.; Gilissen, C.; van Zelst-Stasms, W.; Spruijt, L.; Kets, C.M.; Zhang, J.; Venselaar, H.; et al. Identification of novel candidate genes for early-onset colorectal cancer susceptibility. PLoS Genet. 2016, 12, e1005880. [Google Scholar] [CrossRef] [PubMed]
- Heald, B.; Mester, J.; Rybicki, L.; Orloff, M.S.; Burke, C.A.; Eng, C. Frequent gastrointestinal polyps and colorectal adenocarcinomas in prospective series of PTEN mutation carries. Gastroenterology 2010, 139, 1927–1933. [Google Scholar] [CrossRef] [PubMed]
- McCleary, N.; Sato, K.; Nishihara, R.; Inamura, K.; Morikawa, T.; Zhang, X.; Wu, K.; Yamauchi, M.; Kim, S.A.; Sukawa, Y.; et al. Prognostic utility of molecular factors by age at diagnosis of colorectal cancer. Clin. Cancer Res. 2015, 22, 1489–1498. [Google Scholar] [CrossRef] [PubMed]
- Gryfe, R.; Kim, H.; Hsieh, E.; Aronson, M.; Holowati, E.; Bull, S.; Redston, M.; Gallinger, S. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N. Engl. J. Med. 2000, 342, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Lin, P.C.; Lin, C.C.; Lan, Y.T.; Lin, H.H.; Lin, C.H.; Yang, S.H.; Liang, W.Y.; Chen, W.S.; Jiang, J.K.; et al. Molecular and clinicopathological differences by age at the diagnosis of colorectal cancer. Int. J. Mol. Sci. 2017, 18, 1441. [Google Scholar] [CrossRef] [PubMed]
- Kothari, N.; Teer, J.K.; Abott, A.; Srikumar, T.; Zhang, Y.; Yoder, S.J.; Brohl, A.S.; Kim, R.; Reed, D.R.; Shibata, D. Increased incidence of FBXW7 and POLE proofreading domain mutations in young adult colorectal cancers. Cancer 2016, 122, 2828–2835. [Google Scholar] [CrossRef] [PubMed]
- Jandova, Y.; Xu, W.; Nifonsam, V. Sporadic early-onset colon cancer expresses unique molecular features. J. Surg. Res. 2016, 204, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.T.; Pai, R.K.; Rybicki, L.A.; Dimaio, M.A.; Limaye, M.; Jayachandran, P.; Koong, A.C.; Kunz, P.A.; fisher, G.A.; Ford, J.M.; et al. Clinicopathologic and molecular features of sporadic early-onset colorectal adenocarcinoma with frequent signet ring cell differentiation, rectal and sigmoid involvement, and adverse morphologic features. Mod. Pathol. 2012, 25, 1128–1139. [Google Scholar] [CrossRef] [PubMed]
- Yeo, H.; Abelson, J.S.; Zheng, X.; Yantiss, R.; Shah, M.A. Early-onset colorectal cancer is distinct from traditional colorectal cancer. Clin. Colorectal Cancer 2017, 16, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Rindi, G.; Arnold, R.; Bosman, F.T.; Carneiro, F.; Hruban, R.; Theise, N. Nomenclature and classification od neuroendocrine neoplasms of the digestive system. In WHO Classification of Tumors of the Digestive System, 4th ed.; International Agency for Research on Cancer (IARC): Lyon, France, 2010; pp. 13–14. [Google Scholar]
- Koenig, A.; Krug, S.; Mueller, D.; Barth, P.J.; Koenig, U.; Scharf, M.; Ellenrieder, V.; Michi, P.; Moll, R.; Homayunfar, K.; et al. Clinicopathological hallmarks and biomarkers of colorectal neuroendocrine neoplasms. PLoS ONE 2017, 12, e0188876. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, N.; Ohishi, Y.; Takahashi, S.; Nakamura, K.; Tanaka, M.; Oki, E.; Takayanagi, R.; Oda, Y. Molecular characterization of colorectal neuroendocrine carcinoma; similarities with adenocarcinoma rather than neuroendocrine tumor. Hum. Pathol. 2015, 46, 1890–1900. [Google Scholar] [CrossRef] [PubMed]
- Jesinghaus, M.; Konukiewitz, B.; Keller, G.; Kloor, M.; Steiger, K.; Reiche, M.; Penzel, R.; Endris, V.; Arsenic, R.; Hermann, G.; et al. Colorectal mixed adenoneuroendocrine carcinomas and neuroendocrine carcinomas are genetically closely related to colorectal adenocarcinomas. Mod. Pathol. 2017, 30, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Ha, S.Y.; Kim, S.T.; Kim, H.C.; Heo, J.S.; Park, Y.S.; Lauwers, G.; Lee, J.; Kim, K.M. Identification of the BRAF V600E mutation in gastroenteropancreatic neuroendocrine tumors. Oncotarget 2016, 7, 4024–4035. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Klempner, S.J.; Gershenhorm, B.; Tran, P.; Lee, T.K.; Erlander, M.G.; Gowen, K.; Schrock, A.B.; Morosini, D.; Ross, J.S.; Miller, V.A.; et al. BRAFV600E mutations in high-grade colorectal neuroendocrine tumors may predict responsiveness to BRAF-MEK combination therapy. Cancer Discov. 2016, 6, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Olevian, D.C.; Nikiforova, M.N.; Chiosea, S.; Sun, W.; Bahary, N.; Kuan, S.F.; Pai, R.K. Colorectal poorly differentiated neuroendocrine carcinomas frequently exhibit BRAF mutations and are associated with poor overall survival. Hum. Pathol. 2016, 49, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Woischke, C.; Schaaf, C.W.; Yang, H.M.; Vieth, M.; Veits, L.; Geddert, H.; Markl, B.; Stommer, P.; Schaeffer, D.F.; Frolich, M.; et al. In-depth mutational analyses of colorectal neuroendocrine carcinomas with adenoma or adenocarcinoma components. Mod. Pathol. 2017, 30, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Zaaimi, Y.; Aparicio, T.; Laurent-Puig, P.; Taieb, J.; Zaanan, A. Advanced small bowel adenocarcinoma: Molecular characteristics and therapeutic perspectives. Clin. Res. Hepatol. Gastroenterol. 2015, 40, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Laforest, A.; Aparicio, T.; Zaanan, A.; Silva, F.P.; Didelot, A.; Desbeaux, A.; Le Corre, D.; Benhaim, L.; Pallier, K.; Aust, D.; et al. ERBB2 as a potential therapeutic target in small bowel adenocarcinoma. Eur. J. Cancer 2014, 50, 1740–1746. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Zhang, Z.; Dai, B.; Wei, Q.; Liu, J.; Liu, Y.; Liu, Y.; He, L.; Zhou, D. Whole-exome sequencing of duodenal adenocarcinoma identifies recurrent Wnt/ β-catenin signaling pathway mutations. Cancer 2016, 122, 1689–1696. [Google Scholar] [CrossRef] [PubMed]
- Schrock, A.B.; Devoe, C.E.; McWillims, R.; Sun, J.; Aparicio, T.; Stephens, P.J.; Ross, J.S.; Wilson, R.; Milelr, V.A.; Ali, S.M.; et al. Genomic profiling of small-bowel adenocarcinoma. JAMA Oncol. 2017, 3, 1546–1553. [Google Scholar] [CrossRef] [PubMed]
- Imamura, K.; Yamauchi, M.; Nishihara, R.; Kim, S.; Mima, K.; Sukawa, Y.; Li, T.; Yusanari, M.; Zhang, X.; Wu, K.; et al. Prognostic significance and molecular features of signet-ring cell and mucinous components in colorectal carcinoma. Ann. Surg. Oncol. 2015, 22, 1226–1235. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Brahmandam, M.; Cantor, M.; Namgyal, C.; Kawasaki, T.; Kirkner, G.; Meyerhardt, J.A.; Loda, M.; Fuchs, C.S. Distinct molecular features of colorectal carcinoma with signet cell component and colorectal carcinoma with mucinous component. Mod. Pathol. 2006, 19, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Wang, X.; Gao, J.; Li, J.; Qi, C.; Li, Y.; Li, Z.; Shen, L. Clinicopathologic and molecular features of colorectal adenocarcinoma with signet-ring cell component. PLoS ONE 2016, 11, e0156659. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, S.; Onguru, O. BRAF mutation in colorectal carcinomas with signet ring cell component. Cancer Biol. Med. 2017, 14, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Alvi, M.A.; Loughrey, M.B.; Dunne, P.; McQuaid, S.; Turkington, R.; Fuchs, M.A.; McGready, C.; Bingham, V.; Pang, B.; Moore, W.; et al. Molecular profiling of signet ring cell colorectal cancer provides a strong rationale for genomic targeted and immune checkpoint inhibitor therapies. Br. J. Cancer 2017, 117, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Bellan, A.; Cappellesso, R.; Lo Mele, M.; Peraro, L.; Balsamo, L.; Lanza, C.; Fassan, M.; Rugge, M. Early signet cell carcinoma arising from colonic adenoma: The molecular profiling supports the adenoma-carcinoma sequence. Hum. Pathol. 2016, 50, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Hamburger, A.W.; Salmon, S.E. Primary bioassay of human tumor stem cells. Science 1977, 197, 461–463. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1977, 3, 730–737. [Google Scholar] [CrossRef]
- Sell, S.; Pierce, G.B. Maturation arrest of stem cell differentiation is a common pathway for the cellular origin of teratocarcinomas and epithelial cancers. Lab. Investig. 1994, 70, 6–22. [Google Scholar] [PubMed]
- Lobo, N.A.; Shimono, Y.; Qian, D.; Clarke, M.F. The biology of cancer stem cells. Annu. Rev. Cell Dev. Biol. 2007, 23, 675–699. [Google Scholar] [CrossRef] [PubMed]
- Corbeil, D.; Roper, K.; Hellwig, A.; Tavian, M.; Miraglia, S.; Watt, S.M.; Simmons, P.J.; Peault, B.; Buck, D.W.; Huttner, W.B. The human AC133 hematopoietic stem cell antigen is also expressed in epithelial cells and targeted to plasma membrane protrusions. J. Biol. Chem. 2000, 275, 5512–5520. [Google Scholar] [CrossRef] [PubMed]
- Corbeil, D.; Roper, K.; Fargeas, C.A.; Joester, A.; Huttner, W.B. Prominin: A story of cholesterol, plasma membrane protrusions and human pathology. Traffic 2001, 2, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Weigmann, A.; Corbeil, D.; Hellwig, A.; Huttner, W.B. Prominin, a novel microvilli-specific polytopic membrane protein of the apical surface of epithelial cells, is targeted to plasmalemmal protrusions of non-epithelial cells. Proc. Natl. Acad. Sci. USA 1997, 94, 12425–12430. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitani, L.; Lombardi, D.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumor growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Shmelkov, S.V.; Butler, J.M.; Hooper, A.T.; Hormigo, A.; Kushner, J.; Milde, T.; St Clair, R.; Baljevic, M.; White, I.; Jin, D.K.; et al. CD133 expression is not restricted to stem cells, and both CD133+ and CD133− metastatic colon cancer cells initiate tumors. J. Clin. Investig. 2008, 118, 2111–2120. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; Todaro, M.; de Sousa Mello, F.; Sprick, M.R.; Kemper, K.; Perez Alea, M.; Richel, D.J.; Stassi, G.; Medema, J.P. Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. Proc. Natl. Acad. Sci. USA 2008, 105, 13427–13432. [Google Scholar] [CrossRef] [PubMed]
- Horst, D.; Kriegl, L.; Engel, J.; Kichner, T.; Jung, A. CD133 expression is an independent prognostic marker for low survival in colorectal cancer. Br. J. Cancer 2008, 99, 1285–1289. [Google Scholar] [CrossRef] [PubMed]
- Horst, D.; Kriegl, L.; Engel, J.; Jung, A.; Kichner, T. CD133 and nuclear beta-catenin: The marker combination to detect high risk cases of low stage colorectal cancer. Eur. J. Cancer 2009, 45, 2034–2040. [Google Scholar] [CrossRef] [PubMed]
- Artells, R.; Moreno, I.; Diaz, T.; Martínez, F.; Gel, B.; Navarro, A.; Ibeas, R.; Moreno, J.; Monzó, M. Tumor CD133 mRNA expression and clinical outcome in surgically resected colorectal cancer patients. Eur. J. Cancer 2010, 46, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Horst, D.; Scheel, S.K.; Liebmann, S.; Neumann, J.; Maatz, S.; Kirchner, T.; Jung, A. The cancer stem cell marker CD133 has high prognostic impact but unknown functional relevance for the metastasis of human colon cancer. J. Pathol. 2009, 219, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Song, X.; Chen, Z.; Li, X.; Li, M.; Liu, H.; Li, J. CD133 expression and the prognosis of colorectal cancer: A systematic review and meta-analysis. PLoS ONE 2013, 8, e56380. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, T.; Walters, J.; Bo, P.; Than, E.B.; Jung, K.L.; Chen, K.Y.; Panerelli, N.; Milsom, J.; Augenlitcht, L.; Lipkin, S.M.; et al. NOTCH signaling regulates asymmetric cell fate of fast- and slow-cycling colon cancer initiating cells. Cancer Res. 2016, 76, 3411–3421. [Google Scholar] [CrossRef] [PubMed]
- Kemper, K.; Sprick, M.R.; de Bree, M.; Scopelliti, A.; Vermeulen, L.; Hoek, M.; Zeilstra, J.; Pals, S.T.; Mehmet, H.; Stassi, G.; et al. The AC133 epitope, but not the CD133 protein, is lost upon cancer stem cell differentiation. Cancer Res. 2010, 70, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Kemper, K.; Versloot, M.; Cameron, K.; Colak, S.; de Sousa e Melo, F.; de Jong, J.H.; Bleackley, J.; Vermeulen, L.; Versteeg, R.; Koster, J.; et al. Mutations in the Ras-Raf axis underlie the prognostic value of CD133 in colorectal cancer. Clin. Cancer. Res. 2012, 18, 3132–3141. [Google Scholar] [CrossRef] [PubMed]
- Marhaba, R.; Klingbeil, P.; Nuebel, T.; Nazarenko, I.; Buechler, M.W.; Zoeller, M. CD44 and EpCAM cancer-initiating cell markers. Curr. Mol. Med. 2008, 8, 784–804. [Google Scholar] [CrossRef] [PubMed]
- Orian-Rousseau, V. CD44, a therapeutic target for metastasising tumours. Eur. J. Cancer 2010, 46, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Wielenga, V.J.; van der Neut, R.; Offerhaus, G.J.; Pails, S.T. CD44 glycoproteins in colorectal cancer: Expression, function, and prognostic value. Adv. Cancer Res. 2000, 77, 169–187. [Google Scholar] [PubMed]
- Kim, H.R.; Wheeler, M.A.; Wilson, C.M.; Iida, J.; Eng, D.; Simpson, M.A.; McCarthy, J.B.; Bullard, K.M. Hylaronan facilitates invasion of colon carcinoma cells in vitro via interaction with CD44. Cancer Res. 2004, 64, 4569–4576. [Google Scholar] [CrossRef] [PubMed]
- Dalerba, P.; Dylla, S.J.; Park, I.K.; Liu, R.; Wang, X.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M.; et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163. [Google Scholar] [CrossRef] [PubMed]
- Chu, P.; Clanton, D.J.; Snipas, T.S.; Lee, J.; Mitchell, E.; Nguyen, M.L.; Hare, E.; Peach, R.J. Characterization of a subpopulation of colon cancer cells with stem cell-like properties. Int. J. Cancer 2009, 124, 1312–1321. [Google Scholar] [CrossRef] [PubMed]
- Zelistra, J.; Joosten, S.P.; van Andel, H.; Tolg, C.; Berns, A.; Snoek, M.; van de Wetering, M.; Spaargaren, M.; Clevers, H.; Pals, S.T. Stem cells CD44v isoform promotes intestinal cancer formation in APC(Min) mice downstream of Wnt signaling. Oncogene 2013, 33, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Zeilstra, J.; Joosten, S.P.; Doktwer, M.; Verweil, E.; Spaargaren, M.; Pals, S.T. Deletion of the WNT target and cancer stem cell marker CD44 in Apc(min/+) mice attenuates intestinal tumorigenesis. Cancer Res. 2008, 68, 3655–3661. [Google Scholar] [CrossRef] [PubMed]
- Joosten, J.; Zeilstra, J.; van Andel, H.; Mijnals, R.C.; Zaunbrecher, J.; Duivevoorden, A.; van de Wetering, M.; Clevers, H.; Spaargaren, M.; Spaargaren, M.; et al. MET signaling mediates intestinal crypt-villus development, regeneration, and adenoma formation and is promoted by stem cell CD44 isoforms. Gastroenterology 2017, 153, 1040–1053. [Google Scholar] [CrossRef] [PubMed]
- Todaro, M.; Gaggianesi, M.; Catalano, V.; Benfante, A.; Iovino, F.; Biffoni, M.; Apuzzo, T.; Sperduti, I.; Volpe, S.; Cocorullo, G.; et al. CD44v6 is a marker of costitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell 2014, 14, 342–356. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, K.; Sasaki, E.; Kimura, K.; Komori, K.; Shimizu, F.; Yataba, Y.; Aoki, M. HNRPLL, a newly identified colorectal cancer metastasis suppressor, modulates alternative splicing of CD44 during epithelial-mesenchymal transition. Gut 2017, gutjnl-2016-312927. [Google Scholar] [CrossRef]
- Afify, A.; Durbin-Johnson, B.; Virdi, A.; Jess, H. The expression of CD44v6 in colon: From colon to malignant. Ann. Diagn. Pathol. 2016, 20, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Yuan, Y.; Xia, Y.; Achilefu, S. Monoclonal antibody CD188 binds a carbohydrate epitope expressed on the surface of both colorectal cancer stem cells and their differentiated progeny. Clin. Cancer Res. 2008, 14, 7461–7469. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pang, R.; Law, W.L.; Chu, A.; Poon, J.; Lam, C.; Chow, A.; Ng, L.; Cheung, L.; Lan, X.; Lan, H.; et al. A subpopulation of CD26+ cancer stem cells with metastatic capacity in human colorectal cancer. Cell Stem Cell 2010, 6, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.H.; NIyer, D.N.; Lam, C.S.; Ng, L.; Wong, S.K.M.; Lee, H.S.; Wan, T.; Man, J.; Chow, A.K.M.; Poon, R.T.; et al. Emergence of CD26+ cancer stem cells with metastatic properties in colorectal carcinogenesis. Int. J. Mol. Sci. 2017, 18, e1106. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Chen, L.; Ma, Z.; Du, Z.; Zhao, Z.; Hu, Z.; Li, Q. Isolation and phenotypic characterization of colorectal cancer stem cells with organ-specific metastatic potential. Gastroenterology 2013, 145, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.M.; Wei, D.; Gao, W.C.; Xu, Y.T.; Hu, Z.Q.; Ma, Z.Y.; Gao, C.F.; Zhu, X.Y.; LI, Q.Q. TPO-induced metabolic reprogramming drives liver metastasis of colorectal cancer CD110+ tumor-initiating cells. Cell Stem Cell 2015, 17, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Munz, M.; Baeuerle, P.A.; Gires, O. The emerging role of EpCAM in cancer and stem cell signalling. Cancer Res. 2009, 69, 5627–5629. [Google Scholar] [CrossRef] [PubMed]
- Gires, O.; Klein, C.A.; Baeuerle, P.A. On the abundance of EpCAM on cancer stem cells. Nat. Rev. Cancer 2009, 9, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Ohata, H.; Ishiguro, T.; Aihara, Y.; Sato, A.; Sakai, H.; Sekine, S.; Taniguchi, H.; Akasu, T.; Fujita, S.; Nakagama, H.; et al. Induction of stem-like cell regulator CD44 by Rho kinase inhibition contributes to the maintenance of colon cancer-initiating cells. Cancer Res. 2012, 72, 5101–5110. [Google Scholar] [CrossRef] [PubMed]
- Itzkowitz, S.H.; Yio, X. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: The role of inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G7–G17. [Google Scholar] [CrossRef] [PubMed]
- Carpentino, J.E.; Hynes, M.J.; Appelman, H.D.; Zheng, T.; Steindler, D.A.; Scott, E.W.; Huang, E.H. Aldehyde dehydrogenase-expressing colon stem cells contribute to tumorigenesis in the transition from colitis to cancer. Cancer Res. 2009, 69, 8208–8215. [Google Scholar] [CrossRef] [PubMed]
- Yeung, T.M.; Gandhi, S.C.; Wilding, J.L.; Muschel, R.; Bodmer, W.F. Cancer stem cells from colorectal cancer-derived cell lines. Proc. Natl. Acad. Sci. USA 2010, 107, 3722–3727. [Google Scholar] [CrossRef] [PubMed]
- Swindall, A.F.; Londono-Joshi, A.; Schultz, M.J.; Fineberg, N.; Buchsbaum, D.J.; Bellis, S.L. ST6Gal-1 protein expression is upregulated in human epithelial tumors and correlates with stem cell markers in normal tissues and colon cancer cell lines. Cancer Res. 2013, 73, 2368–2378. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.; El-Deizy, W. Calcein-effluxin human colon cancer cells are enriched for self-renewal capacity and depend on beta-catenin. Oncotarget 2012, 4, 184–191. [Google Scholar]
- Dieter, S.; Ball, C.; Hoffmann, C.; Nowrouzi, A.; Herbst, F.; Zavidij, O.; Abel, U.; Arens, A.; Weichert, W.; Brand, K.; et al. Distinct types of tumor-initiating cells from human colon cancer tumors and metastases. Cell Stem Cell 2011, 9, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Giessler, K.; Kleinheinz, K.; Huebschmana, D.; Balaisubramanian, G.P.; Dubash, T.D.; Dieter, S.M.; Siegel, C.; Herbst, F.; Weber, S.; Hoffmann, C.M.; et al. Genetic subclone architecture of tumor-clone initiating cells in colorectal cancer. J. Exp. Med. 2017, 214, 2073–2087. [Google Scholar] [CrossRef] [PubMed]
- Merlos-Suarez, A.; Baniga, F.M.; Jung, P.; Iglesias, M.; Céspedes, M.V.; Rossell, D.; Sevillano, M.; Hernando-Momblona, X.; da Silva-Diz, V.; Muñoz, P.; et al. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell 2011, 8, 511–524. [Google Scholar] [CrossRef] [PubMed]
- De Sousa E Melo, F.; Colak, S.; Buikhuisen, J.; Koster, J.; Cameron, K.; de Jong, J.H.; Tuynman, J.B.; Prasetyanti, P.R.; Fessler, E.; van den Bergh, S.P.; et al. Methylation of cancer-stem-cell-associated Wnt target genes predicts poor prognosis in colorectal cancer patients. Cell Stem Cell 2011, 9, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Ziskin, J.L.; Dunlop, D.; Yaylaoglu, M.; Fodor, I.K.; Forrest, W.F.; Patel, R.; Ge, N.; Hutchins, G.G.; Pine, J.K.; Quirke, P.; et al. In situ validation of an intestinal stem cell signature in colorectal cancer. Gut 2012, 62, 1012–1023. [Google Scholar] [CrossRef] [PubMed]
- Dame, M.K.; Attili, D.; McClintock, D.D.; Dedhia, P.H.; Ouillette, P.; Hardt, O.; Chin, A.M.; Xue, X.; Laliberte, J.; Katz, E.L. Identification, isolation and characterization of human LGR5+ colon adenoma cells. Development 2018, 145, dev153049. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.L.; Zeng, Z.; Adileh, M.; Jacobo, A.; Li, C.; Vakiani, E.; Hua, G.; Zhang, L.; Haimovitz-Friedman, A.; Fuks, Z.; et al. Logarithmic expansion of LGR5+ cells in human colorectal cells. Cell Signal. 2017, 42, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Geng, L.; Wang, D.; Yi, H.; Palmon, G.; Wang, J. R-Spondin 1/LGR5 activates TGF β signaling and suppresses colon cancer metastasis. Cancer Res. 2017, 77, 6589–6602. [Google Scholar] [CrossRef] [PubMed]
- Cortina, C.; Turon, G.; Stork, D.; Herando-Momblona, X.; Sevillano, M.; Aguilera, M.; Tosi, S.; Merlos-Surez, A.; Attolini, C.S.; Sancho, E.; et al. A genome editing approach to study cancer stem cells in human tumors. EMBO Mol. Med. 2017, 9, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, M.; Ohta, Y.; Nishikori, S.; Matano, M.; Takano, A.; Fujii, M.; Date, S.; Sugimoto, S.; Kanai, T.; Sato, T. Visualization and targeting of LGR5+ human colon cancer stem cells. Nature 2017, 585, 187–192. [Google Scholar] [CrossRef] [PubMed]
- De Sousa e Melo, F.; Kurtova, A.; Harnoss, J.; Kljavin, N.; Hoeck, J.; Hung, J.; Anderson, J.E.; Storm, E.; Modrosan, Z.; Koeppen, H.; et al. A distinct role for Lgr5+ stem cells in primary and metastatic colon cancer. Nature 2017, 543, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Kreso, A.; O’Brien, C.A.; van Galen, P.; Gan, O.I.; Notta, F.; Brown, A.M.; Ng, K.; Ma, J.; Wienholds, E.; Dunant, C.; et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science 2013, 339, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Bu, P.; Chen, K.Y.; Chen, Y.H.; Wang, L.; Walters, J.; Shin, Y.J.; Goerger, J.P.; Sun, J.; Witherspoon, M.; Rakhilin, N.; et al. A microRNA miR-34a-regulated bimodal switch targets NOTCH in colon cancer stem cells. Cell Stem Cell 2013, 12, 602–615. [Google Scholar] [CrossRef] [PubMed]
- Bu, P.; Wang, L.; Chen, K.Y.; Srinivasan, T.; Lakshminarasimha Murty, P.K.; Tung, K.L.; Varanko, A.K.; Chen, H.J.; Ai, Y.; King, S.; et al. A miR-34a-Numb feedforward loop triggered by inflammation regulates asymmetric stem cell division in intestine and colon cancer. Cell Stem Cell 2016, 18, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, P.J. Role of ADAM10 in intestinal crypt homeostasis and tumorigenesis. Bichem. Biophys. Acta Mol. Cell Res. 2017, 1864, 2228–2239. [Google Scholar] [CrossRef] [PubMed]
- Fre, S.; Pallavi, S.K.; Huyghe, M.; Lae, K.P.; Janssen, S.; Robine, S.; Artavanis-Tsakonas, S.; Louvard, D. Notch and Wnt signals cooperatively control cell proliferation and tumorigenesis in the intestine. Proc. Natl. Acad. Sci. USA 2009, 106, 6309–6314. [Google Scholar] [CrossRef] [PubMed]
- Van Es, J.H.; Van Gijn, M.E.; Riccio, O.; Van Den Born, M.; Vooijs, M.; Begthel, H.; Cozijnsen, M.; Robine, S.; Winton, D.J.; Radtke, F.; et al. NOTCH/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 2005, 435, 959–963. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.H.; Feng, Y.; Rakshit, R.; Fearon, E.R.; Dempsey, P.J. Cell-autonomous ADAM 10 signaling is required for adenoma initiation after APC mutation. Gastroenterology 2013, 144, S51–S52. [Google Scholar] [CrossRef]
- Kobayashi, S.; Yamada-Okabe, H.; Suzuki, M.; Natori, O.; Kato, A.; Matsubara, K.; Jau Chen, Y.; Yamazaki, M.; Funahashi, S.; Yoshida, K.; et al. LGR5-positive colon cancer stem cells interconvert with drug–resistant LGR5-negative cells and are capable of tumor reconstitution. Stem Cells 2012, 30, 2631–2644. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Ye, X.; Fan, F.; Xia, L.; Bhattacharya, R.; Bellister, S.; Tozzi, F.; Sceusi, E.; Zhou, Y.; Tachibana, I.; et al. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged-1. Cancer Cell 2013, 23, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Wilson, G.; Huang, B.; Peng, M.; Teng, G.; Zhang, D.; Zhang, R.; Ebert, M.P.; Chen, J.; Wong, B.C.; et al. Silencing of Jagged1 inhibits cell growth and invasion in colorectal cancer. Cell Death Dis. 2014, 5, e1170. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Gadkai, R.A.; Ramakanth, S.V.; Padmanabhan, K.; Madhumathi, D.S.; Dedvi, L.; Appaji, L.; Aster, J.C.; Rangarajan, A.; Dighe, R.R. A novel monoclonal antibody against Notch1 targets leukemia-associated mutant Notch1 and depletes therapy resistant cancer stem cells in solid tumors. Sci. Rep. 2015, 5, 11012. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ishiguro, H.; Okubo, T.; Kuwabara, Y.; Kimura, M.; Mitsui, A.; Sugito, N.; Ogawa, R.; Katada, T.; Tanaka, T.; Shiozaki, M.; et al. NOTCH1 activates the Wnt/β-catenin signaling pathway in colon cancer. Oncotarget 2017, 8, 60378–60389. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Heredia, J.M.; Lucena-Cacace, A.; Verdugo-Sivianes, E.M.; Perez, M.; Carnero, A. The cargo protein MAP17 (PDZK1IP1) regulates the cancer stem cell pool activating the Notch pathway by abducting NUMB. Clin. Cancer Res. 2017, 23, 3871–3883. [Google Scholar] [CrossRef] [PubMed]
- Irshad, S.; Bansal, M.; Guarnieri, P.; Davis, H.; Zen, A.; Baran, B.; Pinna, C.; Rahman, H.; Biswas, S.; Bardella, C.; et al. Bone morphogenetic protein crosstalk in poor-prognosis, mesenchymal subtype colorectal cancer. J. Pathol. 2017, 242, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Vu, T.; Yuan, G.; Datta, P.K. STRAp promotes stemness of human colorectal cancer via epigenetic regulation of the NOTCH pathway. Cancer Res. 2017, 77, 5464–5478. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, Y.; Seno, H.; Fukuoka, A.; Ueo, T.; Yamaga, Y.; Maruno, T.; Nakanishi, N.; Kanda, K.; Komekado, H.; Kawada, M.; et al. Dclk1 distinguishes between tumor and normal stem cells in the intestine. Nat. Genet. 2013, 45, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Middelhoff, M.; Westphalen, C.B.; Hayakawa, Y.; Yan, K.S.; Gershon, M.D.; Wang, T.C.; Quante, M. Dclk1-expressing Tuft cells: Critical modulators of the intestinal niche? Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 313, G285–G299. [Google Scholar] [CrossRef] [PubMed]
- McKinley, E.T.; Sui, Y.; Al-Kofahi, Y.; Millis, B.A.; Tyska, M.J.; Roland, J.T.; Santamaria-Pang, A.; Ohland, C.L.; Jobin, C.; Franklin, J.L.; et al. Optimized multiplex immunofluorescence single-cell analysis reveals Tuft cell heterogeneity. JCI Insight 2017, 2, 93487. [Google Scholar] [CrossRef] [PubMed]
- Westphalen, B.; Asfaha, S.; Hayakawa, Y.; Takemoto, Y.; Lukin, D.J.; Nuber, A.H.; Brandtner, A.; Setlik, W.; Remotti, H.; Muley, A.; et al. Long-lived intestinal cells serve as colon cancer-initiating cells. J. Clin. Investig. 2014, 124, 1283–1295. [Google Scholar] [CrossRef] [PubMed]
- Chandrakesan, P.; Wygant, N.; May, R.; Qu, D.; Chinthalapally, H.R.; Sureban, S.M.; Ali, N.; Lightfoot, S.A.; Umar, S.; Houchen, C.W. DCLK1 facilitates intestinal tumor growth via enhancing pluripotency and epithelial mesenchymal transition. Oncotarget 2014, 5, 9269–9280. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wang, S.; Sun, Q.; Yang, Z.; Liu, M.; Tang, H. DCLK1 promotes epithelial-mesenchymal transition via the PI3K/AKT/NF-kB pathway in colorectal cancer. Int. J. Cancer 2018, 142, 2068–2079. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, M.; Hisamori, S.; Oshima, N.; Sato, F.; Shimono, Y.; Sakai, Y. miR-137 regulates the tumorigenicity of colon cancer stem cells through the inhibition of DCLK1. Mol. Cancer Res. 2016, 14, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Kantara, C.; O’Connell, M.; Sarkar, S.; Moya, S.; Ullrich, R.; Singh, P. Curcumin promotes autophagic survival of a subset of colon cancer stem cells, which are ablated by DCLK1-siRNA. Cancer Res. 2014, 74, 2487–2498. [Google Scholar] [CrossRef] [PubMed]
- Chandrakesan, P.; Yao, J.; Qu, D.; May, R.; Weygant, N.; Ge, Y.; Ali, N.; Sureban, S.; Guda, M.; Vega, K.; et al. Dclk1, a tumor stem cell marker, regulates pro-survival signaling and self-renewal of intestinal tumor cells. Mol. Cancer 2017, 16, 30. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, M.R.; Sarkar, S.; Luthra, G.K.; Okugawa, Y.; Toiyama, Y.; Gajjar, A.H. Epigenetic changes and alternate promoter usage by human colon cancers for expressing DCLK1-isoforms. Sci. Rep. 2015, 5, 14983. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; O’Connell, M.; Okugawa, Y.; Lee, B.S.; Toiyama, V.; Kusunoki, M.; Dabovai, R.D.; Goel, A.; Singh, P. FOXD3 regulates CSC marker, DCLK1-S, and invasive potential: Prognostic implications in colon cancer. Mol. Cancer Res. 2017, 15, 1678–1691. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Popov, V.L.; O’Connell, M.; Stevenson, H.L.; Lee, B.S.; Obeid, R.A.; Luthra, G.; Singh, P. A novel antibody against cancer stem cell biomarker, DCLK1-S, is potentially useful for assessing colon cancer risk after screening colonoscopy. Lab. Investig. 2017, 97, 1245–1261. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.J.; Edwards, R.; Tucci, S.; Bu, P.; Milsom, J.; Lee, S.; Edelmann, W.; Gümüs, Z.H.; Shen, X.; Lipkin, S. Chemokine 25-induced signaling suppresses colon cancer invasion and metastasis. J. Clin. Investig. 2012, 122, 3184–3196. [Google Scholar] [CrossRef] [PubMed]
- Asfaha, S.; Hayakawa, Y.; Muley, A.; Stokes, S.; Graham, T.A.; Ericksen, R.E.; Westphalen, C.B.; von Burstin, J.; Mastracci, T.L.; Worthley, D.L.; et al. Krt19+/Lgr5− cells are radioresistant cancer-initiating stem cells in the colon and intestine. Cell Stem Cell 2015, 16, 627–638. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.; Kreso, A.; Ryan, P.; Hermans, K.G.; Gibson, L.; Wang, Y.; Tsatsanis, A.; Gallinger, S.; Dick, J.E. ID1 and ID3 regulate the self-renewal of human colon cancer-initiating cells through p21. Cancer Cell 2012, 21, 777–792. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.J.; Jang, J.B.; Lee, H.Y.; Park, S.R.; Kim, J.Y.; Nam, J.S.; Hong, I.S. The Wnt/β-catenin signaling/Id2 cascade mediates the effects of hypoxia on the hierarchy of colorectal-cancer stem cells. Sci. Rep. 2016, 6, 22966. [Google Scholar] [CrossRef] [PubMed]
- Kreso, A.; Van Galen, P.; Pedley, N.M.; Lima-Fernandes, E.; Frelin, C.; Davis, T.; Cao, L.; Baiazitov, R.; Du, W.; Sydorenko, N.; et al. Self-renewal as a therapeutic target in human colorectal cancer. Nat. Med. 2014, 20, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Leng, Z.; Thao, K.; Xia, Q.; Tan, J.; Yue, Z.; Chen, J.; Xi, H.; Li, J.; Zheng, H. Kruppel-like factor 4 acts as an oncogene in colon cancer stem cell-enriched spheroid cells. PLoS ONE 2013, 8, e56082. [Google Scholar] [CrossRef] [PubMed]
- Maddox, J.; Shakya, A.; South, S.; Shelton, D.; Andersen, J.N.; Chidester, S.; Kang, J.; Gligorich, K.M.; Jones, D.A.; Spangrude, G.J.; et al. Transcription factor Oct1 is a somatic and cancer stem cell determinant. PLoS Genet. 2012, 8, e1003048. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.L.; Yang, M.H.; Tsai, M.L.; Lan, H.Y.; Su, S.H.; Chang, S.C.; Teng, H.W.; Yang, S.H.; Lan, Y.T.; Chiou, S.H.; et al. SNAIL regulates interleukin-8 expression, stem cell-like activity, and tumorigenicity of human colorectal carcinoma cells. Gastroenterology 2011, 141, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.S.; Huang, C.C.; Pan, M.R.; Chen, H.H.; Hung, W.C. Prospero homeobox 1 promotes epithelial-mesenchymal transition in colon cancer cells by inhibiting E-cadherin via miR-9. Clin. Cancer Res. 2012, 18, 6416–6425. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.Q.; Brabletz, T.; Fearon, E.; Willis, A.L.; Hu, C.Y.; Li, X.Y.; Weiss, S.J. Canonical Wnt suppressor, Axin2, promotes colon carcinoma oncogenic activity. Proc. Natl. Acad. Sci. USA 2012, 109, 11312–11317. [Google Scholar] [CrossRef] [PubMed]
- Goto, N.; Ueo, T.; Fukuda, A.; Kawada, K.; Sakai, Y.; Miyoshi, H.; Taketo, M.M.; Chiba, T.; Seno, H. Distinct roles of HES1 in normal stem cells and tumor stem-like cells of the intestine. Cancer Res. 2017, 77, 3442–3454. [Google Scholar] [CrossRef] [PubMed]
- Nakata, T.; Shimizu, H.; Nagata, S.; Ito, G.; Fujii, S.; Suzuki, K.; Kawamoto, A.; Ishibashi, F.; Kuno, R.; Anzai, S.; et al. Indispensable role of Notch ligand-dependent signaling in the proliferation and stem cell niche maintenance of APC-deficient intestinal tumors. Biochem. Biophys. Res. Commun. 2017, 482, 1296–1303. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Zhang, Y.; Wang, S.; Liu, Y.; Zheng, L.; Yang, J.; Huang, W.; Ye, Y.; Luo, W.; Xiao, D. Hes1 is involved in the self-renewal and tumorigenicity of stem-like cancer cells in colon cancer. Sci. Rep. 2014, 4, 3963. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.W.; Liao, M.Y.; Lin, W.W.; Wang, Y.P.; Lu, T.Y.; Wu, H.C. Epitehlial cell adhesion molecule regulates tumor initiation and tumorigenesis via activating reprogramming factors and epithelial-mesenchymal transition gene expression in colon cancer. J. Biol. Chem. 2012, 287, 39449–39459. [Google Scholar]
- Zubeldia, I.G.; Bleau, A.M.; Redrado, M.; Serrano, D.; Agliano, A.; Gil-Puig, C.; Vidal-Vanaclocha, F.; Lecanda, J.; Calvo, A. Epithelial to mesenchymal transition and cancer stem cell phenotypes leading to liver metastasis are abrogated by the nobel TGFbeta1-targeting pepetides P17 and P144. Exp. Cell Res. 2013, 319, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Okabe, H.; Watanabe, M.; Ishimoto, T.; Iwatsuki, M.; Baba, Y.; Tanaka, Y.; Kurashige, J.; Miyamoto, Y.; Baba, H. CD44v6 expression is related to mesenchymal phenotype and poor prognosis in patients with colorectal cancer. Oncol. Rep. 2013, 29, 1570–1578. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Park, Y.S.; Kim, H.J.; Kim, C.H.; Lim, S.W.; Huh, J.W.; Lee, J.H.; Kim, H.R. CD44 enhnaces the epithelial-mesenchymal transition in association with colon cancer invasion. Int. J. Oncol. 2012, 41, 211–218. [Google Scholar] [PubMed]
- Rosenbluh, J.; Nijhawan, D.; Cox, E.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C.; et al. Βeta-catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 2012, 151, 1457–1473. [Google Scholar] [CrossRef] [PubMed]
- Tenbaum, S.; Ordonez-Moran, P.; Puig, I.; Chicote, I.; Arqués, O.; Landolfi, S.; Fernández, Y.; Herance, J.R.; Gispert, J.D.; Mendizabal, L.; et al. Βeta-catenin confers resistance to PI3K and AKT inhibitors and subverts FOXO3a to promote metastasis in colon cancer. Nat. Med. 2012, 18, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. YAP/TAZ incorporation in the β-catenin destruction complex orchestrates the Wnt response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Diamantopoulou, Z.; White, G.; Fadlullah, M.; Dreger, M.; Pickering, K.; Maltas, J.; Ashton, G.; MacLeod, R.; Baillie, G.; Kouskoff, V.; et al. TIAM1 antagonizes TAZ/YAP both in the destruction complex in the cytoplasm and in the nucleus to inhibit invasion of intestinal epithelial cells. Cancer Cell 2017, 31, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Regan, J.L.; Schumacher, D.; Standte, S.; Steffen, A.; Haybaeck, J.; Keilholz, U.; Schweiger, C.; Glob-Schwerzl, N.; Mumberg, D.; Henderson, D.; et al. Non-canonical Hedgehog signaling is a positive regulator of the WNT pathway and is required for the survival of colon cancer stem cells. Cell Rep. 2017, 21, 2813–2828. [Google Scholar] [CrossRef] [PubMed]
- Masuda, M.; Uno, Y.; Ohbayashi, N.; Ohata, H.; Mimata, A.; Kukimoto-Niino, M.; Moriyama, H.; Kashimoto, S.; Inoue, T.; Goto, N.; et al. TNIK inhibition abrogates colorectal cancer stemness. Nat. Cummun. 2016, 7, 12586. [Google Scholar] [CrossRef] [PubMed]
- Giraud, J.; Failla, L.; Pascussi, J.M.; Legerquist, E.; Ollier, J.; Finetti, P.; Bertucci, F.; Ya, C.; Gasmi, I.; Bourgaux, J.F.; et al. Autocrine secretion of progastrin promotes the survival and self-renewal of colon acncre stem-like cells. Cancer Res. 2016, 76, 3618–3628. [Google Scholar] [CrossRef] [PubMed]
- Prieur, A.; Cappellini, M.; Habif, G.; Lefranc, M.P.; Mazard, T.; Morency, E.; Pascussi, J.M.; Flacelerie, M.; Cahuzac, N.; Vire, B.; et al. Targeting the Wnt pathway and cancer stem cells with anti-progastrin humanized antibodies as a potential treatment for KRSA-mutated colorectal cancer. Clin. Cancer Res. 2017, 23, 5267–5280. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.J.; McCoy, M.J.; Hemming, C.; Bulsara, M.K.; Iacopetta, B.; Platell, C.F. Objective analysis of cancer stem cell marker expression using immunohistochemistry. Pathology 2017, 49, 24–29. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Miller, T.J.; McCoy, M.J.; Hemmings, C.; Bulsara, M.K.; Iacopetta, B.; Platell, C.F. The prognostic value of cancer stem-like markers SOX2 and CD133 in stage III colon cancer is modified by expression of the immune-related markers FoxP3, PD-L1 and CD3. Pathology 2017, 49, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Shaked, H.; Hofseth, L.; Chumanevich, A.; Chumanevich, A.A.; Wang, J.; Wang, Y.; Taniguchi, K.; Guma, M.; Shenouda, S.; Clevers, H.; et al. Chronic epithelial NF-kB activation accelerates APC loss and intestinal tumor initiation through iNOS upregulation. Proc. Natl. Acad. Sci. USA 2012, 109, 14007–14012. [Google Scholar] [CrossRef] [PubMed]
- Schiwitalla, S.; Fingerle, A.; Cammareri, P.; Nebelsiek, T.; Göktuna, S.I.; Ziegler, P.K.; Canli, O.; Heijmans, J.; Huels, D.J.; Moreaux, G.; et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem cell-like properties. Cell 2013, 152, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Putoczki, T.L.; Thiem, S.; Loving, A.; Busuttil, R.A.; Wilson, N.J.; Ziegler, P.K.; Nguyen, P.M.; Preaudet, A.; Farid, R.; Edwards, K.M.; et al. Interleukin-11 is the dominant IL-6 family cytokine during gastrointestinal tumorigenesis and can be targeted therapeutically. Cancer Cell 2013, 24, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.; Wang, K.; Mucida, D.; Stewart, C.A.; Schnabl, B.; Jauch, D.; Taniguchi, K.; Yu, G.Y.; Osterreicher, C.H.; Hung, K.E.; et al. Adenoma-linked barrier defects and barrier products drives IL-23/IL-17-mediated tumor growth. Nature 2012, 491, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, A.; Fisher, R.; Butterworth, E.; Pi, L.; Chang, L.J.; Appelman, H.D.; Chang, M.; Scott, E.W.; Huang, E.H. Transition from colitis to cancer: High Wnt activity sustains the tumor-initiating potential of colon cancer stem cell precursors. Cancer Res. 2012, 72, 5091–5100. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.; Gagliani, N.; Zenewicz, L.; Huber, F.J.; Bosurgi, L.; Hu, B.; Hedl, M.; Zhang, W.; O’Connor, W., Jr.; Murphy, A.J.; et al. IL-22BP is regulated by the inflammasomes and modulates tumorigenesis in the intestine. Nature 2012, 491, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Lin, Y.; Nagarsheth, N.; Peng, D.; Zhao, L.; Zhao, E.; Vatan, L.; Szeliga, W.; Dou, Y.; Owens, S.; et al. IL-22+ CD4+ T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity 2014, 40, 772–784. [Google Scholar] [CrossRef] [PubMed]
- Basak, O.; Beumer, J.; Wiebrands, K.; Seno, H.; van Oudenaarden, A.; Clevers, H. Induced quiescence of LGR5+ stem cells in intestinal organoids enables differentiation of hormone producing enteroendocrine cells. Cell Stem Cell 2017, 20, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Nakakuta-Ddamba, A.; Tobias, J.; Jensen, S.T.; Lenguer, C.J. Mouse label-retaining cells are molecularly and functionally distinct from reserve intestinal stem cells. Gastroenterology 2016, 151, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Gribble, F.M.; Reimann, F. Enteroendocrine cells: Chemosensors in the intestinal epithelium. Ann. Rev. Physiol. 2016, 78, 277–299. [Google Scholar] [CrossRef] [PubMed]
- Haber, A.; Biton, M.; Rogel, N.; Herbst, R.H.; Shekkar, K.; Smillie, C.; Burgin, G.; Delorey, T.M.; Howitt, M.R.; Katz, Y.; et al. A single-cell survey of the small intestinal epithelium. Nature 2017, 551, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Bancz, M.S.; Kanwar, R.; Kulkami, A.A.; Boora, G.K.; Metge, F.; Kipp, B.R.; Zhang, L.; Thorland, E.C.; Minn, K.T.; Tendu, R.; et al. The genomic landscape of small intestine neuroendocrine tumors. J. Clin. Investig. 2013, 123, 2502–2508. [Google Scholar] [CrossRef] [PubMed]
- Francis, J.M.; Kiezun, A.; Ramos, A.H.; Serra, S.; Pedamallu, C.S.; Qian, Z.R.; Banck, M.S.; Kanwar, R.; Kulkarni, A.A.; Karpathakis, A.; et al. Somatic mutation of CDKN1B in small intestine neuroendocrine tumors. Nat. Genet. 2013, 45, 1483–1486. [Google Scholar] [CrossRef] [PubMed]
- Kleist, B.; Poetsch, M. Neuroendocrine differentiation: The mysterious fellow of colorectal cancer. World J. Gastroenterol. 2015, 21, 11740–11747. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Giel-Moloney, M.; Riudi, G.; Leiter, A.B. Enteroendocrine precursors differentiate independently of Wnt and form serotonin expressing adenomas in response to active beta-catenin. Proc. Natl. Acad. Sci. USA 2007, 104, 11328–11333. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.M.; Laird, P.W.; Park, P.J. The landscape of microsatellite instability un colorectal and endometrial cancer genomes. Cell 2013, 155, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Graham, T.A.; Sottoriva, A. Measuring cancer evolution from the genome. J. Pathol. 2017, 241, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Stickler, J.H.; Loree, J.M.; Ahronian, L.G.; Parikh, A.R.; Niedwiecki, D.; Lima Pereira, A.A.; McKinney, M.; Korn, W.M.; Atreya, C.E.; Banks, K.C.; et al. Genomic landscape of cell-free DNA in patients with colorectal cancer. Cancer Discov. 2018, 8, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Alwers, E.; Jia, M.; Kloor, M.; Blaker, H.; Brenner, H.; Hoffmeister, M. Associations between molecular classifications of colorectal cancer and patient survival: A systematic review. Clin. Gatstroenterol. Hepatol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Isella, C.; Brundu, F.; Bellomo, S.; Galimi, F.; Zanella, E.; Porporato, R.; Petti, C.; Fiori, A.; Ozzan, F.; Senetta, R.; et al. Selective analysis of cancer-cell intrinsic transcriptional traits defines novel clinically relevant subtypes of colerectal cancer. Nat. Commun. 2017, 8, 15107. [Google Scholar] [CrossRef] [PubMed]
- Alderdice, M.; Richman, S.D.; Gollins, S.; Stewart, J.P.; Hurt, C.; Adams, R.; McCorry, A.M.; Roddy, A.C.; Vimalachandran, D.; Isella, C.; et al. Prospective patient stratification into robust cancer-cell intrinsic subtypes from colorectal cancer biopsies. J. Pathol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Sagaert, X.; Vanstapel, A.; Verbeek, S. Tumor heterogeneity in colorectal cancer: What do we know so far? Pathobiology 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Ubink, I.; Elias, S.G.; Moelans, C.B.; Laclé, M.M.; van Grevenstein, W.; van Diest, P.J.; Borel Rinkes, I.; Kranenburg, O. A novel diagnostic tool for selecting patients with mesenchymal-type colon cancer reveals intratumor subtype heterogeneity. J. Natl. Cancer Inst. 2017, 109, djw303. [Google Scholar] [CrossRef] [PubMed]
- Strum, K. Colorectal adenoma. N. Engl. J. Med. 2016, 374, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Meier, B.; Caca, K.; Fischer, A.; Schmidt, A. Endoscopic management of colorectal adenomas. Ann. Gastroenterol. 2017, 30, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Schoen, R.E.; Pinsky, P.F.; Weissfeld, J.L.; Yococki, L.A.; Church, T.; Laiyemo, A.O.; Bresalier, R.; Andriole, G.L.; Buys, S.S.; Crawford, E.D.; et al. Colorectal-cancer incidence and mortality with screening flexible sigmoidoscopy. N. Engl. J. Med. 2012, 366, 2345–2357. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, R.; Wu, K.; Lochhead, P.; Morikawa, T.; Liao, X.; Qian, S.R.; Inamura, K.; Kim, S.A.; Kuchiba, A.; Yamauchi, M.; et al. Long-term colorectal-cancer incidence and mortality after lower endoscopy. N. Engl. J. Med. 2013, 369, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Holme, O.; Loberg, M.; Kalager, M.; Bretthauer, M.; Hernan, M.A.; Aas, E.; Eide, T.J.; Skovlund, E.; Schneede, J.; Tveit, K.M.; et al. Effect of flexible sigmoidoscopy screening on colorectal cancer incidence and moretality: A randomizef clinical trial. JAMA 2014, 312, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Holme, O.; Schoen, R.E.; Senore, C.; Segnan, N.; Hoff, G.; Loberg, M.; Bretthauer, M.; Adami, H.O.; Kalager, M. Effectiveness of flexible sigmoidoscopy screening in men and women and different age groups: Pooled analysis of randomized trials. Br. Med. J. 2017, 356, i6673. [Google Scholar] [CrossRef] [PubMed]
- Atkin, W.; Wooldrage, K.; Parin, D.M.; Kralji-Hans, I.; MacRae, E.; Shah, U.; Duffy, S.; Cross, A.J. Long-term effects of once-only flexible sigmoidoscopy screening after 17 years of follow-up: The UK Flexible Sigmoidoscopy Screening randomized controlled trial. Lancet 2017, 389, 1299–1311. [Google Scholar] [CrossRef]
- Zauber, A.G.; Winawer, S.J.; O’Brien, M.J.; Lansdorp-Vogelaar, I.; van Ballegooijen, M.; Hankey, B.F.; Shi, W.; Bond, H.J.; Schapiro, M.; Panish, J.F.; et al. Colonoscopy polypectomy and long-term prevention of colorectal-cancer deaths. N. Engl. J. Med. 2012, 366, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Loberg, M.; Kalager, M.; Holme, O.; Hoff, G.; Adami, H.O.; Bretthauer, M. Long-etrm colorectal-cancer mortality after adenoma removal. N. Engl. J. Med. 2014, 371, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Atkin, W.; Wooldrage, K.; Brenner, A.; Martin, J.; Shah, U.; Perera, S.; Lucas, F.; Brown, J.P.; Kralj-Hans, I.; Greli, P.M. Adenoma surveillance and colorectal cancer incidence: A retrospective, multicenter, cohort study. Lancet Oncol. 2017, 18, 823–834. [Google Scholar] [CrossRef]
- Sepulveda, A.R.; Hamilton, S.R.; Allegra, C.J.; Grody, W.; Cushman-Vokoun, A.M.; Funkhouser, W.K.; Kopetz, S.E.; Lieu, C.; Lindor, N.M.; Minsky, B.D.; et al. Molecular biomarkers for the evaluation of colorectal cancer: Guideline from the American Society fo Clinical Pathology, College of American Pathologists, Association for Molecular Pathology, and the American Society of Clinical Oncology. J. Clin. Oncol. 2017, 35, 1453–1486. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Custem, E.; McDermott, R.; Hill, A.; et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.H.L.; Segelov, E.; Wong, R.S.; Smith, A.; Herbertson, R.A.; Li, B.T.; Tebbutt, N.; Price, T.; Pavlakin, N. Epidermal growth factor receptor (EGFR) inhibitors for metastatic colorectal cancer. Cochrane Database Syst. Rev. 2017, 6, CD007047. [Google Scholar] [CrossRef] [PubMed]
- Richman, S.D.; Southward, K.; Chanbers, P. HER2 overexpression and amplification as a potential therapeutic target in colorectal cancer: Analysis of 3256 patients enrolled in the QUASAR, FOCUS and PICCOLO colorectal cancer trial. J. Pathol. 2016, 238, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Haisworth, J.D.; Meric-Bernstam, F.; Swanton, C.; Hurwitz, H.; Spigel, D.R.; Sweeney, C.; Burris, H.; Bose, R.; Yoo, B.; Stein, A.; et al. Targeted therapy for advanced solid tumors on the basis of molecular profiles: Results from MyPathway, an open-lebel, phase IIa multiple basket study. J. Clin. Oncol. 2018, 36, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, H.; Raghav, K.P.S.; Burris, H.A. Pertuzumab + Trastuzumab for HER2-amplified/overexpressed metastatic colorectal cancer (mCRC): Interim data from MyPathway. J. Clin. Oncol. 2017, 35 (Suppl. 4), 676. [Google Scholar] [CrossRef]
- Oost, K.C.; van Voorthijsen, L.; Fumagalli, A.; lindeboom, R.G.H.; Sprangers, J.; Omerzu, M.; Rodriguez-Colman, M.J.; Heinz, M.C.; Verlaan-Klink, I.; Maurice, M.M.; et al. Specific labeling of stem cell activity in human colorectal organoid using ASCL2-responsive minigene. Cell Rep. 2018, 22, 1600–1614. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, S.; Ohta, Y.; Fuji, M.; Matano, M.; Shimokawa, M.; Nauki, K.; Date, S.; Nishikori, S.; Nakazato, Y.; Nakamura, T.; et al. Reconstruction of the human colon epithelium in vivo. Cell Stem Cell 2018, 22, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Romentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Canellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel Iii, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGF β attenuates tumour response to PD-L1 blockade contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Vogelstein, B. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Li, L.; Vogelstein, B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 2017, 355, 1330–1334. [Google Scholar] [CrossRef] [PubMed]
- Blokzijl, F.; de Ligt, J.; Jager, M.; Sasselly, V.; Roerink, S.; Sasaki, N.; Huch, M.; Boymans, S.; Kruijk, E.; Prins, P.; et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 2016, 538, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Pitmon, E.; Wang, K. Microbiome, inflammation and colorectal cancer. Semin. Immunol. 2017, 32, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Fang, L.; Lee, M.H. Dysbiosis of gut microbiota in promoting the development of colorectal cancer. Gastroenterol. Rep. 2018, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hussan, H.; Clinton, S.K.; Roberts, K.; Bailey, M.T. Fusobacterium’s link to colorectal neoplasia sequenced: A systematic review and future insights. World J. Gastroenterol. 2017, 23, 8626–8650. [Google Scholar] [CrossRef] [PubMed]
- Repass, J.; Reproducibility Project: Cancer Biology. Replication study: Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. eLife 2018, 13, e25801. [Google Scholar] [CrossRef]
- Dejea, C.M.; Fathi, P.; Craig, J.M.; Boleij, A.; Taddese, R.; Geius, A.L.; Wu, X.; De Stefano Shields, C.E.; Hechenbleikner, E.M.; Huso, D.L.; et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 2018, 359, 592–597. [Google Scholar] [CrossRef] [PubMed]













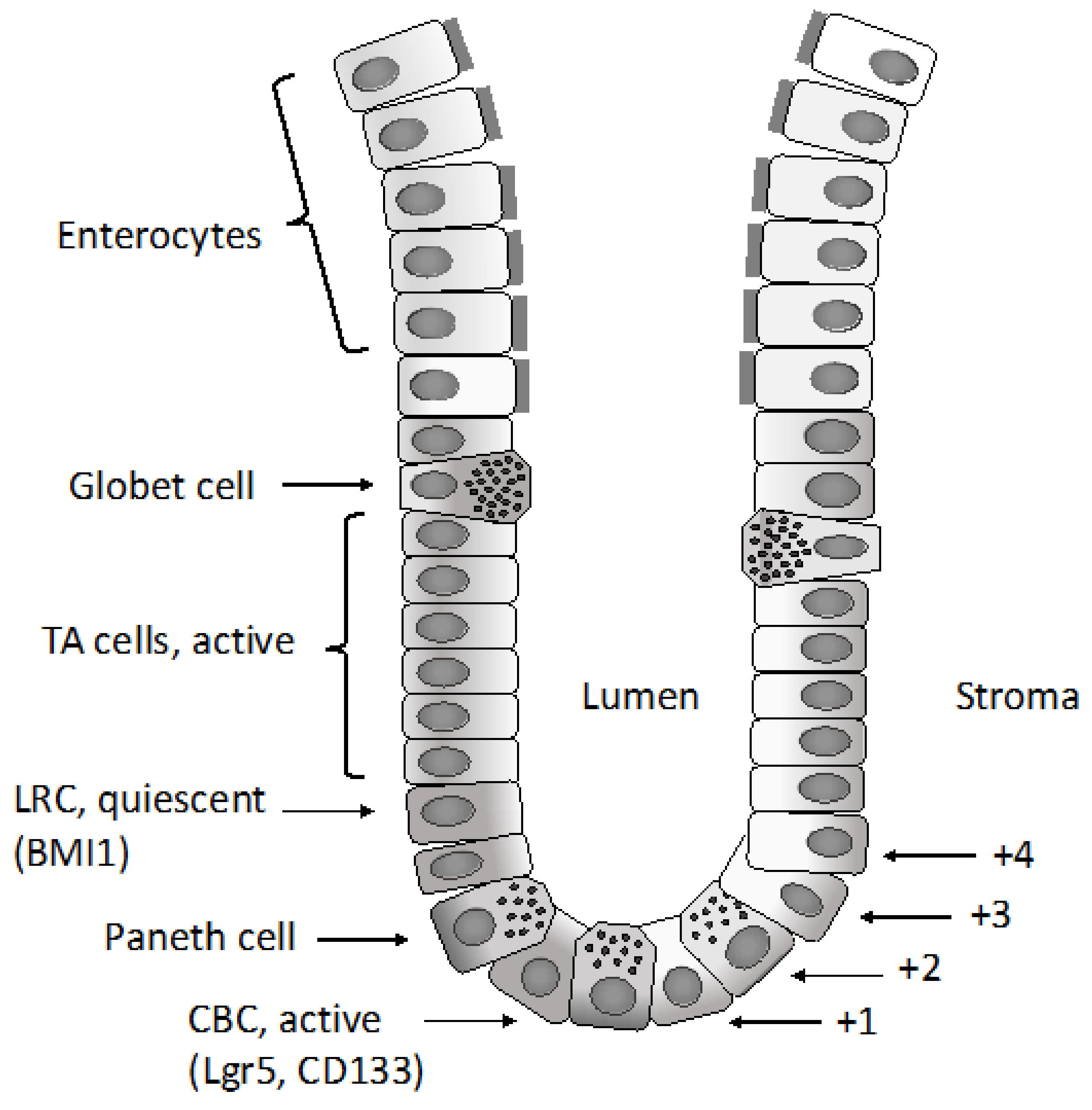{kind=link}
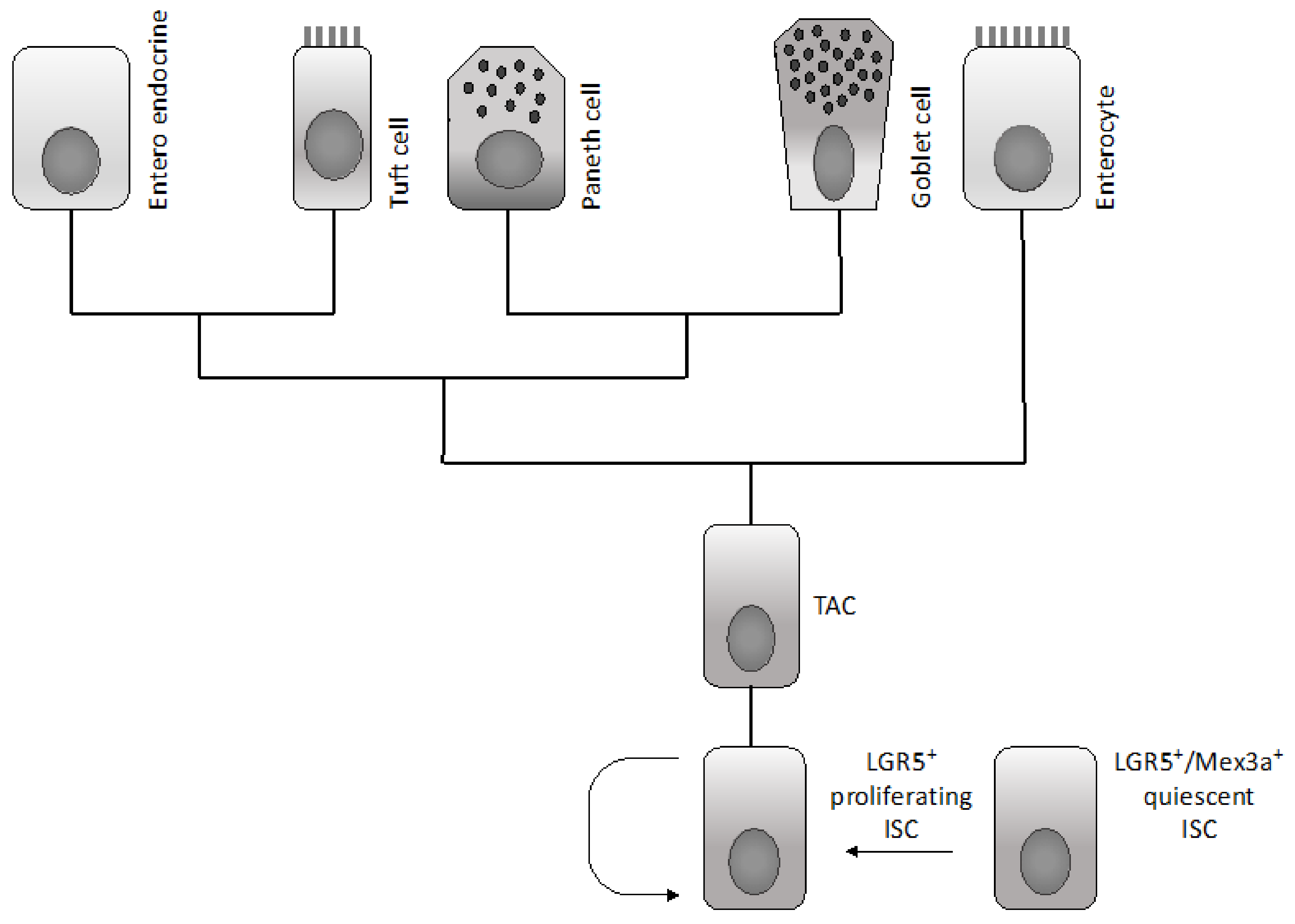{kind=link}
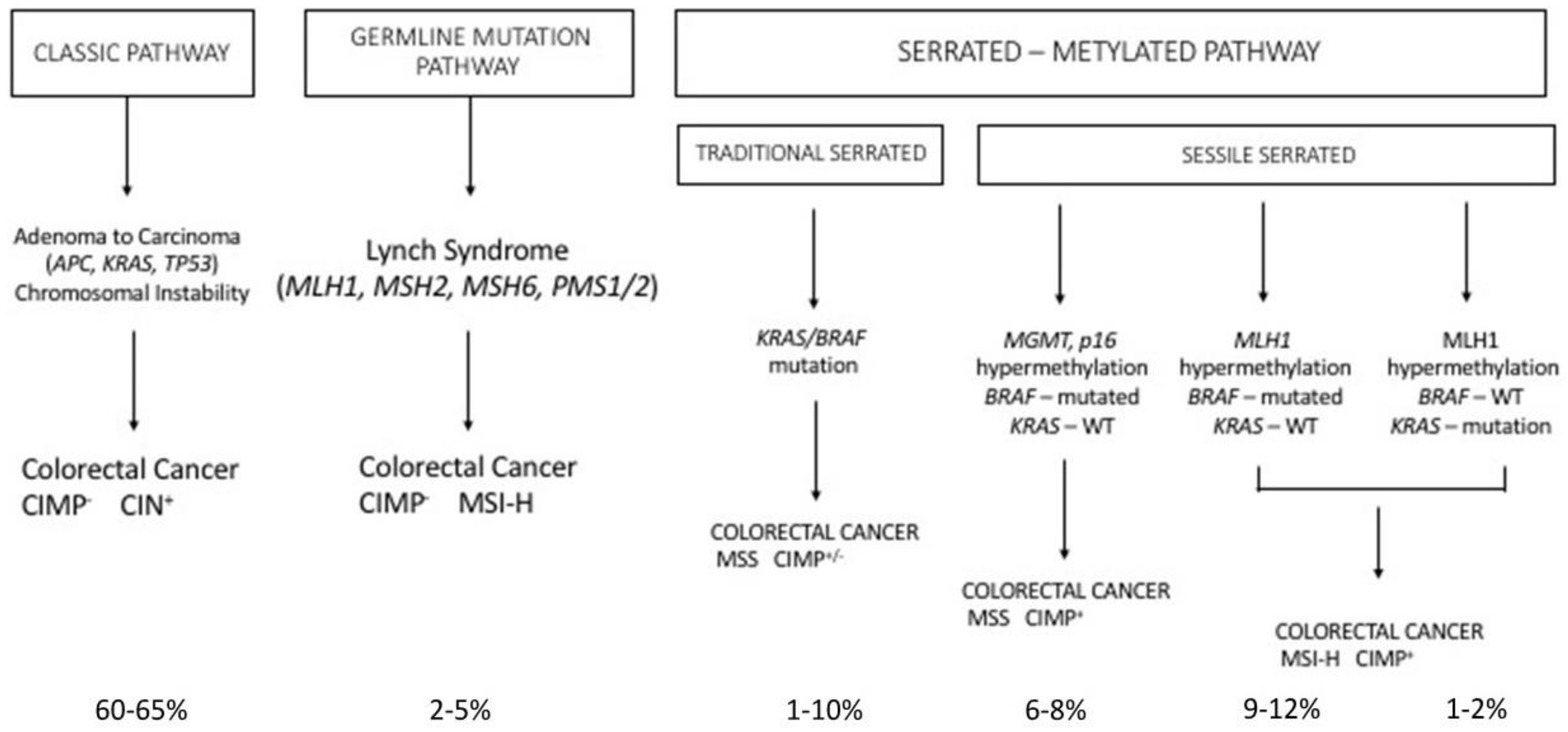{kind=link}
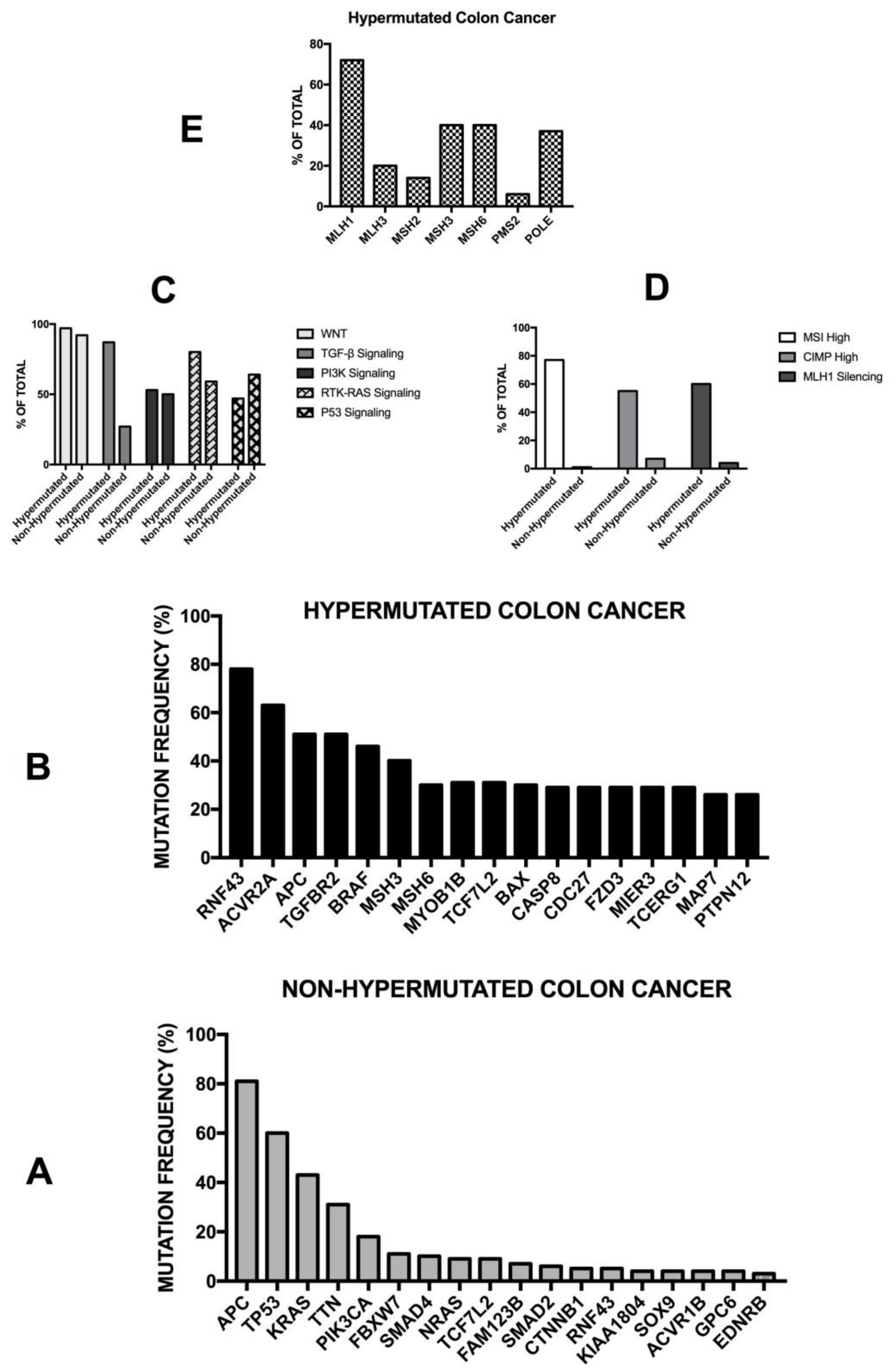{kind=link}
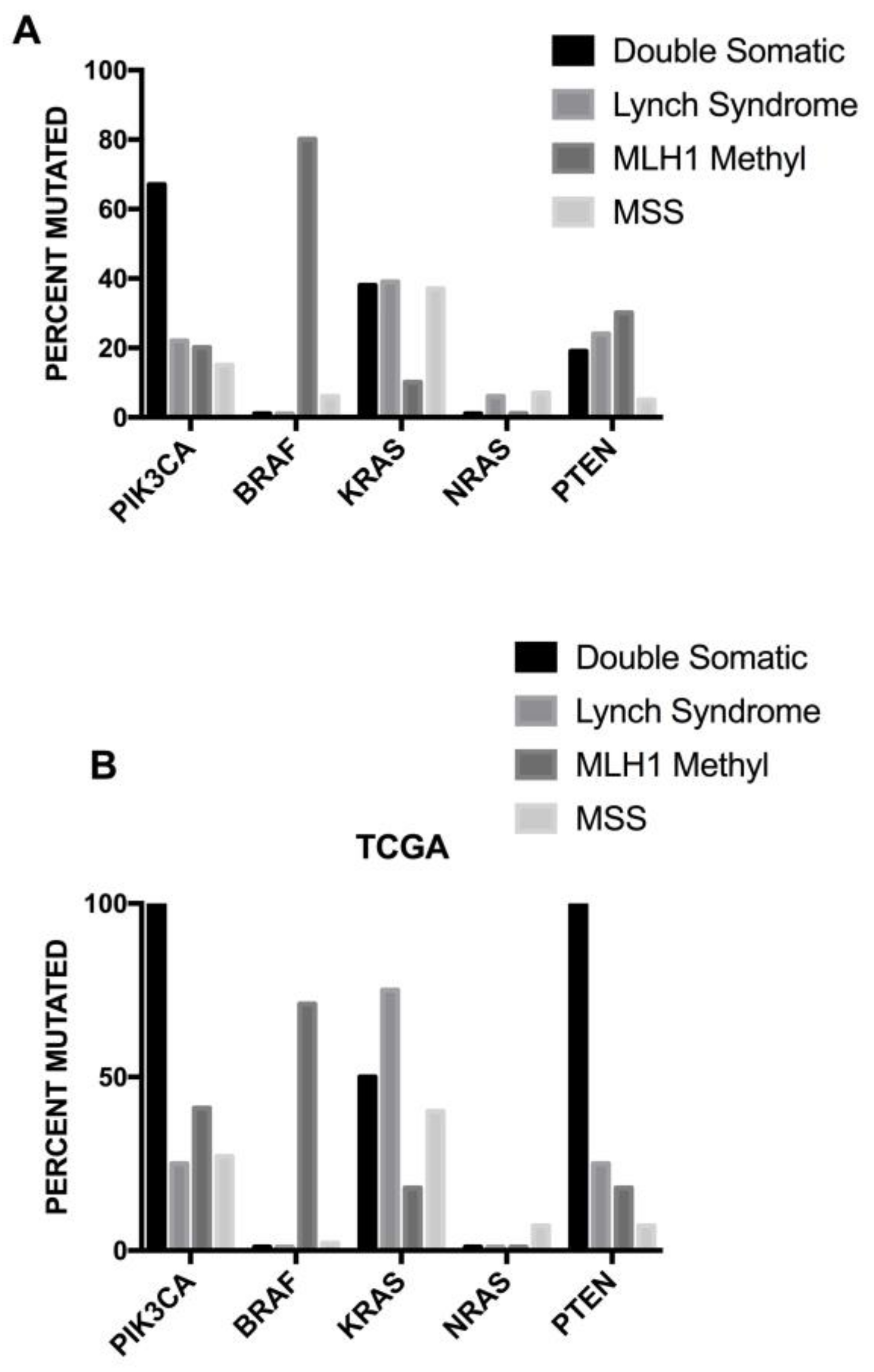{kind=link}
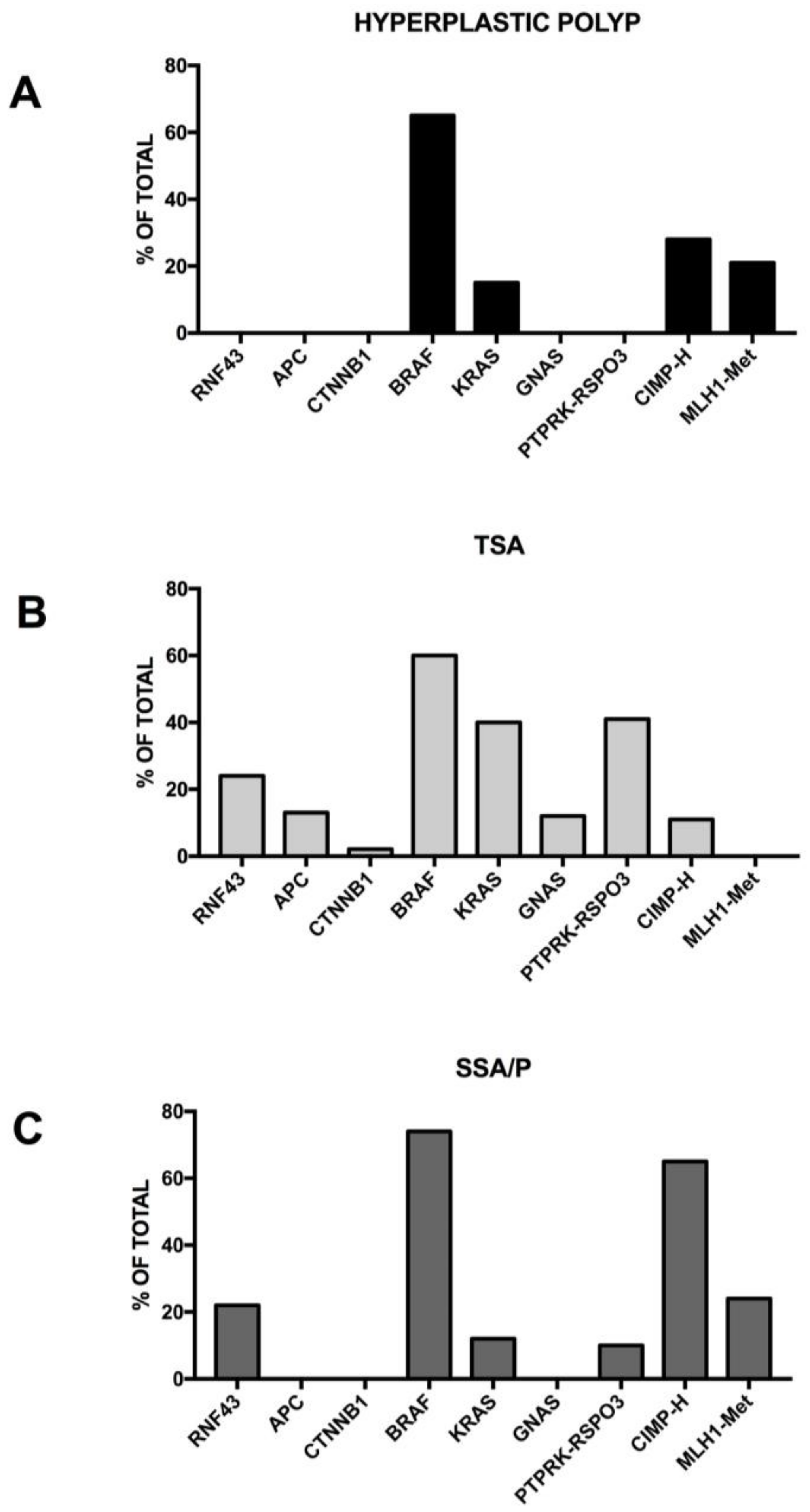{kind=link}
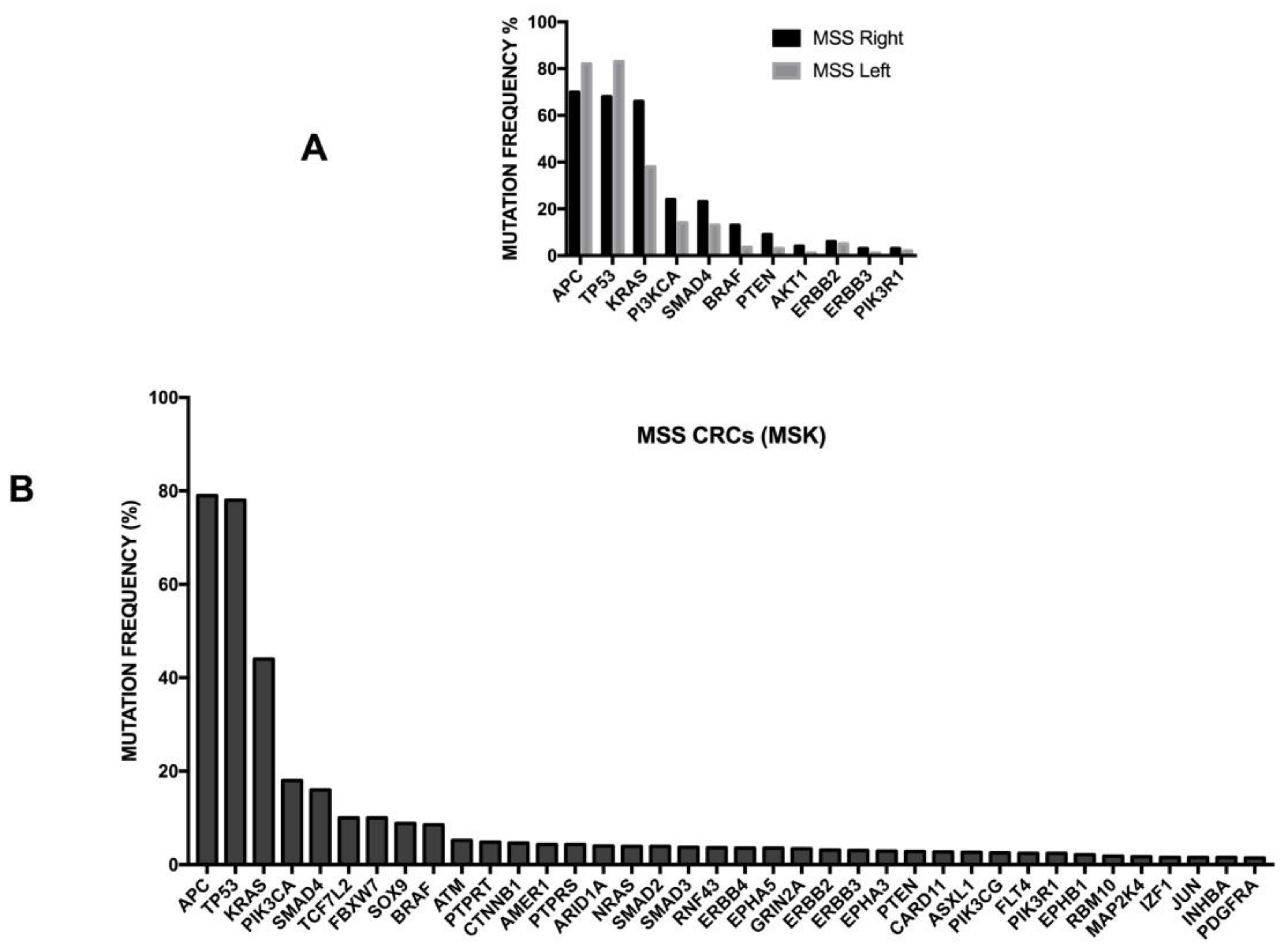{kind=link}
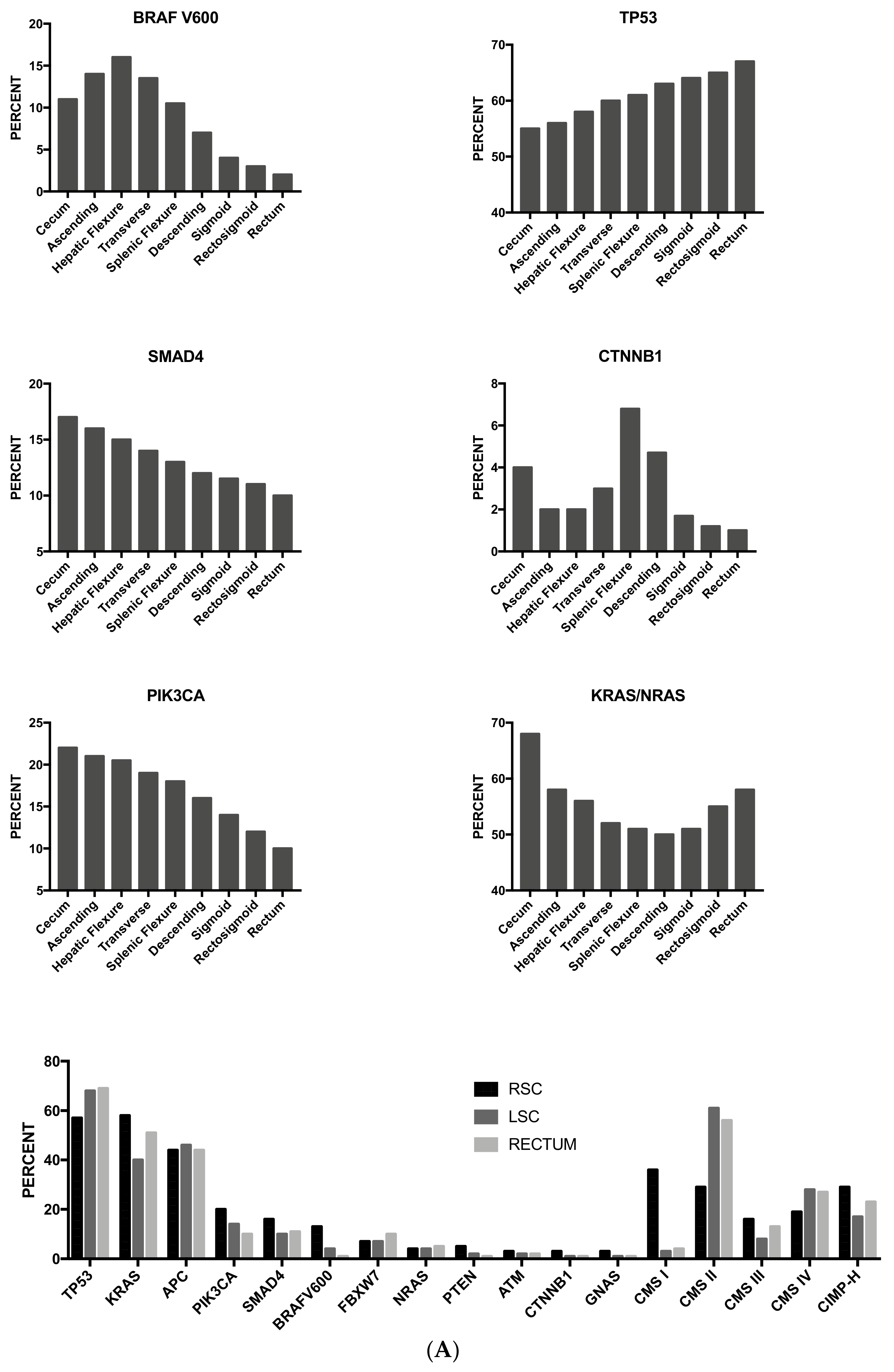{kind=link}
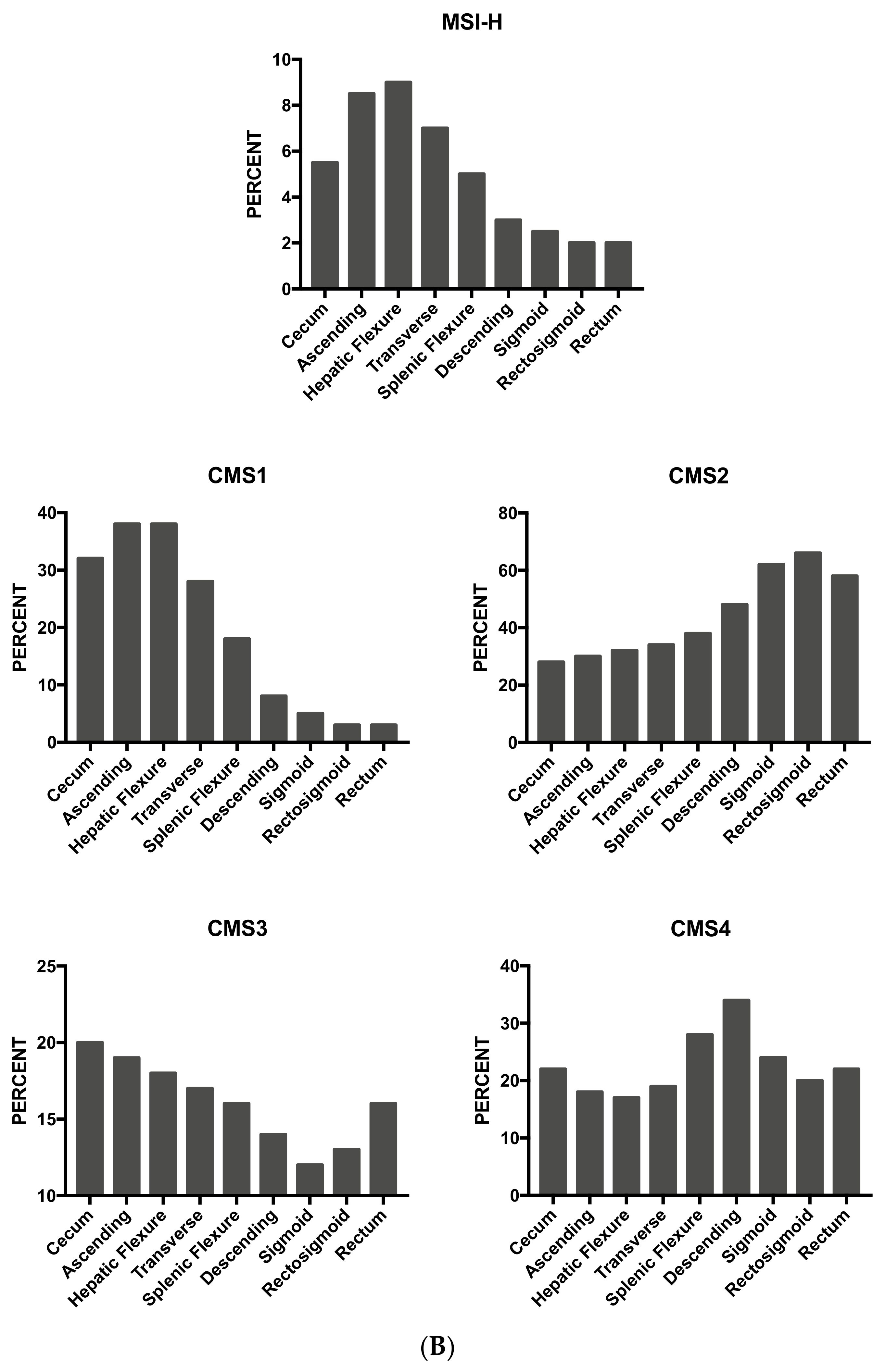{kind=link}
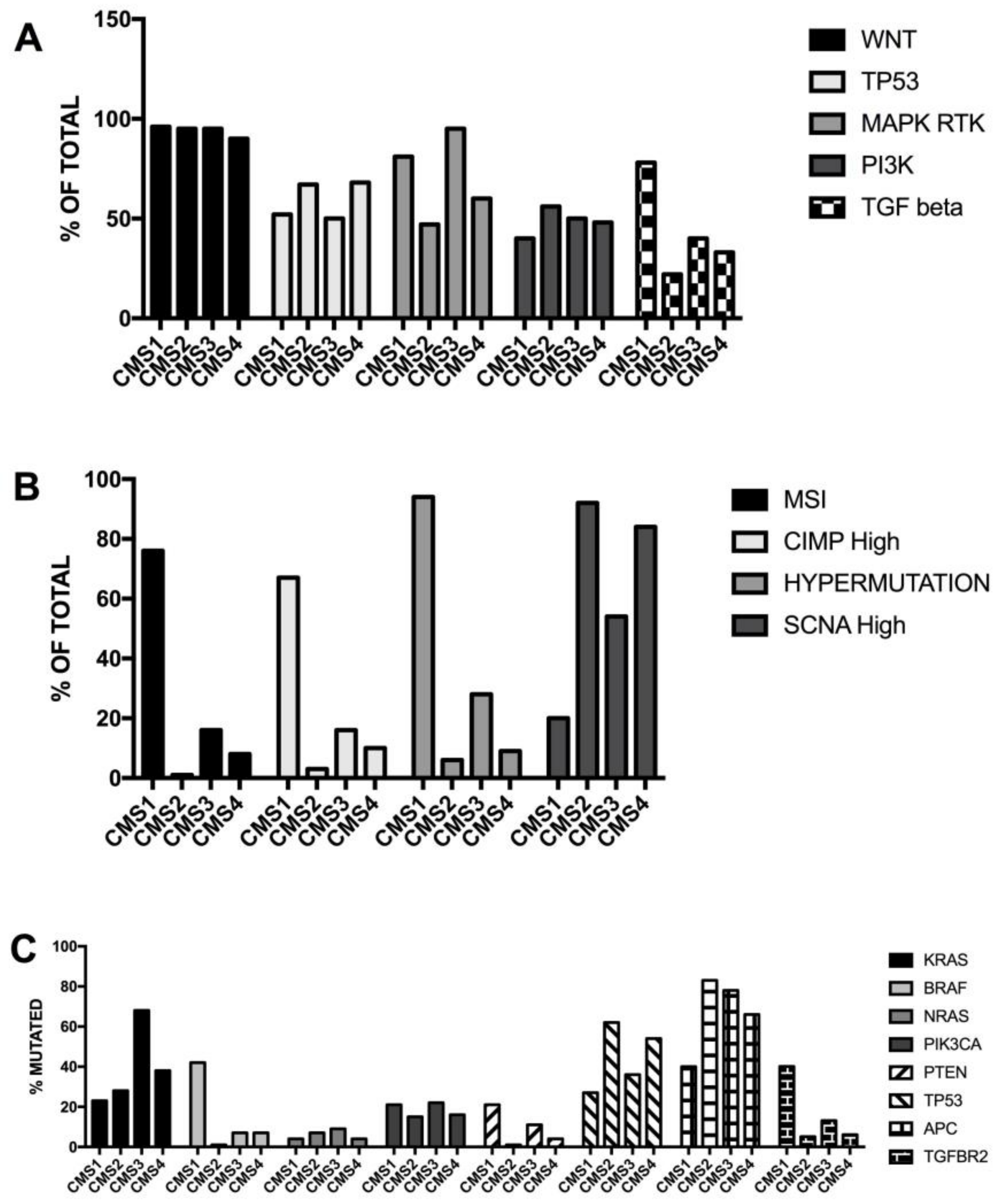{kind=link}
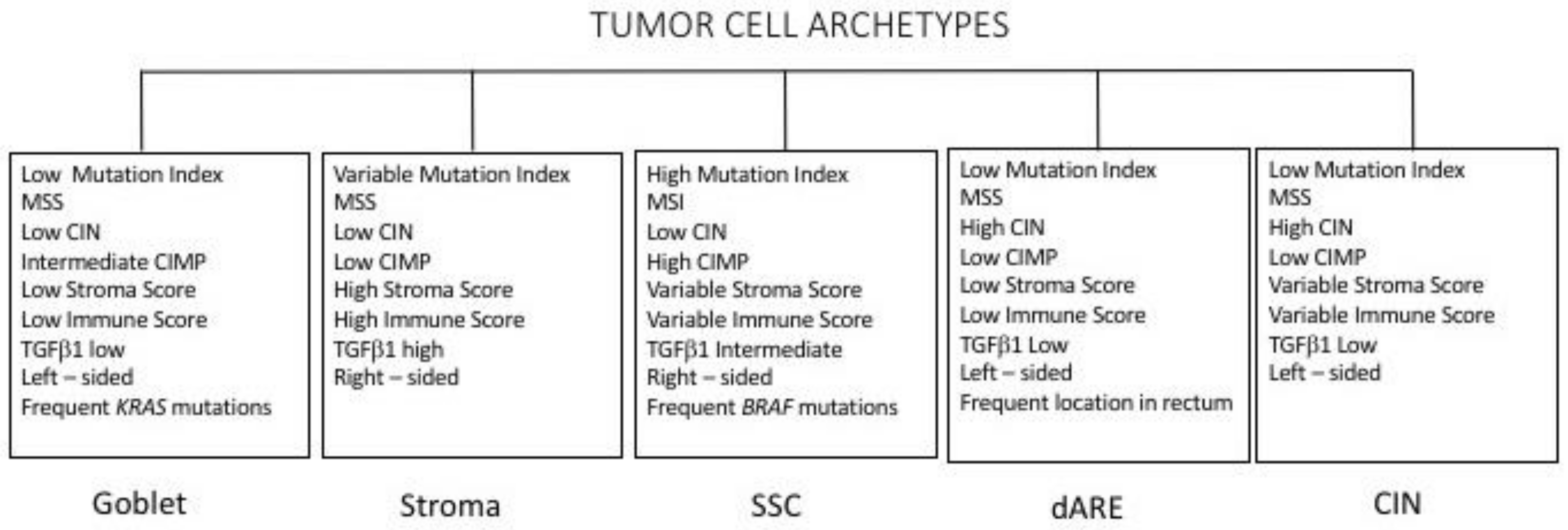{kind=link}
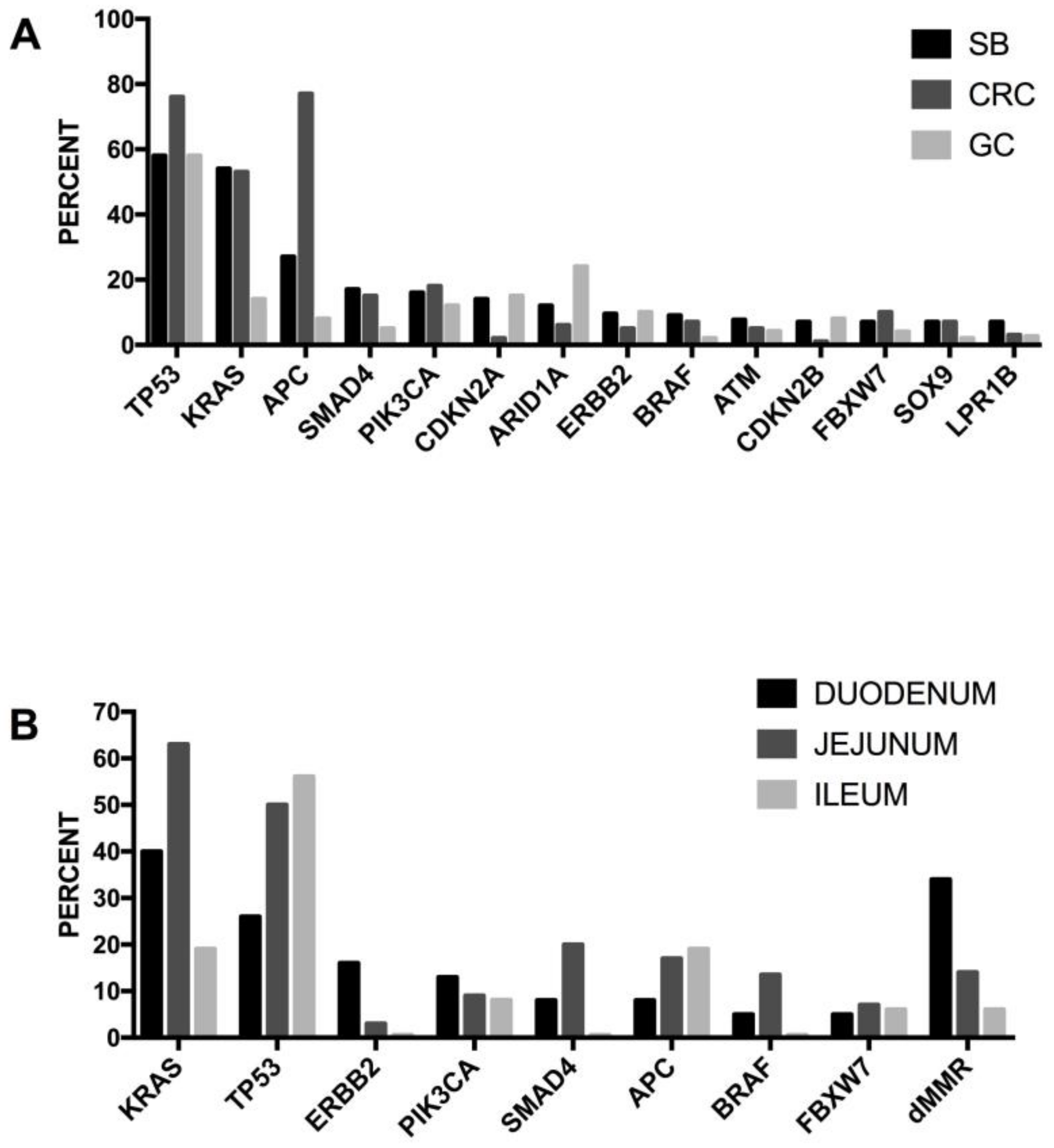{kind=link}
| Syndrome | Inheritance | Genes | Functions | Phenotype | Risk of CRC | Frequency in CRC |
|---|---|---|---|---|---|---|
| Lynch Syndrome | Dominant | MLH-1, MSH-2, MSH-6, PMS-2, EPCAM | DNA mismatch repair. The mutations determine defects in DNA repair and high microsatellite DNA instability | Early-onset colorectal cancer, high penetrance, increased risk of extra-intestinal cancers. No increase in the frequency of adenomatous polyps. MMR-deficient crypt foci are observed | 50–80% | 2–4% |
| Familial Adenomatous Polyposis | Dominant | APC (truncating nonsense or frameshift mutations) | WNT signaling | Phenotype strong: >1000 polyps (mutations 1250–12,654); phenotype mild (mutations in 5′ and 3′). APC loss is an initiating event in adenoma development. Polyp formation is preceded by aberrant foci formation | 100% | <1% |
| MUTYH-Associated Polyposis | Recessive | MUTYH (biallelic germline mutations) | DNA base excision repair protein for repair of mismatches | Mild phenotype with <100 polyps | 43–100% | <1% |
| Proofreading Polymerase-Associated Polyposis | Dominant | POLD1 (polymerase δ1), POLE (polymerase ε). In mutants, proofreading activity is lost, while polymerase activity is maintained. | Proofreading and repair of polymerase errors during DNA replication | <100 intetsinal polyps; early-onset CRC and extracolonic tumors. CRCs, POLE-mutated are mutually exclusive with MMR deficiency and have increased CD8+ lymphocyte infiltration. | High | 1% |
| Familial Juvenile Polyposis | Dominant | SMAD4, BMPR1 | TGF-β signaling | Multiple juvenile polyps found in the colon and stomach. In patients with SMAd4 mutations, association with hereditary hemorrhagic teleangectasia. | 39–68% | |
| Peutz-Jeghers Syndrome | Dominant | STK11 | Multiple signaling pathways | Mucocutaneous melanin hyper-pigmentation, multiple intestinal hamartomatous polyps (masses of connective tissue, covered by intestinal epithelium). | 39% | |
| Hereditary Mixed Polyposis | Dominant | GREM1 | Bone morphogenetic protein antagonist; Transforming growth factor-β signaling | Multiple polyps found in the colon. Adenomas are the most frequent, both tubular and villous. | 20% | |
| Cowden Syndrome or PTEN hamartoma tumor syndrome (PHTS) | Dominant | PTEN (Decreased PTEN protein expression) | Negative regulation of AKT signaling. PTEN loss causes chromosome instability. | Few colon polyps, frequent hamartomatous polyps, presence of intramucosal lipomas. | 9–16% | |
| CHEK2 | Dominant | CHEK2 (CHEK2 1100 delC variant, observed in 4% on non-FAP-related hereditary polyposis) | Negative regulation of cyclin-dependent kinases | CHEK2 is observed in 4–5% of cases of hereditary nonpolyposis colorectal cancer. CHEK2 is a multiorgan cancer susceptibility gene. | Moderate | |
| Serrated Polyposis Syndrome | Dominant | RNF43 (25%) | Negative regulation of the WNT/β-catenin signaling pathway | Few (<5) to >20 serrated polyps occurring proximal to the sigmoid colon. | <50% | <1% |
| NTHL1-Associated Adenomatous Polyposis | Recessive | NTHL1 (DNA glycosylase gene) | Base excision repair | 1–>50 polyps; development of CRCs mismatch repair-proficient | Unknown |
| Tumor Subtype | Proportion | Main Genomic Features | Genetic Drivers | Tumor Location (Proximal (P) or Distal (D)) | Precursor Lesions | Gene Expression Signature | Therapeutic Targeting | Prognosis |
|---|---|---|---|---|---|---|---|---|
| CMS1 Hypermutated | 14% | Hypermutated 95% MSI+ 70% CIMP+ 65% CANs+ 20% Frequent Mutations: BRAF 40% APC 35% TP53 30% KRAS 25% | BRAF | 74% P 26% D | Serrated | Strong immune activation; High PD1 activation; NK cell and TH1 infiltration; Low stromal infiltration | Immune Response (sensitivity to immune check inhibitors); HSP90 | Intermediate (worse prognosis after relapse); these tumors tended to be diagnosed at less advanced stages (I–II) |
| CMS2 Canonical | 40% | Hypermutated 2% MSI+ 2% CIMP+ 4% CNAs+ 96% Frequent Mutations: BRAF 0% APC 80% TP53 70% KRAS 30% | APC | 20% P 80% D | Tubular | High expression of WNT and MYC targets; epithelial differentiation; upregulation of Src and cell cycle pathways; upregulation of the miR 17–92 cluster (MYC target); very low immune infiltration and activation; | EGFR (sensitivity to anti-EGFR MAbs); HER2 | Good (superior survival rates also after relapse, with some long-term survivors) |
| CMS3 Metabolic | 10% | Hypermutated 30% MSI+ 15% CIMP+ 21% CNAs+ 54% Frequent Mutations: BRAF 10% APC 75% TP53 30% KRAS 70% | KRAS | 55% P 45% D | Unknown | upregulation of multiple metabolic signatures (sugar, amino acids, fatty acids, nitrogen); epithelial differentiation; low immune infiltration and activation; very low stromal infiltration; | Intermediate | |
| CMS4 Mesenchymal | 25% | Hypermutated 2% MSI+ 3% CIMP+ 9% CNAs+ 86% Frequent Mutations: BRAF 5% APC 65% TP53 55% KRAS 40% | miR-200 (downregulation) TGF-β pathway | 34% P 66% D | Serrated | upregulation of genes implicated in epithelial-to-mesenchymal transition and of signatures associated with the activation of TGF-β signaling, angiogenesis matrix remodeling pathways and the complement-mediated inflammatory system. High stromal infiltration. High VEGF/VEGFR and integrin β3 pathways. | PDGFRA; KIT; HSP90 | Poor (worse overall survival and relapse- free survival); these tumors tended to be diagnosed at more advanced stages (III–IV) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Testa, U.; Pelosi, E.; Castelli, G. Colorectal Cancer: Genetic Abnormalities, Tumor Progression, Tumor Heterogeneity, Clonal Evolution and Tumor-Initiating Cells. Med. Sci. 2018, 6, 31. https://doi.org/10.3390/medsci6020031
Testa U, Pelosi E, Castelli G. Colorectal Cancer: Genetic Abnormalities, Tumor Progression, Tumor Heterogeneity, Clonal Evolution and Tumor-Initiating Cells. Medical Sciences. 2018; 6(2):31. https://doi.org/10.3390/medsci6020031
Chicago/Turabian StyleTesta, Ugo, Elvira Pelosi, and Germana Castelli. 2018. "Colorectal Cancer: Genetic Abnormalities, Tumor Progression, Tumor Heterogeneity, Clonal Evolution and Tumor-Initiating Cells" Medical Sciences 6, no. 2: 31. https://doi.org/10.3390/medsci6020031
APA StyleTesta, U., Pelosi, E., & Castelli, G. (2018). Colorectal Cancer: Genetic Abnormalities, Tumor Progression, Tumor Heterogeneity, Clonal Evolution and Tumor-Initiating Cells. Medical Sciences, 6(2), 31. https://doi.org/10.3390/medsci6020031