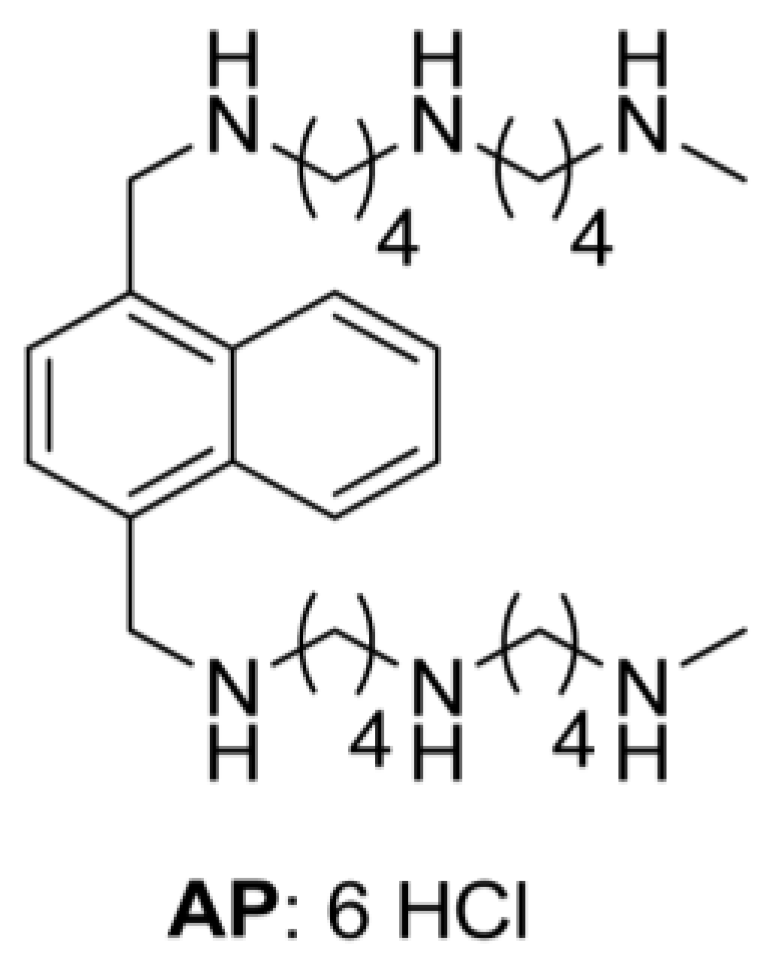
A Novel Polyamine-Targeted Therapy for BRAF Mutant Melanoma Tumors



Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. 3D Spheroid Culture
2.3. Cell Viability Assay
2.4. Radiolabeled Spermidine Transport Assays
2.5. Statistical Analysis
3. Results
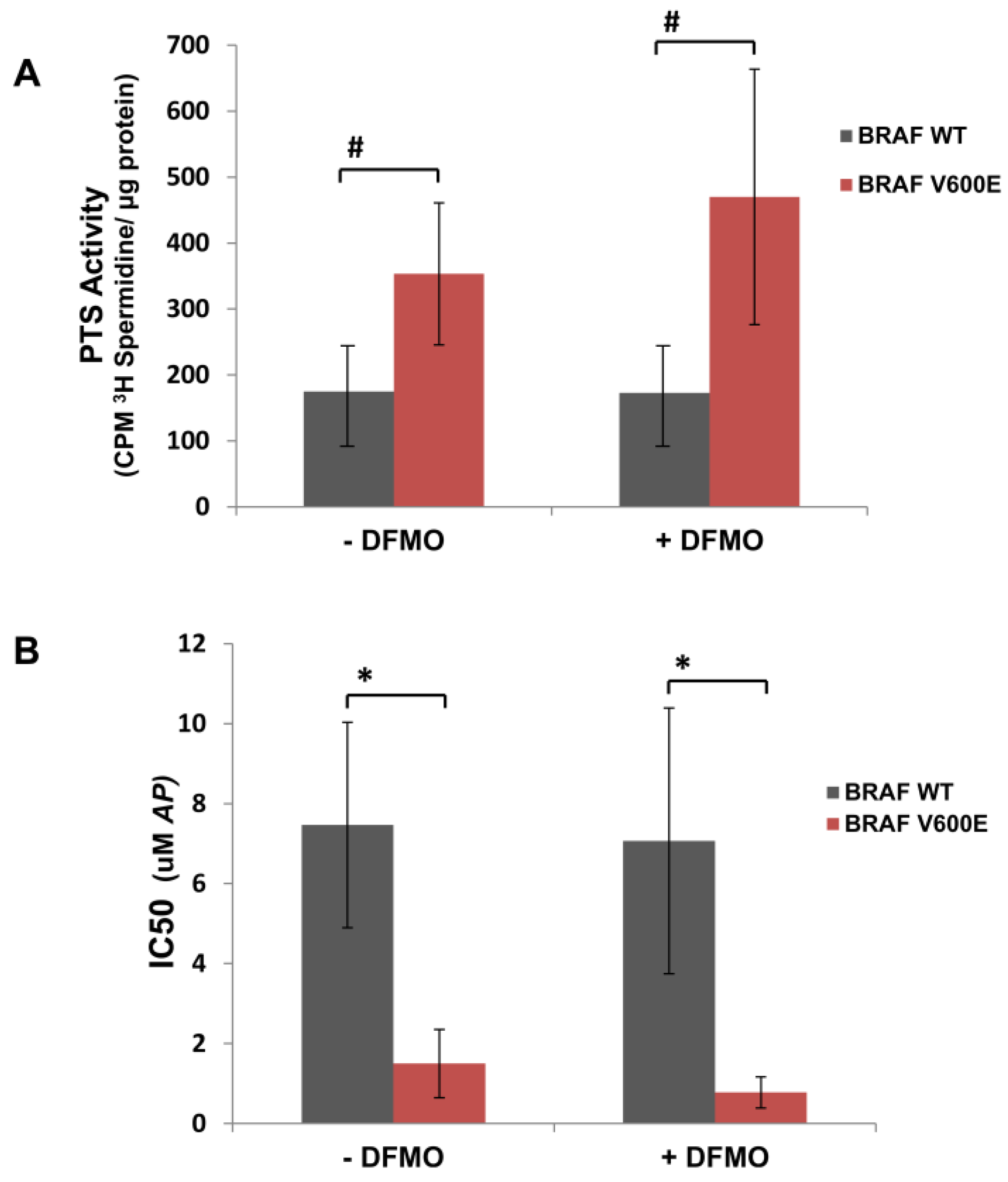
3.1. Human Mutant BRAFV600E Melanoma Cells Are More Sensitive to Cytotoxic Effects of AP Than BRAFWT Melanoma Cells
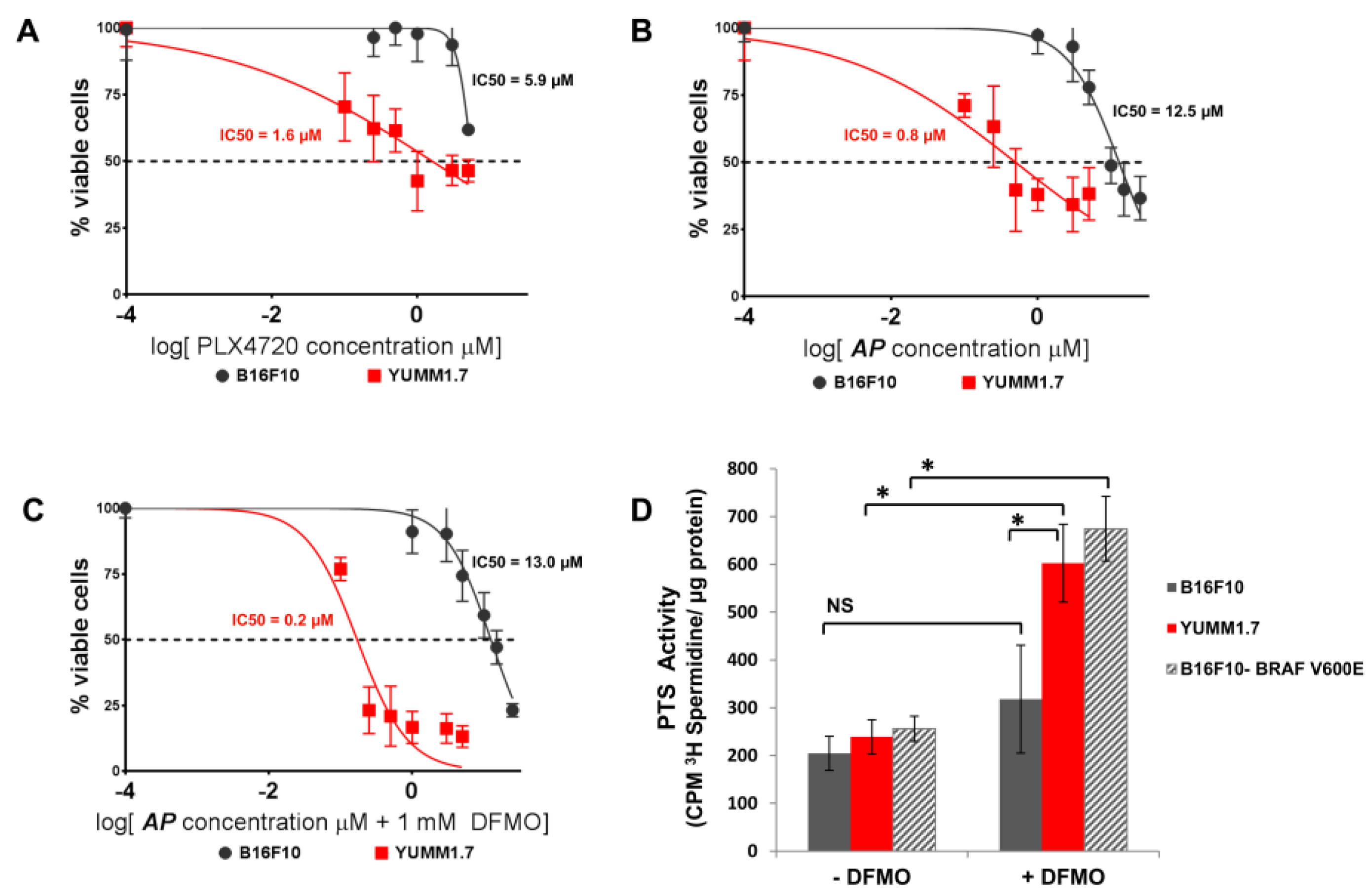
3.2. AP Is More Cytotoxic to BRAFV600E Murine Melanoma Cells Than BRAFWT Melanoma Cells
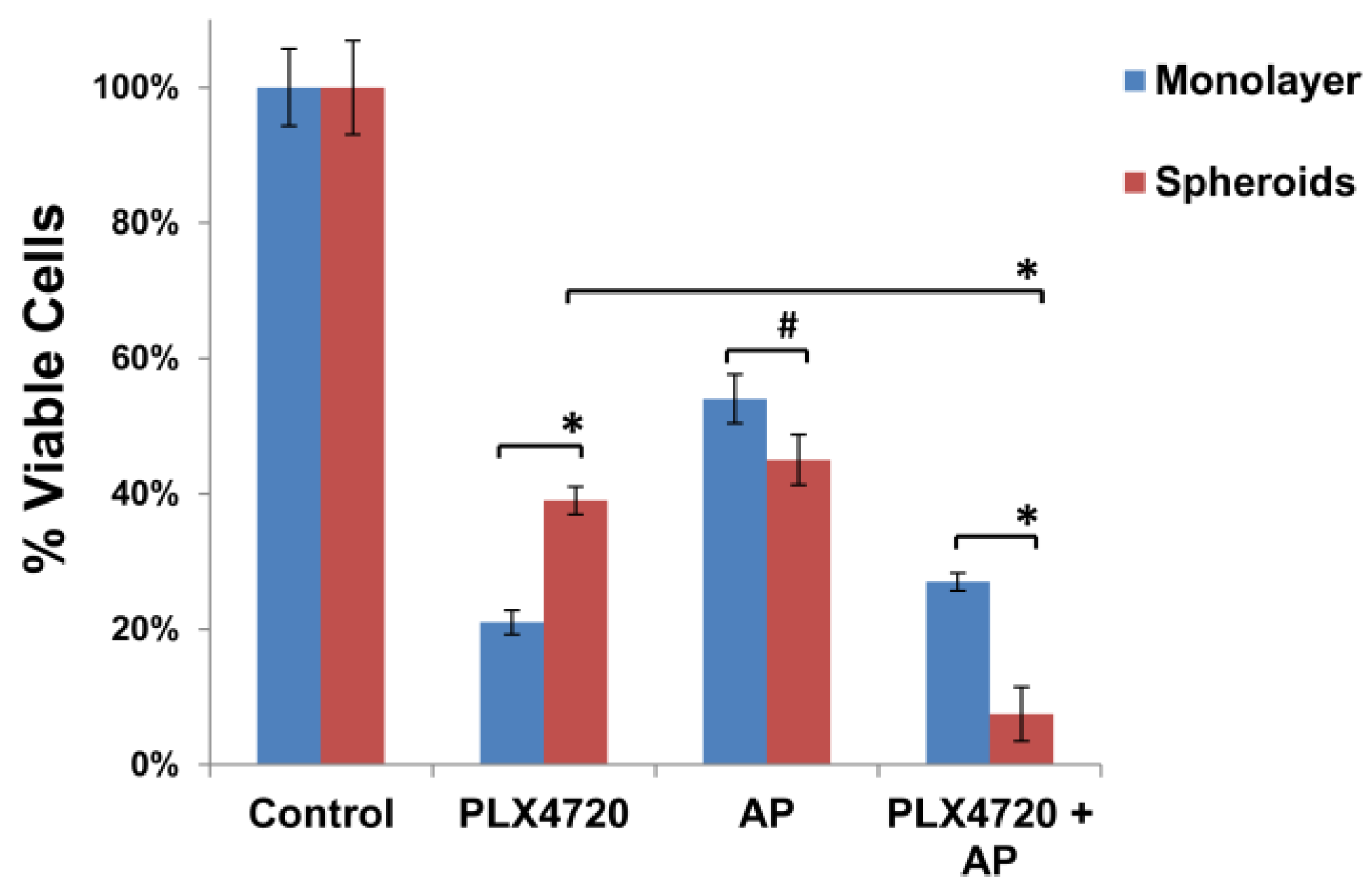
3.3. Increased Resistance of Spheroid Melanoma Cells to PLX4720 Is Overcome with AP Co-Treatment
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Solit, D.B.; Rosen, N. Resistance to BRAF inhibition in melanomas. N. Engl. J. Med. 2011, 364, 772–774. [Google Scholar] [CrossRef] [PubMed]
- Haq, R.; Fisher, D.E. Targeting melanoma by small molecules: Challenges ahead. Pigment Cell Melanoma Res. 2013, 26, 464–469. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H., Jr.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Bello-Fernandez, C.; Packham, G.; Cleveland, J.L. The ornithine decarboxylase gene is a transcriptional target of c-Myc. Proc. Natl. Acad. Sci. USA 1993, 90, 7804–7808. [Google Scholar] [CrossRef]
- Casero, R.A., Jr.; Marton, L.J. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat. Rev. Drug Discov. 2007, 6, 373–390. [Google Scholar] [CrossRef]
- Forshell, T.P.; Rimpi, S.; Nilsson, J.A. Chemoprevention of B-cell lymphomas by inhibition of the Myc target spermidine synthase. Cancer Prev. Res. 2010, 3, 140–147. [Google Scholar] [CrossRef]
- Origanti, S.; Shantz, L.M. Ras transformation of RIE-1 cells activates cap-independent translation of ornithine decarboxylase: Regulation by the Raf/MEK/ERK and phosphatidylinositol 3-kinase pathways. Cancer Res. 2007, 67, 4834–4842. [Google Scholar] [CrossRef]
- Poulin, R.; Casero, R.A.; Soulet, D. Recent advances in the molecular biology of metazoan polyamine transport. Amino Acids 2012, 42, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Bachrach, U.; Seiler, N. Formation of acetylpolyamines and putrescine from spermidine by normal and transformed chick embryo fibroblasts. Cancer Res. 1981, 41, 1205–1208. [Google Scholar] [PubMed]
- Chang, B.K.; Libby, P.R.; Bergeron, R.J.; Porter, C.W. Modulation of polyamine biosynthesis and transport by oncogene transfection. Biochem. Biophys. Res. Commun. 1988, 157, 264–270. [Google Scholar] [CrossRef]
- Roy, U.K.; Rial, N.S.; Kachel, K.L.; Gerner, E.W. Activated K-RAS increases polyamine uptake in human colon cancer cells through modulation of caveolar endocytosis. Mol. Carcinog. 2008, 47, 538–553. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A.E. Polyamine metabolism and its importance in neoplastic growth as a target for chemotherapy. Cancer Res. 1988, 48, 759–774. [Google Scholar] [PubMed]
- Tabor, C.W.; Tabor, H. Polyamines. Ann. Rev. Biochem. 1984, 53, 749–790. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A.E. Recent advances in the biochemistry of polyamines in eukaryotes. Biochem. J. 1986, 234, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Gerner, E.W.; Meyskens, F.L., Jr. Polyamines and cancer: Old molecules, new understanding. Nat. Rev. Cancer 2004, 4, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Wallace, H.M.; Fraser, A.V.; Hughes, A. A perspective of polyamine metabolism. Biochem. J. 2003, 376, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Seiler, N.; Delcros, J.G.; Moulinoux, J.P. Polyamine transport in mammalian cells. An update. Int. J. Biochem. Cell Biol. 1996, 28, 843–861. [Google Scholar] [CrossRef]
- Muth, A.; Kamel, J.; Kaur, N.; Shicora, A.C.; Ayene, I.S.; Gilmour, S.K.; Phanstiel, O. Development of polyamine transport ligands with improved metabolic stability and selectivity against specific human cancers. J. Med. Chem. 2013, 56, 5819–5828. [Google Scholar] [CrossRef] [PubMed]
- Phanstiel, O.; Kaur, N.; Delcros, J.G. Structure-activity investigations of polyamine-anthracene conjugates and their uptake via the polyamine transporter. Amino Acids 2007, 33, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Kramer, D.L.; Miller, J.T.; Bergeron, R.J.; Khomutov, R.; Khomutov, A.; Porter, C.W. Regulation of polyamine transport by polyamines and polyamine analogs. J. Cell. Physiol. 1993, 155, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, J.A.; Keller, U.B.; Baudino, T.A.; Yang, C.; Norton, S.; Old, J.A.; Nilsson, L.M.; Neale, G.; Kramer, D.L.; Porter, C.W.; et al. Targeting ornithine decarboxylase in Myc-induced lymphomagenesis prevents tumor formation. Cancer Cell 2005, 7, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Schayowitz, A.; Bertenshaw, G.; Jeffries, E.; Schatz, T.; Cotton, J.; Villanueva, J.; Herlyn, M.; Krepler, C.; Vultur, A.; Xu, W. Functional profiling of live melanoma samples using a novel automated platform. PLoS ONE 2012, 7, e52760. [Google Scholar] [CrossRef] [PubMed]
- Alhonen-Hongisto, L.; Seppanen, P.; Janne, J. Intracellular putrescine and spermidine deprivation induces increased uptake of the natural polyamines and methylglyoxal bis(guanylhydrazone). Biochem. J. 1980, 192, 941–945. [Google Scholar] [CrossRef] [PubMed]
- Obenauf, A.C.; Zou, Y.; Ji, A.L.; Vanharanta, S.; Shu, W.; Shi, H.; Kong, X.; Bosenberg, M.C.; Wiesner, T.; Rosen, N.; et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature 2015, 520, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, Y.; Waki, A.; Yoshida, K.; Kakezuka, A.; Kobayashi, M.; Namiki, H.; Kuroda, Y.; Kiyono, Y.; Yoshii, H.; Furukawa, T. The use of nanoimprinted scaffolds as 3D culture models to facilitate spontaneous tumor cell migration and well-regulated spheroid formation. Biomaterials 2011, 32, 6052–6058. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.M.; Cukierman, E. Modeling tissue morphogenesis and cancer in 3D. Cell 2007, 130, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Haycock, J.W. 3D cell culture: A review of current approaches and techniques. In 3D Cell Culture: Methods and Protocols; Springer: Berlin, Germany, 2011; pp. 1–15. [Google Scholar]
- Qin, Y.; Roszik, J.; Chattopadhyay, C.; Hashimoto, Y.; Liu, C.; Cooper, Z.A.; Wargo, J.A.; Hwu, P.; Ekmekcioglu, S.; Grimm, E.A. Hypoxia-driven mechanism of vemurafenib resistance in melanoma. Mol. Cancer Ther. 2016, 15, 2442–2454. [Google Scholar] [CrossRef] [PubMed]
- Cufi, S.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Martin-Castillo, B.; Vellon, L.; Menendez, J.A. Autophagy positively regulates the CD44+ CD24-/low breast cancer stem-like phenotype. Cell Cycle 2011, 10, 3871–3885. [Google Scholar] [CrossRef] [PubMed]
- Mirzoeva, O.K.; Hann, B.; Hom, Y.K.; Debnath, J.; Aftab, D.; Shokat, K.; Korn, W.M. Autophagy suppression promotes apoptotic cell death in response to inhibition of the PI3K—mTOR pathway in pancreatic adenocarcinoma. J. Mol. Med. 2011, 89, 877–889. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Galluzzi, L.; Kepp, O.; Marino, G.; Michaud, M.; Vitale, I.; Maiuri, M.C.; Kroemer, G. Oncosuppressive functions of autophagy. Antioxid. Redox Signal. 2011, 14, 2251–2269. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Marino, G.; Bennetzen, M.V.; Eisenberg, T.; Megalou, E.; Schroeder, S.; Cabrera, S.; Benit, P.; Rustin, P.; Criollo, A.; et al. Spermidine and resveratrol induce autophagy by distinct pathways converging on the acetylproteome. J. Cell Biol. 2011, 192, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Dallavalle, S.; Giannini, G.; Alloatti, D.; Casati, A.; Marastoni, E.; Musso, L.; Merlini, L.; Morini, G.; Penco, S.; Pisano, C.; et al. Synthesis and cytotoxic activity of polyamine analogues of camptothecin. J. Med. Chem. 2006, 49, 5177–5186. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Davis, A.; Yu, S.; Ahmed, K. Response of cancer cells to molecular interruption of the CK2 signal. Mol. Cell Biochem. 2001, 227, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Svensson, K.J.; Welch, J.E.; Kucharzewska, P.; Bengtson, P.; Bjurberg, M.; Pahlman, S.; Ten Dam, G.B.; Persson, L.; Belting, M. Hypoxia-mediated induction of the polyamine system provides opportunities for tumor growth inhibition by combined targeting of vascular endothelial growth factor and ornithine decarboxylase. Cancer Res. 2008, 68, 9291–9301. [Google Scholar] [CrossRef] [PubMed]
- Mozdzan, M.; Szemraj, J.; Rysz, J.; Stolarek, R.A.; Nowak, D. Anti-oxidant activity of spermine and spermidine re-evaluated with oxidizing systems involving iron and copper ions. Int. J. Biochem. Cell Biol. 2006, 38, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Pouyssegur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Keith, B.; Simon, M.C. Hypoxia-inducible factors, stem cells, and cancer. Cell 2007, 129, 465–472. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, M.P.; Marchbank, K.; Webster, M.R.; Valiga, A.A.; Kaur, A.; Vultur, A.; Li, L.; Herlyn, M.; Villanueva, J.; Liu, Q. Hypoxia induces phenotypic plasticity and therapy resistance in melanoma via the tyrosine kinase receptors ROR1 and ROR2. Cancer Discov. 2013, 3, 1378–1393. [Google Scholar] [CrossRef] [PubMed]
- Pucciarelli, D.; Lengger, N.; Takáčová, M.; Csaderova, L.; Bartosova, M.; Breiteneder, H.; Pastorekova, S.; Hafner, C. Hypoxia increases the heterogeneity of melanoma cell populations and affects the response to vemurafenib. Mol. Med. Rep. 2016, 13, 3281–3288. [Google Scholar] [CrossRef] [PubMed]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Roesch, A.; Vultur, A.; Bogeski, I.; Wang, H.; Zimmermann, K.M.; Speicher, D.; Korbel, C.; Laschke, M.W.; Gimotty, P.A.; Philipp, S.E.; et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1Bhigh cells. Cancer Cell 2013, 23, 811–825. [Google Scholar] [CrossRef] [PubMed]
- Schwab, L.P.; Peacock, D.L.; Majumdar, D.; Ingels, J.F.; Jensen, L.C.; Smith, K.D.; Cushing, R.C.; Seagroves, T.N. Hypoxia inducible factor-1α promotes primary tumor growth and tumor-initiating cell activity in breast cancer. Breast Cancer Res. 2012, 14, R6. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, J.; Zhang, Z.; Zhou, W.; Wang, A.J.; Heddleston, J.M.; Pinna, C.M.; Hubaud, A.; Stadler, B.; Choi, M.; Bar, M. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res. 2011, 71, 4640–4652. [Google Scholar] [CrossRef] [PubMed]
- Mohyeldin, A.; Garzon-Muvdi, T.; Quinones-Hinojosa, A. Oxygen in stem cell biology: A critical component of the stem cell niche. Cell Stem Cell 2010, 7, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Buttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 2009, 11, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Shen, L.; Yu, J.; Wan, H.; Guo, A.; Chen, J.; Long, Y.; Zhao, J.; Pei, G. Rapamycin and other longevity-promoting compounds enhance the generation of mouse induced pluripotent stem cells. Aging Cell 2011, 10, 908–911. [Google Scholar] [CrossRef] [PubMed]
- Strohecker, A.M.; White, E. Targeting mitochondrial metabolism by inhibiting autophagy in BRAF-driven cancers. Cancer Discov. 2014, 4, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.H.; Piao, S.F.; Dey, S.; McAfee, Q.; Karakousis, G.; Villanueva, J.; Hart, L.S.; Levi, S.; Hu, J.; Zhang, G.; et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J. Clin. Investig. 2014, 124, 1406–1417. [Google Scholar] [CrossRef] [PubMed]
- Seiler, N. Thirty years of polyamine-related approaches to cancer therapy. Retrospect and prospect. Part 1. Selective enzyme inhibitors. Curr. Drug Targets 2003, 4, 537–564. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| − DFMO | + DFMO | ||||
|---|---|---|---|---|---|
| Cell Line | BRAF Mutational Status | IC50 (µM AP) | PTS Activity b (cpm 3H Spd/µg Protein) | IC50 (µM AP) | PTS Activity b (cpm 3H Spd/µg Protein) |
| WM983B | V600E | 2.6 | 487 ± 64 | 0.9 | 706 ± 78 * |
| WM3734 | V600E | 2.2 | 444 ± 46 | 1.2 | 654 ± 59 * |
| 1205Lu | V600E | 0.7 | 230 ± 22 | 0.6 | 299 ± 54 |
| WM989 | V600E | 1.2 | 304 ± 28 | 1.0 | 348 ± 48 |
| WM88 | V600E | 0.8 | 302 ± 18 | 0.2 | 343 ± 59 |
| WM3451 | WT | 9.0 | 130 ± 21 | 5.1 | 157 ± 34 |
| WM3743 | WT | 8.9 | 117 ± 23 | 10.9 | 110 ± 25 |
| WM3211 | WT | 4.5 | 278 ± 58 | 5.2 | 251 ± 46 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peters, M.C.; Minton, A.; Phanstiel IV, O.; Gilmour, S.K. A Novel Polyamine-Targeted Therapy for BRAF Mutant Melanoma Tumors. Med. Sci. 2018, 6, 3. https://doi.org/10.3390/medsci6010003
Peters MC, Minton A, Phanstiel IV O, Gilmour SK. A Novel Polyamine-Targeted Therapy for BRAF Mutant Melanoma Tumors. Medical Sciences. 2018; 6(1):3. https://doi.org/10.3390/medsci6010003
Chicago/Turabian StylePeters, Molly C., Allyson Minton, Otto Phanstiel IV, and Susan K. Gilmour. 2018. "A Novel Polyamine-Targeted Therapy for BRAF Mutant Melanoma Tumors" Medical Sciences 6, no. 1: 3. https://doi.org/10.3390/medsci6010003
APA StylePeters, M. C., Minton, A., Phanstiel IV, O., & Gilmour, S. K. (2018). A Novel Polyamine-Targeted Therapy for BRAF Mutant Melanoma Tumors. Medical Sciences, 6(1), 3. https://doi.org/10.3390/medsci6010003