Application of Massively Parallel Sequencing in the Clinical Diagnostic Testing of Inherited Cardiac Conditions

Abstract
:1. Introduction
2. Massively Parallel Sequencing
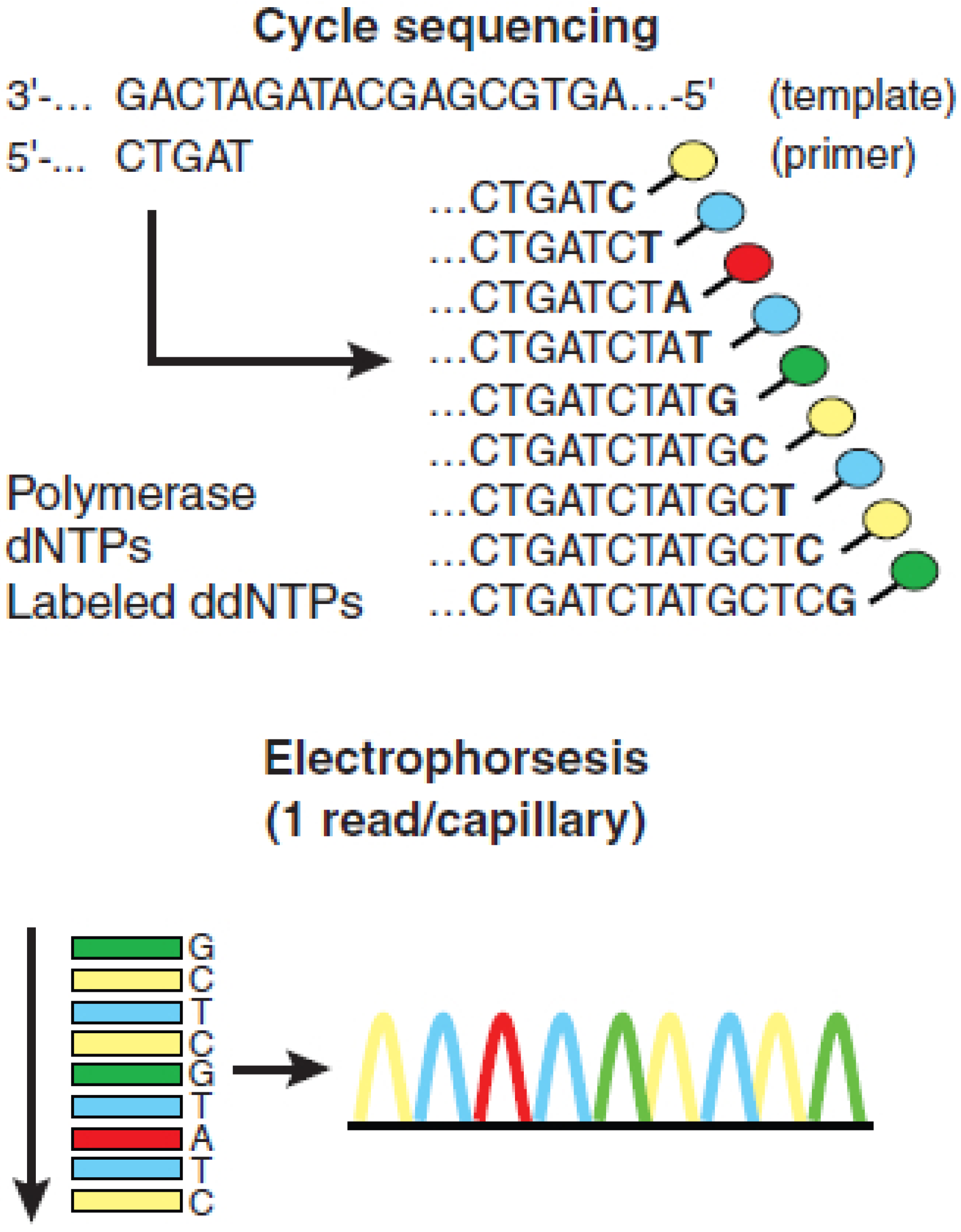
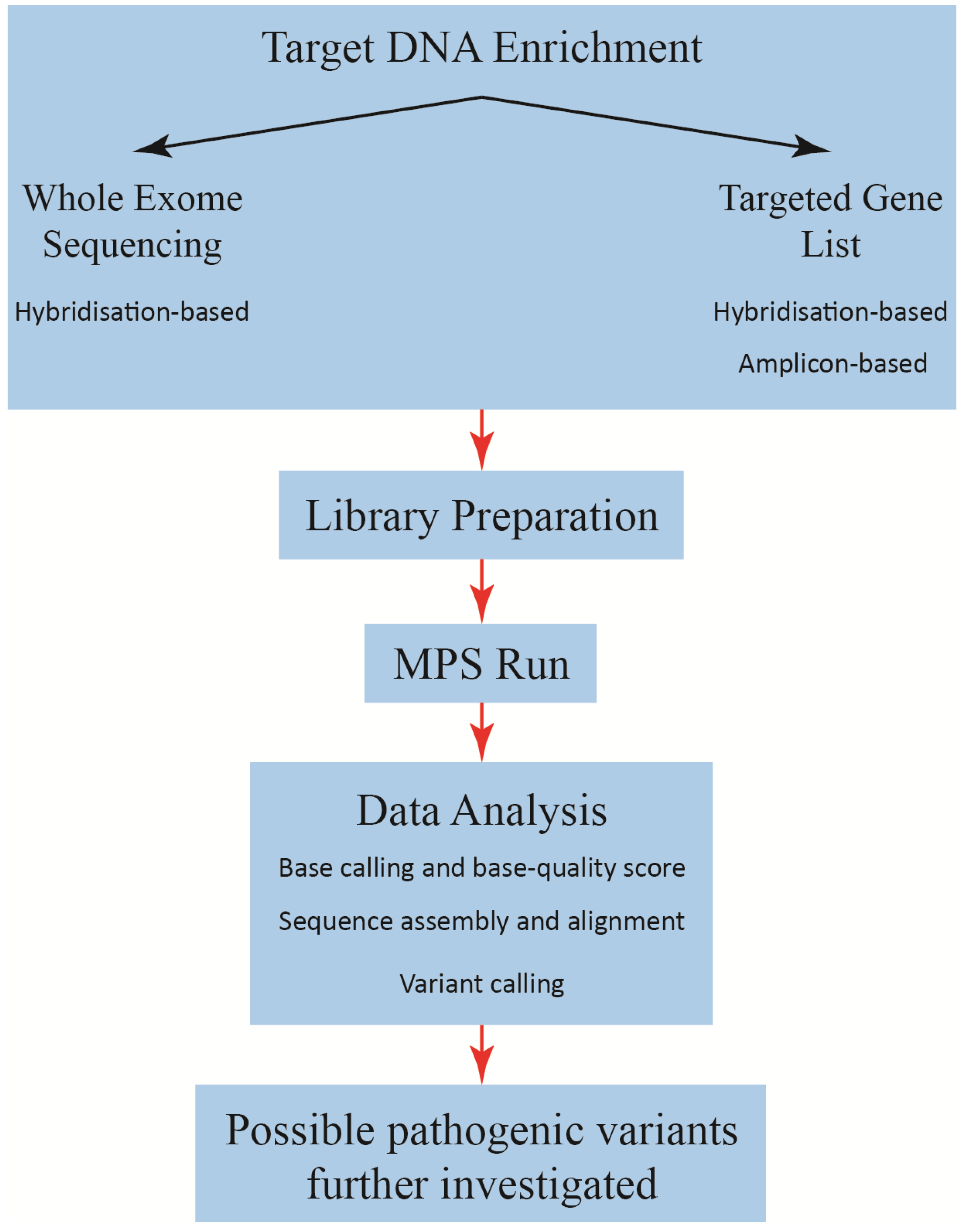
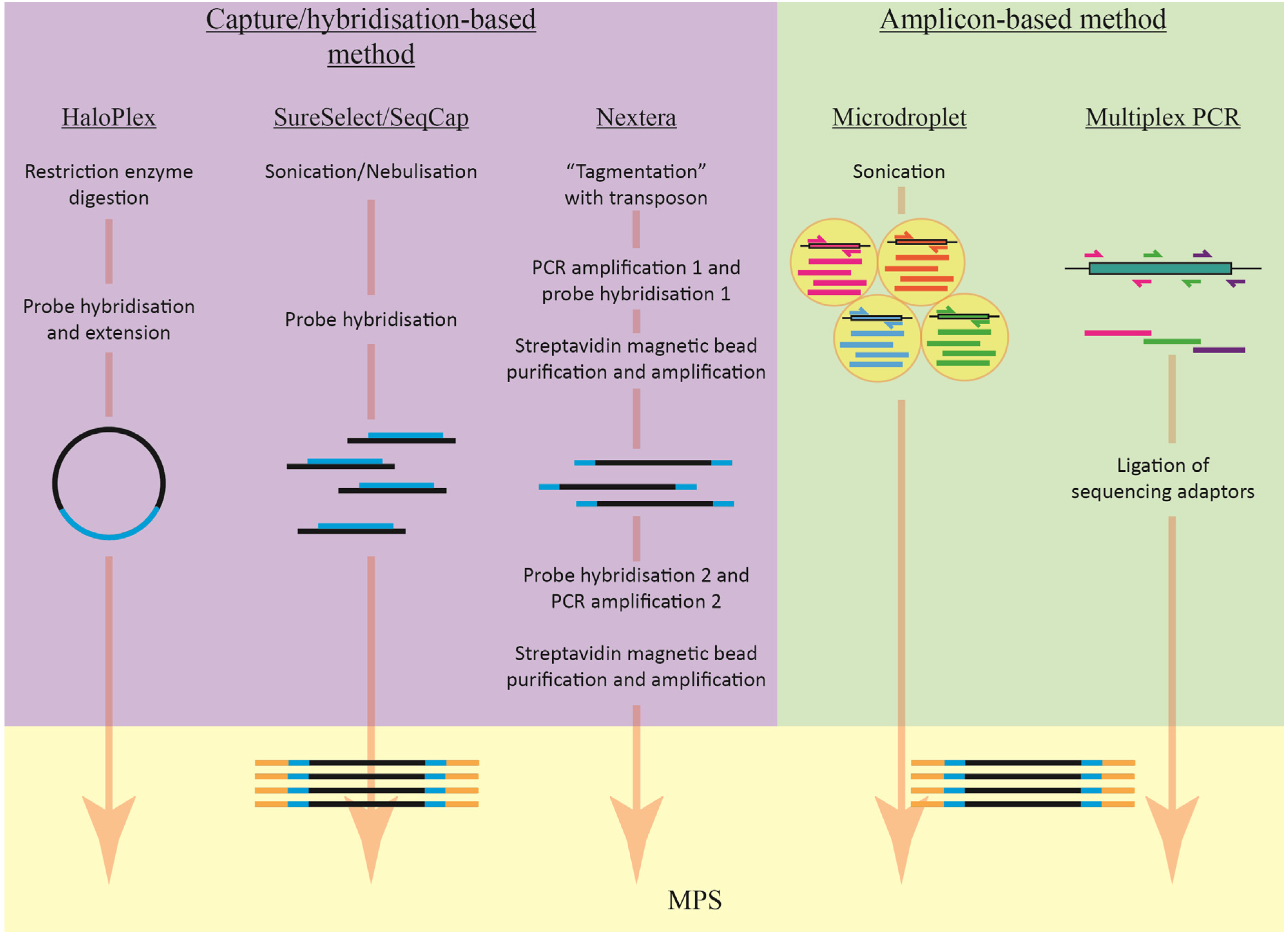
2.1. Target Enrichment Methods
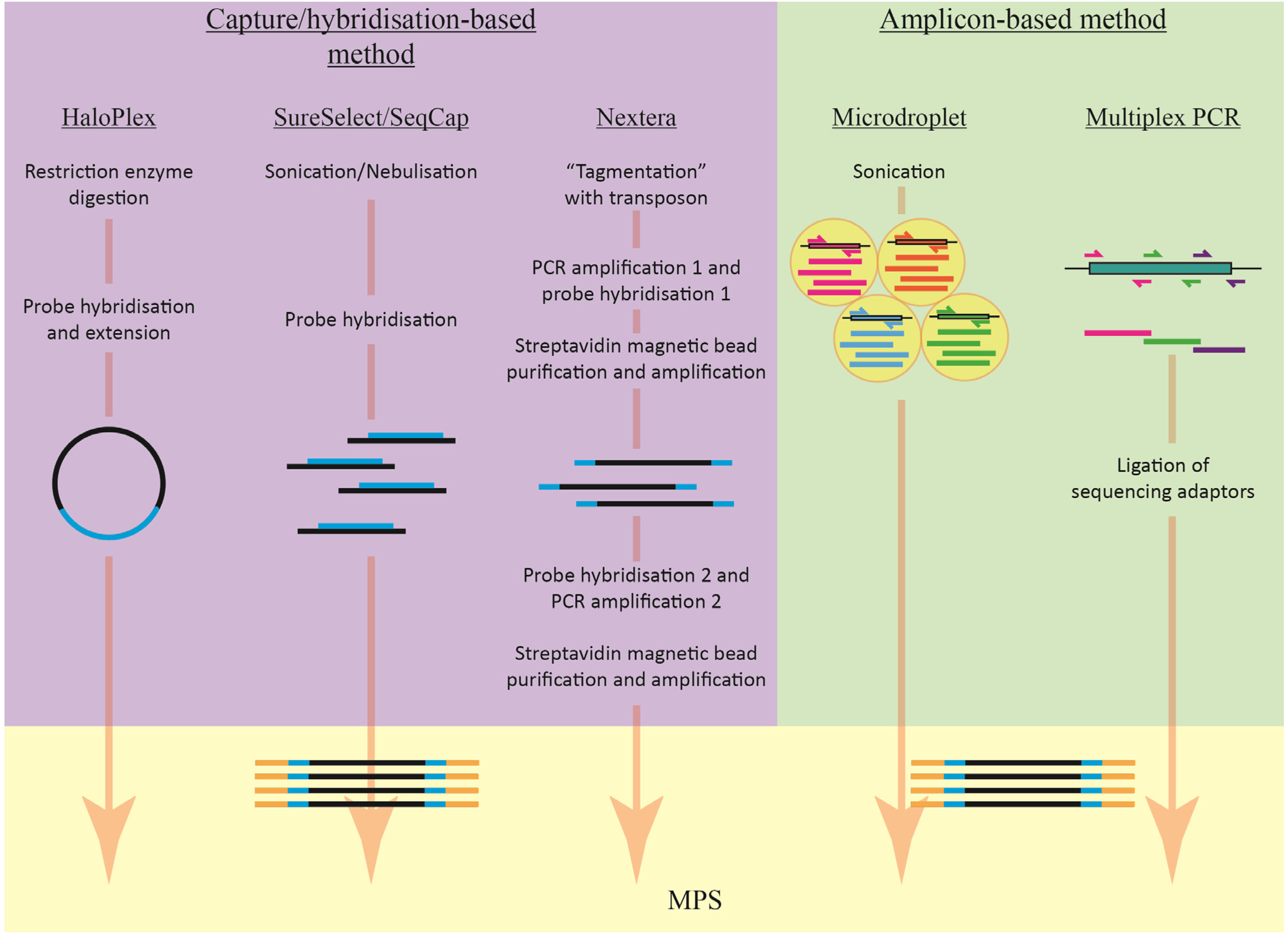
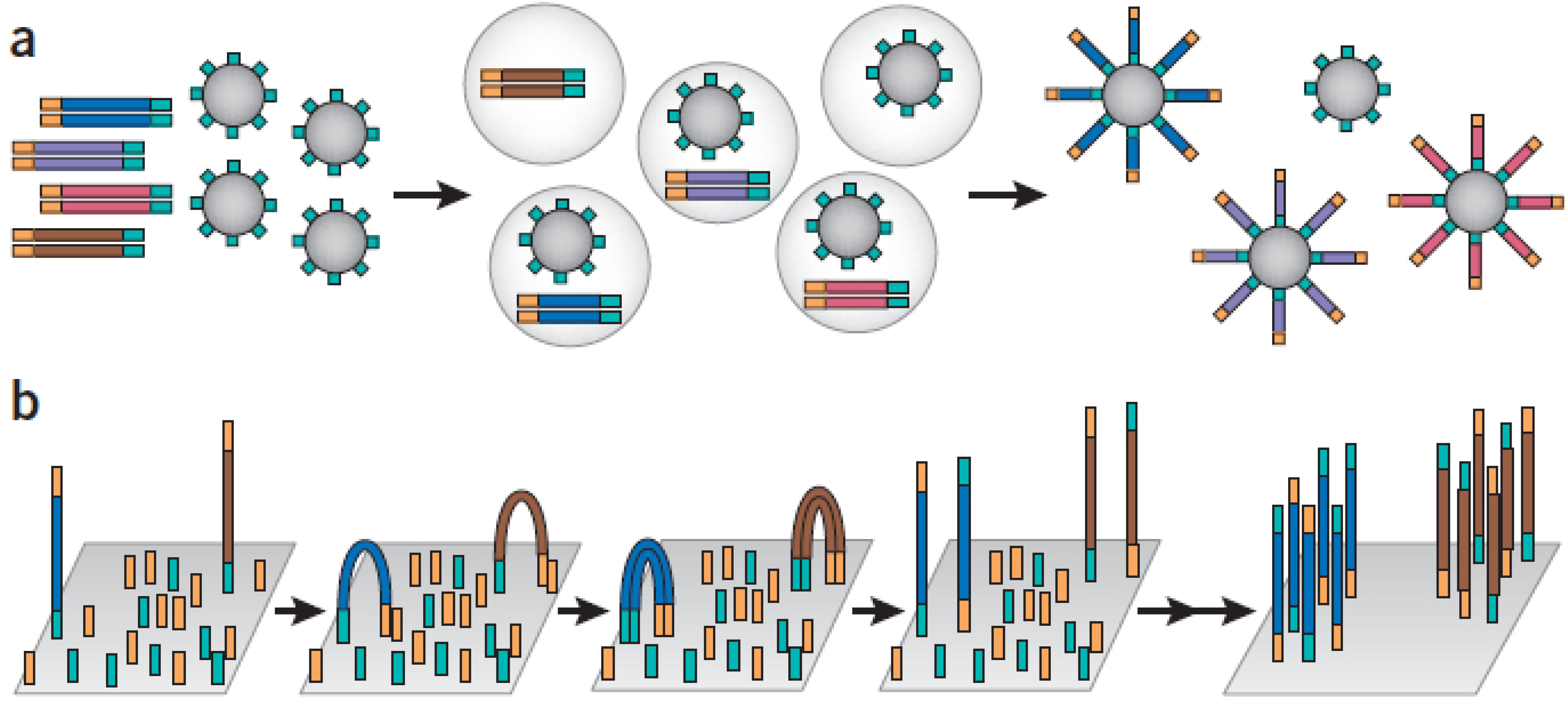
2.1.1. Amplicon-Based Enrichment

| Enrichment system | Company | Amplicon or hybridisation | Target size | # Amplicons | Input DNA |
|---|---|---|---|---|---|
| Ion AmpliSeq DNA Custom Kit | Life Technologies | Amplicon | 5 Mb | 12–6,144 | 10 ng per pool |
| TruSeq Custom Amplicon | Illumina | Amplicon | 4–650 Kb | 16–1,536 | 50 ng |
| Microdroplet PCR Custom gene panel | RainDance | Amplicon | 20,000 | 250 ng | |
| Access Array 48.48 | Fluidigm | Amplicon | 48–480 | 50 ng | |
| SeqCap EZ Choice Library | NimbleGen Roche | Hybridisation | 7–50 Mb | N/A | 500 ng |
| SureSelect Target Enrichment Kit | Agilent Technologies | Hybridisation | 200 kb–24 Mb | N/A | 500 ng–3 μg |
| HaloPlex Target Enrichment Kit | Agilent Technologies | Hybridisation | 1 kb–5 Mb | N/A | 200 ng–250 ng |
| Nextera Rapid Capture Custom Enrichment Kit | Illumina | Hybridisation | 500 kb–15 Mb | N/A | 50 ng |
2.1.2. Capture/Hybridisation-Based Enrichment
2.2. Second Generation Sequencing
| Platform | Amplification method | Chemistry | Read length (bp) | Throughput | Run time | Sequencing homopolymer regions | # Sequence reads/run |
|---|---|---|---|---|---|---|---|
| Roche 454-GS Junior | Emulsion PCR | Pyrosequencing | 200–400 | 35 Mb | 10 h | Prone to errors | >70,000 (amplicon sequencing) |
| Illumina-MiSeq | Bridge PCR | Reversible dye terminator | 35–150 | >120 Mb (single-end sequencing, 1× 35 bp) | 4 h | More accurate | >3.4 million single-end reads |
| >680 Mb (paired-end sequencing, 2× 100 bp) | 19 h | >6.8 million paired-end reads | |||||
| >1 Gb (paired-end sequencing, 2× 150 bp) | 27 h | ||||||
| Life Technologies –IonTorrent | Emulsion PCR | Sequence-by-ligation | 100–200 bp | Chip314: >10 Mb | All three chips take <2 h | More accurate | Chip314 (>1 million wells) |
| Chip316: >100 Mb | Chip316 (>6 million wells) | ||||||
| Chip318: >1 Gb | Chip 318 (>11 million wells); The number of reads is approximately 30%–40% of the available wells for each chip |
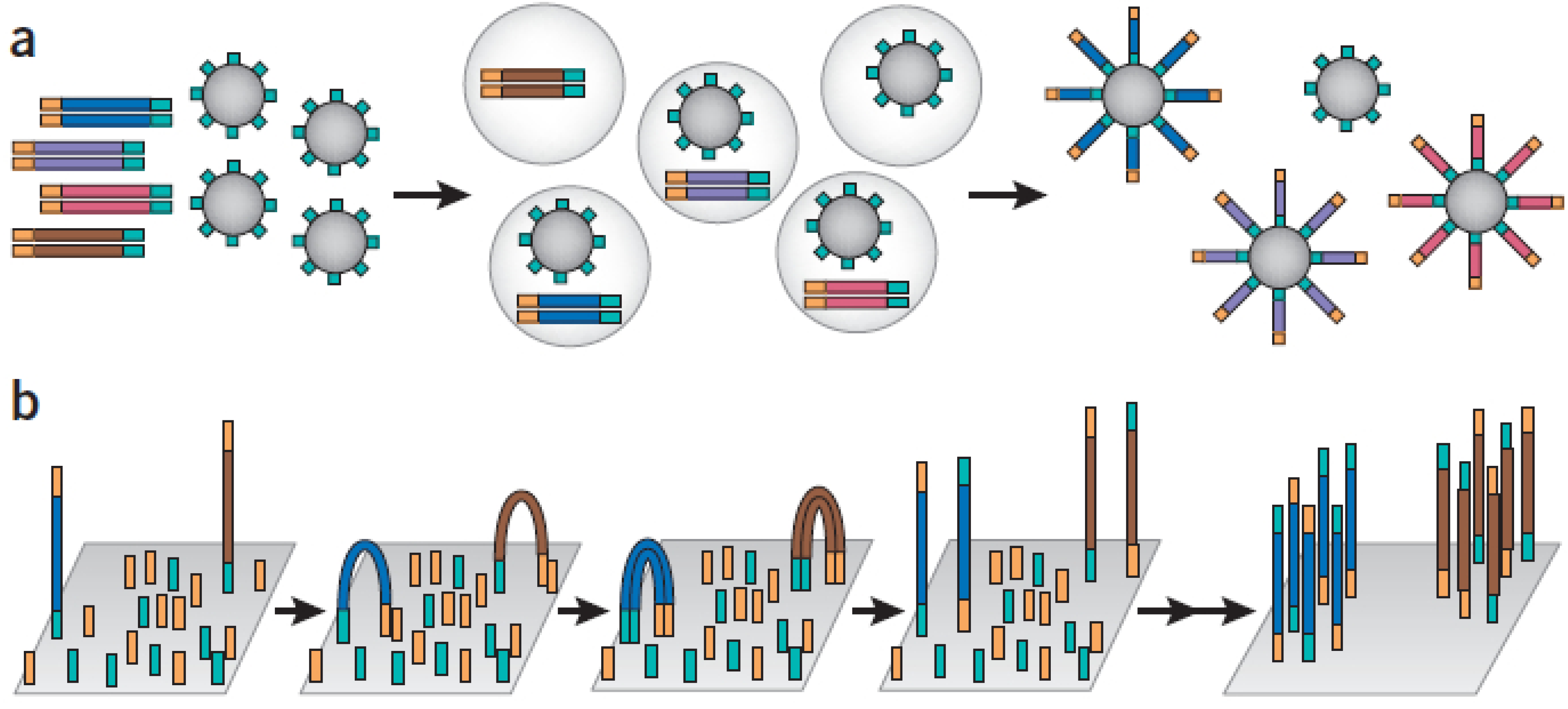
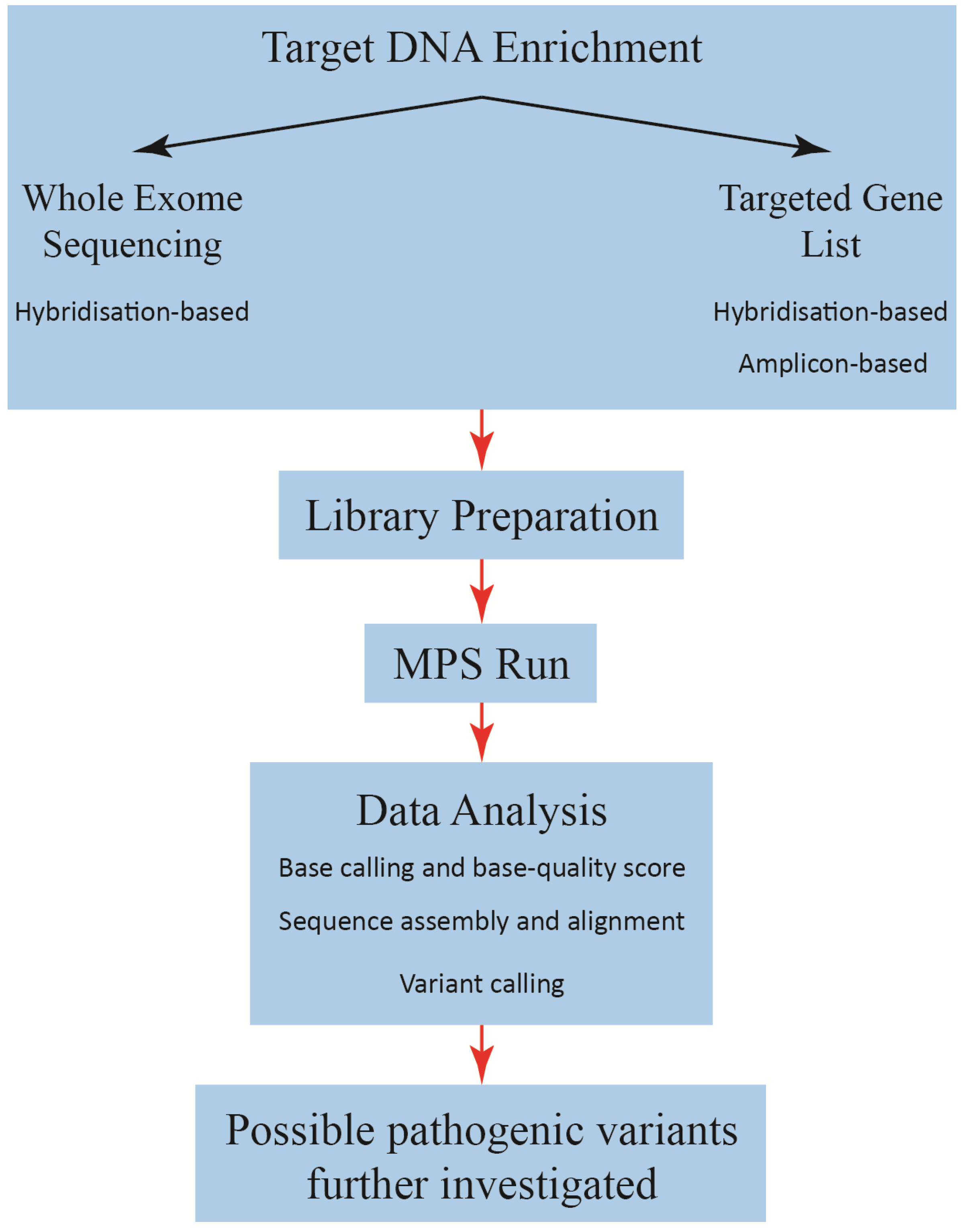
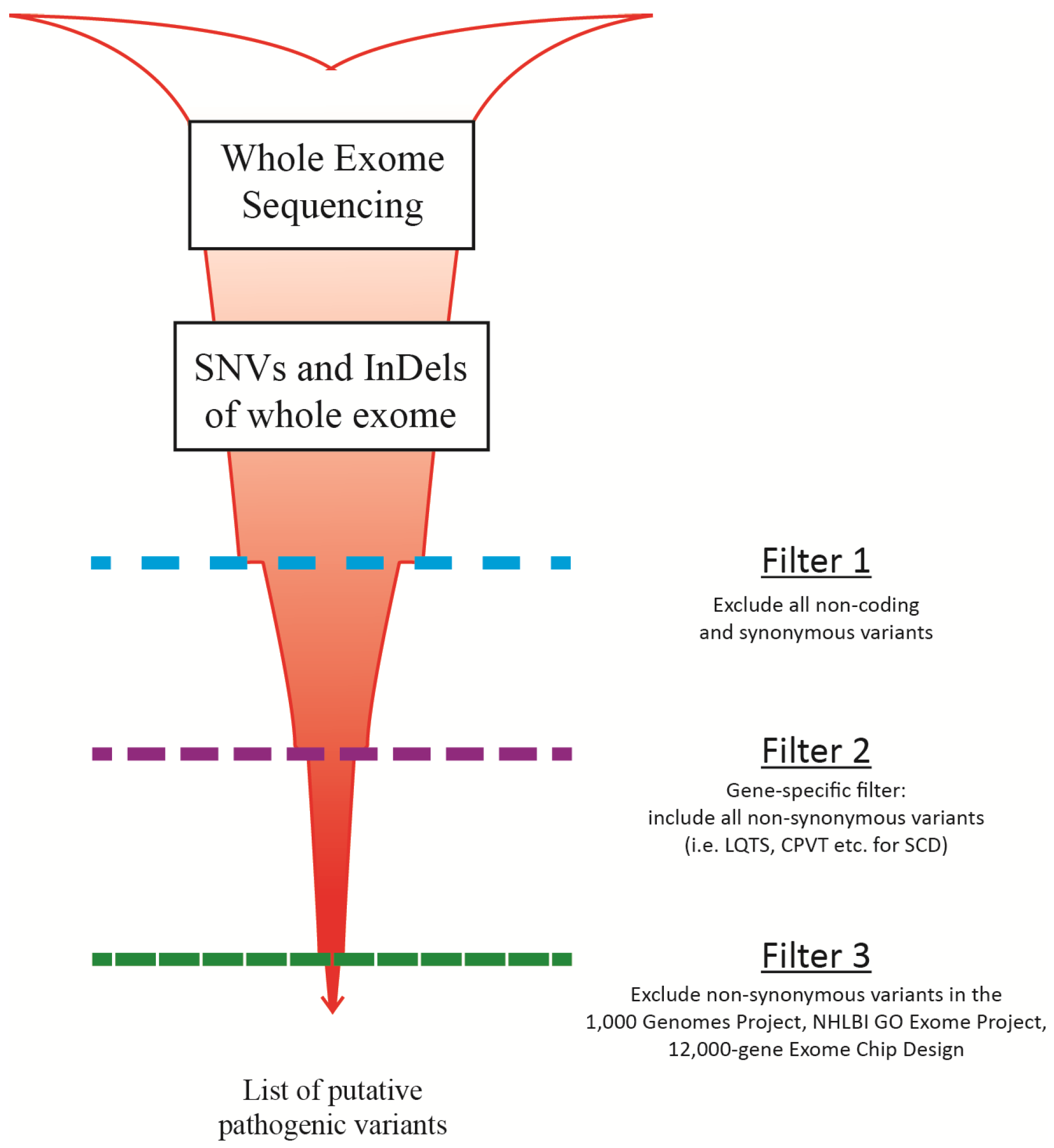
3. Downstream Data Processing
| Program | Functions | URL | Reference |
|---|---|---|---|
| Bowtie2 | Alignment | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml | [64] |
| BWA | Alignment | http://bio-bwa.sourceforge.net | [65] |
| SOAP2 | Alignment | http://soap.genomics.org/cn/soupaligner.html | [66] |
| MAQ | Alignment and assembly | http://maq.sourceforge.net | [67] |
| Novoalign | Alignment | http://www.novocraft.com | [68] |
| SAMtools | Variant calling | http://samtools.sourceforge.net | [62] |
| VARiD | Variant calling | http://compbio.cs.utoronto.ca/varid | [69] |
| VarScan2 | Variant calling | http://varscan.sourceforge.net | [70] |
| GATK Unified Genotyper | Variant calling | http://www.broadinstitute.org/gatk/index.php | [61] |
| SOAPsnp | Variant calling | http://soap.genomics.org.cn/soapsnp.html | [63] |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
False Positive Variants
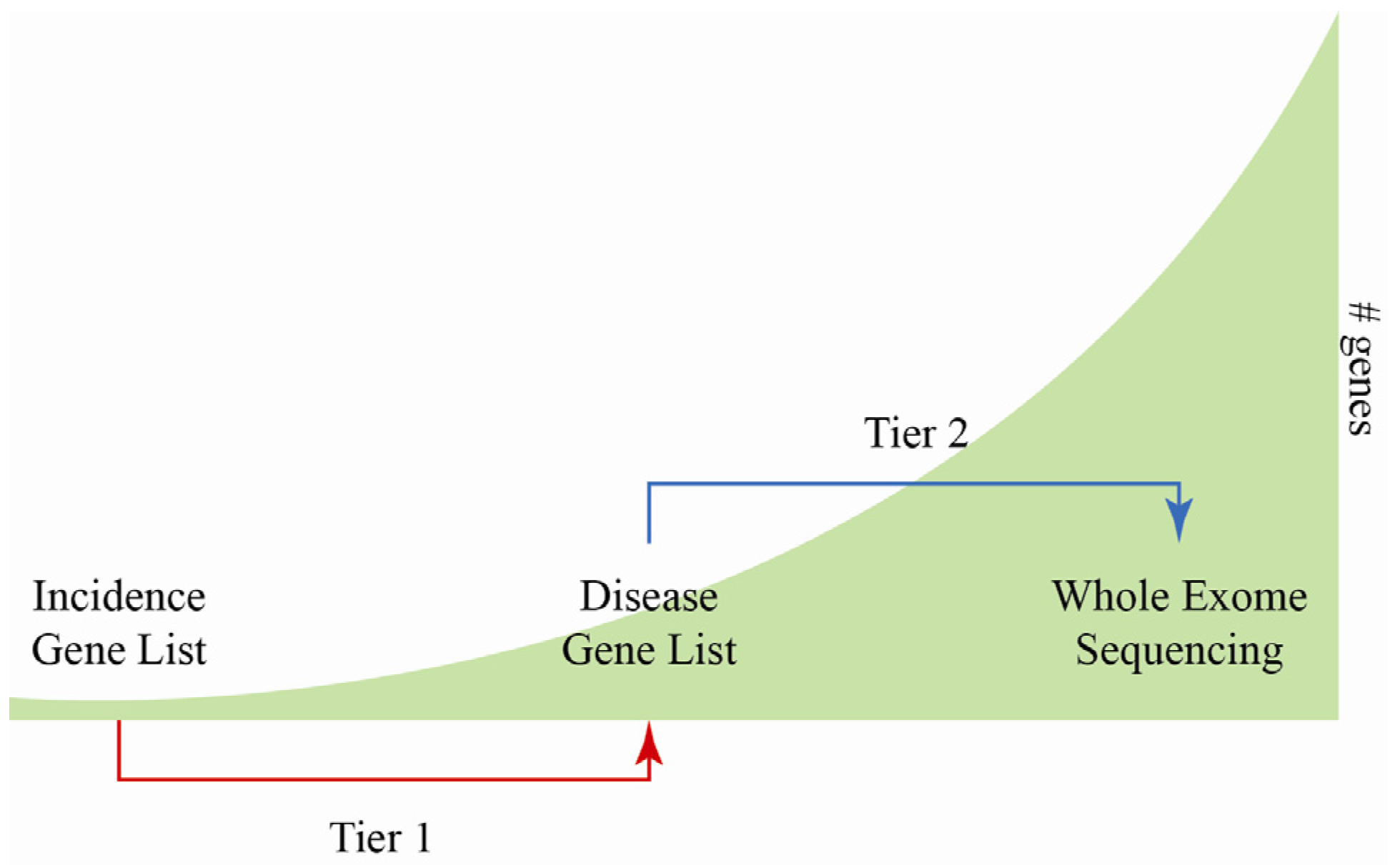
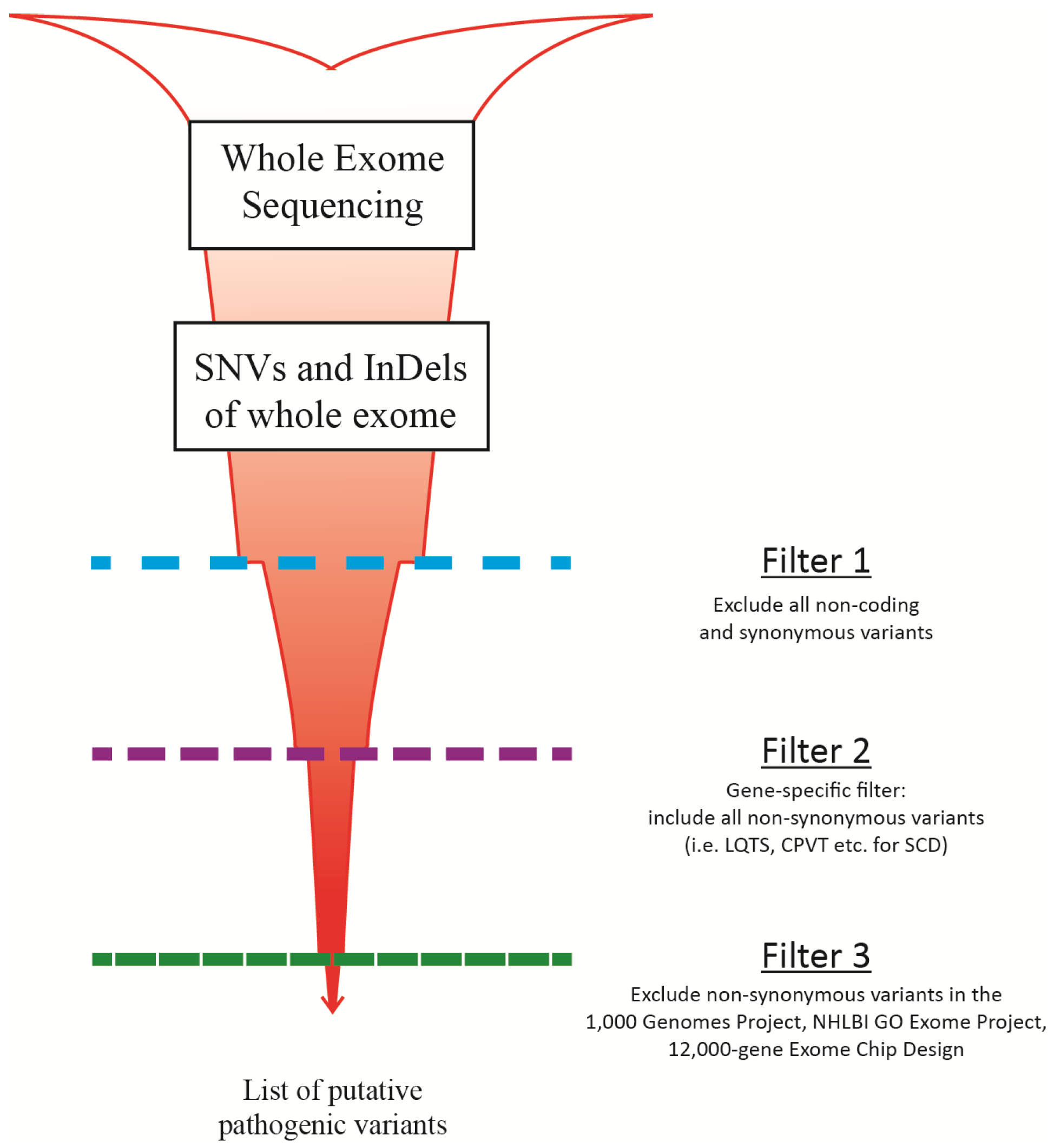
4. Challenges and Limitations of MPS
5. Validation of MPS Method for Diagnostic Use
6. MPS for Sudden Cardiac Death Screening
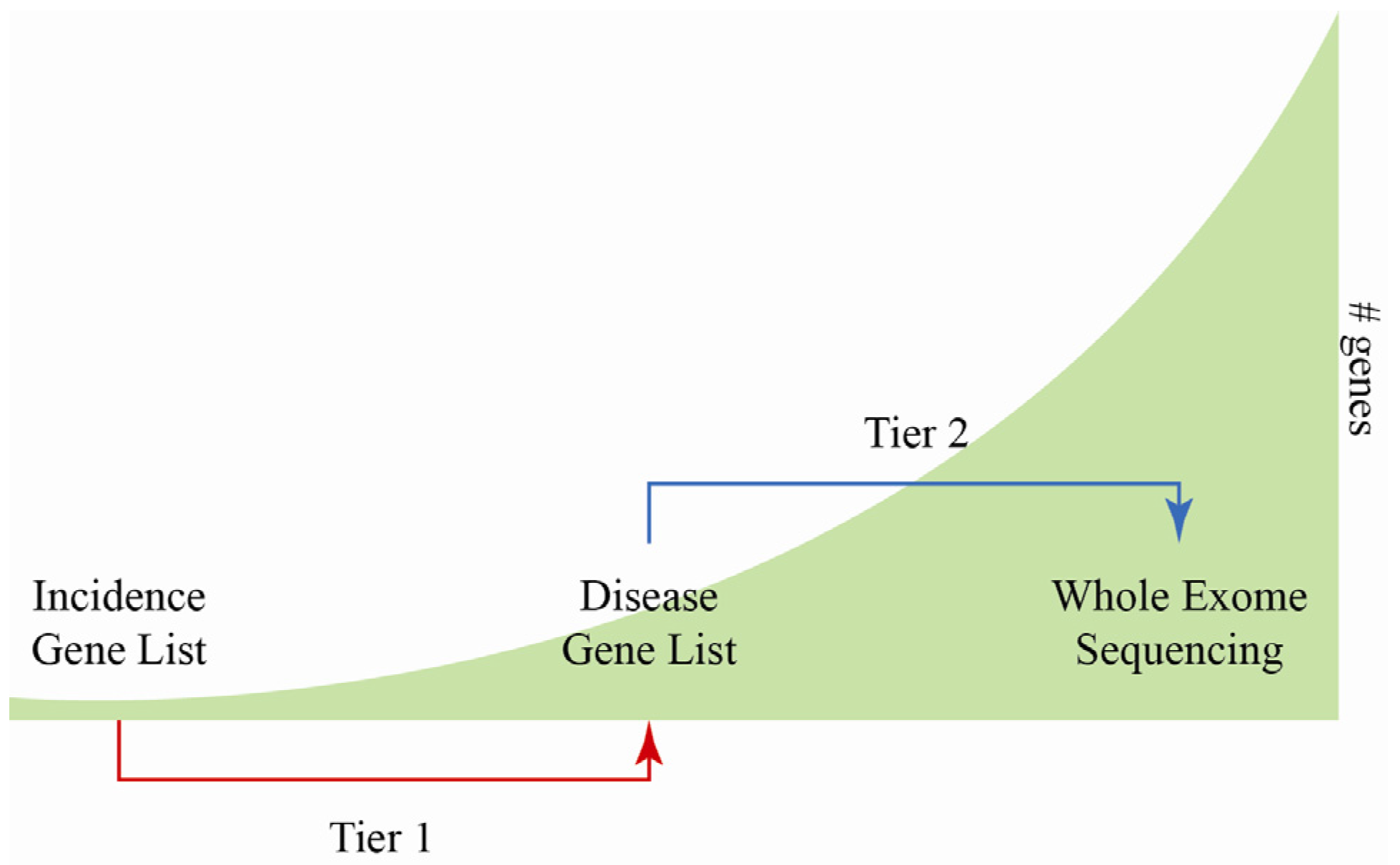
| Gene | Description | HCM | DCM | BrS | LQT | SQT | CPVT |
|---|---|---|---|---|---|---|---|
| BAG3 | BAG family molecular chaperone regulator 3 | 2%–4% [115] | |||||
| CACNA1C | Voltage-dependent L-type calcium channel, α1c subunit | 6%–7% [116] | Rare [117] | Limited data [118] | |||
| CACNB2 | Voltage-dependent L-type calcium channel, β2 subunit | 4%–5% [116] | |||||
| CASQ2 | Calsequestrin-2 precursor | 1%–2% [119] | |||||
| GLA | α-galactosidase A precursor | 0.5%–3% [120,121] | |||||
| KCNA5 | Potassium voltage-gated channel subfamily A, member 5 | ||||||
| KCNE1 | Potassium voltage-gated channel subfamily E, member 1 | Rare [117] | |||||
| KCNE2 | Potassium voltage-gated channel subfamily E, member 2 | Rare [117] | |||||
| KCNH2 | Potassium voltage-gated channel subfamily H, member 2 | 25%–30% [117] | Limited data [118] | ||||
| KCNQ1 | Potassium voltage-gated channel subfamily Q, member 1 | 30%–35% [117,122] | Limited data [118] | ||||
| LMNA | Lamin A/C | 4%–8% [115] | |||||
| MYBPC3 | Myosin-binding protein C, cardiac-type | 15%–30% [123,124 ] | 2%–4% [125 ,126] | ||||
| MYH6 | Myosin heavy-chain 6 | Rare [127 ] | 4% [126 ] | ||||
| MYH7 | Myosin heavy-chain 7 | 15%–30% [123,124] | 4% [125,128] | ||||
| MYL2 | Myosin regulatory light chain 2 | <2% [123,124 ] | |||||
| RBM20 | RNA-binding motif protein 20 | 3%–6% [115] | |||||
| RYR2 | Ryanodine receptor 2 | 50%–55% [119] | |||||
| SCN1B | Sodium channel protein type 1, β subunit | 1%–2% [116] | |||||
| SCN5A | Sodium channel protein type 5, α subunit | 1%–2% [115] | 11%–18% [116] | 5%–10% [117] | |||
| TNNI3 | Troponin I type 3, cardiac type | <2% [123, 124] | Rare [125,126] | ||||
| TNNT2 | Troponin T type 2, cardiac type | 2%–5% [123 ,124] | 3% [125,128] | ||||
| TPM1 | Tropomyosin α1 | 2% [123, 124] | 1%–2% [115,125,126] | ||||
| TTN | Titin | Rare [127] | 15%–25% [115] | ||||
| SCN5A | Sodium channel protein type 5, α subunit | 1%–2% [115] | 11%–18% [116] | 5%–10% [117] | |||
| TNNI3 | Troponin I type 3, cardiac type | <2% [123,124 ] | Rare [125 ,126] | ||||
| TNNT2 | Troponin T type 2, cardiac type | 2%–5% [123 ,124] | 3% [125,128] | ||||
| TPM1 | Tropomyosin α1 | 2% [123 ,124] | 1%–2% [115,125,126] | ||||
| TTN | Titin | Rare [127 ] | 15%–25% [115] |
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Doolan, A.; Semsarian, C.; Langlois, N. Causes of sudden cardiac death in young Australians. Med. J. Aust. 2004, 180, 110–112. [Google Scholar]
- Behr, E.R.; Dalageorgou, C.; Christiansen, M.; Syrris, P.; Hughes, S.; Tome Esteban, M.T.; Rowland, E.; Jeffery, S.; McKenna, W.J. Sudden arrhythmic death syndrome: Familial evaluation identifies inheritable heart disease in the majority of families. Eur. Heart J. 2008, 29, 1670–1680. [Google Scholar] [CrossRef]
- Skinner, J.R.; Crawford, J.; Smith, W.; Aitken, A.; Heaven, D.; Evans, C.A.; Hayes, I.; Neas, K.R.; Stables, S.; Koelmeyer, T.; et al. Prospective, population-based long QT molecular autopsy study of postmortem negative sudden death in 1 to 40 year olds. Heart Rhythm 2011, 8, 412–419. [Google Scholar] [CrossRef]
- Tan, H.L.; Hofman, N.; van Langen, I.M.; van der Wal, A.C.; Wilde, A.A.M. Heritability and diagnostic yield of cardiological and genetic examination in surviving relatives. Circulation 2005, 112, 207–213. [Google Scholar] [CrossRef]
- Boczek, N.J.; Best, J.M.; Tester, D.J.; Giudicessi, J.R.; Middha, S.; Evans, J.M.; Kamp, T.J.; Ackerman, M.J. Exome sequencing and systems biology converge to identify novel mutations in the L-type calcium channel, CACNA1C, linked to autosomal dominant long QT syndrome. Circ. Cardiovasc Genet. 2013, 6, 279–289. [Google Scholar] [CrossRef]
- Next Generation Sequencing-Oxford University Hospitals. Available online: http://www.ouh.nhs.uk/services/referrals/genetics/genetics-laboratories/molecular-genetics-laboratory/next-generation-sequencing.aspx (accessed on 28 January 2014).
- VCGS-Genetic testing for genetic heart conditions: Patient information sheet. Available online: http://www.vcgs.org.au/pathology/downloads/molecular/patient%20information%20sheet%2018_3_13.pdf (accessed on 28 January 2014).
- Maxam, A.M.; Gilbert, W. A new method for sequencing DNA. Proc. Natl. Acad. Sci. USA 1977, 74, 560–564. [Google Scholar] [CrossRef]
- Hunkapiller, T.; Kaiser, R.J.; Koop, B.F.; Hood, L. Large-scale and automated DNA sequence determination. Science 1991, 254, 59–67. [Google Scholar]
- Swerdlow, H.; We, S.L.; Harke, H.; Dovichi, N.J. Capillary gel electrophoresis for DNA sequencing. Laser-induced fluorescence detection with the sheath flow cuvette. J. Chromatogr. 1990, 516, 61–67. [Google Scholar] [CrossRef]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef]
- Meldrum, C.; Doyle, M.A.; Tothill, R.W. Next-generation sequencing for cancer diagnostics: A practical perspective. Clin. Biochem. Rev. 2011, 32, 177–195. [Google Scholar]
- Robinson, P.N.; Krawitz, P.; Mundlos, S. Strategies for exome and genome sequence data analysis in disease-gene discovery projects. Clin. Genet. 2011, 80, 127–132. [Google Scholar] [CrossRef]
- Life Technologies-Ion AmpliSeq Panels. Available online: http://www.lifetechnologies.com/nz/en/home/life-science/sequencing/next-generation-sequencing/ion-torrent-next-generation-sequencing-workflow/ion-torrent-next-generation-sequencing-select-targets/ampliseq-target-selection.html?icid=ampliseq-panels (accessed on 27 January 2014).
- Singh, R.R.; Patel, K.P.; Routbort, M.J.; Reddy, N.G.; Barkoh, B.A.; Handal, B.; Kanagal-Shamanna, R.; Greaves, W.O.; Medeiros, L.J.; Aldape, K.D.; et al. Clinical validation of a next-generation sequencing screen for mutational hotspots in 46 cancer-related genes. J. Mol. Diagn. 2013, 15, 604–622. [Google Scholar]
- Tsongalis, G.J.; Peterson, J.D.; de Abreu, F.B.; Tunkey, C.D.; Gallagher, T.L.; Strausbaugh, L.D.; Wells, W.A.; Amos, C.L. Routine use of the Ion Torrent AmpliSeq™ Cancer Hotspot Panel for identification of clinically actionable somatic mutations. Clin. Chem. Lab. Med. 2013, 13, 1–8. [Google Scholar]
- Illumina-TruSeq Custom Amplicon Guide. Available online: http://supportres.illumina.com/documents/myillumina/b718c350-b3b2–4234-b71a-0b832f14cda3/truseq_custom_amplicon_libraryprep_ug_15027983_b.pdf (accessed on 30 January 2014).
- Chandrasekharappa, S.C.; Lach, F.P.; Kimble, D.C.; Kamat, A.; Teer, J.K.; Donovan, F.X.; Flynn, E.; Sen, S.K.; Thongthip, S.; Sanborn, E.; et al. Massively parallel sequencing, aCGH, and RNA-Seq technologies provide a comprehensive molecular diagnosis of Fanconi anemia. Blood 2013, 121, e138–e148. [Google Scholar] [CrossRef]
- Chang, F.; Li, M.M. Clinical application of amplicon-based next-generation sequencing in cancer. Cancer Genet. 2013. pii:S2210-7762(13)00142-7. [Google Scholar]
- Tewhey, R.; Warner, J.B.; Nakano, M.; Libby, B.; Medkova, M.; David, P.H.; Kotsopoulos, S.K.; Samuels, M.L.; Hutchison, J.B.; Larson, J.W.; et al. Microdroplet-based PCR enrichment for large-scale targeted sequencing. Nat. Biotechnol. 2009, 27, 1025–1031. [Google Scholar] [CrossRef]
- Dames, S.; Chou, L.-S.; Xiao, Y.; Wayman, T.; Stocks, J.; Singleton, M.; Eilbeck, K.; Mao, R. The development of next-generation sequencing assays for the mitochondrial genome and 108 nuclear genes associtted with mitochondrial disorders. J. Mol. Diagn. 2013, 15, 526–534. [Google Scholar] [CrossRef]
- Bonnefond, A.; Philippe, J.; Durand, E.; Muller, J.; Saeed, S.; Arslan, M.; Martínez, R.; de Graeve, F.; Dhennin, V.; Rabearivelo, I.; et al. Highly sensitive diagnosis of 43 monogenic forms of diabetes or obesity, through one step PCR-based enrichment in combination with next-generation sequencing. Diabetes Care 2014, 37, 460–467. [Google Scholar] [CrossRef]
- Valencia, C.A.; Ankala, A.; Rhodenizer, D.; Bhide, S.; Littlejohn, M.R.; Keong, L.M.; Rutkowski, A.; Sparks, S.; Bonnemann, C.; Hegde, M. Comprehensive mutation analysis for congenital muscular dystrophy: A clinical PCR-based enrichment and next-generation sequencing panel. PLoS One 2013, 8, e53083. [Google Scholar] [CrossRef]
- Halbritter, J.; Diaz, K.; Chaki, M.; Porath, J.D.; Tarrier, B.; Fu, C.; Innis, J.L.; Allen, S.J.; Lyons, R.H.; Stefanidis, C.J.; et al. High-throughput mutation analysis in patients with a nephronophthisis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. J. Med. Genet. 2012, 49, 756–767. [Google Scholar] [CrossRef]
- Hollants, S.; Redeker, E.J.W.; Matthijs, G. Microfluidic amplification as a tool for massive parallel sequencing of the familial hypercholesterolemia genes. Clin. Chem. 2012, 58, 717–724. [Google Scholar] [CrossRef]
- Mamanova, L.; Coffey, A.J.; Scott, C.E.; Kozarewa, I.; Turner, E.H.; Kumar, A.; Howard, E.; Shendure, J.; Turner, D.J. Target-enrichment strategies for next-generation sequencing. Nat. Methods 2010, 7, 111–118. [Google Scholar] [CrossRef]
- Hagemann, I.S.; Cottrell, C.E.; Lockwood, C.M. Design of targeted, capture-based, next generation sequencing tests for precision cancer therapy. Cancer Genet. 2013, 206, 420–431. [Google Scholar] [CrossRef]
- NimbleGen-NimbleGen SeqCap EZ Library LR User’s Guide. Available online: http://www.nimblegen.com/products/lit/06560881001_SeqCapEZLibraryLR_Guide_v2p0.pdf (accessed on 28 January 2014).
- Trujillano, D.; Perez, B.; González, J.; Tornador, C.; Navarrete, R.; Escaramis, G.; Ossowski, S.; Armengol, L.; Cornejo, V.; Desviat, L.R.; et al. Accurate molecular diagnosis of phenylketonuria and tetrahydrobiopterin-deficient hyperphenylalaninemias using high-throughput targeted sequencing. Eur. J. Hum. Genet. 2014, 22, 528–534. [Google Scholar] [CrossRef]
- Trujillano, D.; Ramos, M.D.; González, J.; Tornador, C.; Sotillo, F.; Escaramis, G.; Ossowski, S.; Armengol, L.; Casals, T.; Estivill, X. Next generation diagnostics of cystic fibrosis and CFTR-related disorders by targeted multiplex high-coverage resequencing of CFTR. J. Med. Genet. 2013, 50, 455–462. [Google Scholar] [CrossRef]
- Schorderet, D.F.; Iouranova, A.; Favez, T.; Tiab, L.; Escher, P. IROme, a new high-throughput molecular tool for the diagnosis of inherited retinal dystrophies. BioMed Res. Int. 2013, 2013, 198089. [Google Scholar]
- Agilent Technologies-SureSelect Target Enrichment System for Illumina Paired-End Sequencing Library. Available online: https://www.genomics.agilent.com/files/Manual/G3360-90020_SureSelect_Indexing_1.0.pdf (accessed on 28 January 2014).
- Falk, M.J.; Pierce, E.A.; Consugar, M.; Xie, M.H.; Guadalupe, M.; Hardy, O.; Rappaport, E.F.; Wallace, D.C.; LeProust, E.; Gai, X. Mitochondrial disease genetic diagnostics: Optimized whole-exome analysis for all MitoCarta nuclear genes and the mitochondrial genome. Discov. Med. 2012, 14, 389–399. [Google Scholar]
- Mutai, H.; Suzuki, N.; Shimizu, A.; Torii, C.; Namba, K.; Morimoto, N.; Kudoh, J.; Kaga, K.; Kosaki, K.; Matsunaga, T. Diverse spectrum of rare deafness genes underlies early-childhood hearing loss in Japanese patients: A cross-sectional, multi-center next-generation sequencing study. Orphanet J. Rare Dis. 2013, 8, 172. [Google Scholar] [CrossRef]
- Shearer, A.E.; DeLuca, A.P.; Hildebrand, M.S.; Taylor, K.R.; Gurrola, J.N.; Scherer, S.; Scheetz, T.E.; Smith, R.J. Comprehensive genetic testing for hereditary hearing loss using massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 21104–21109. [Google Scholar] [CrossRef]
- Vandrovcova, J.; Thomas, E.R.A.; Atanur, S.S.; Norsworthy, P.J.; Neuwirth, C.; Tan, Y.; Kasperaviciute, D.; Biggs, J.; Game, L.; Mueller, M.; et al. The use of next-generation sequencing in clinical diagnosis of familial hypercholesterolemia. Genet. Med. 2013, 15, 948–957. [Google Scholar] [CrossRef]
- Wooderchak-Donahue, W.; O’Fallon, B.; Furtado, L.; Durtschi, J.; Plant, P.; Ridge, P.; Rope, A.; Yetman, A.; Bayrak-Toydemir, P. A direct comparison of next generation sequencing enrichment methods using an aortopathy gene panel- clinical diagnostics perspective. BMC Med. Genomics 2012, 5, 50. [Google Scholar]
- Agilent Technologies-HaloPlex Target Enrichment System. Available online: http://www.chem.agilent.com/library/usermanuals/Public/G9900-90001.pdf (accessed on 28 January 2014).
- Nextera Rapid Capture Enrichment Guide. Available online: http://supportres.illumina.com/documents/documentation/chemistry_documentation/samplepreps_nextera/nexterarapidcapture/nextera-rapid-capture-enrichment-guide-15037436-f.pdf (accessed on 28 January 2014).
- Parla, J.S.; Iossifov, I.; Grabill, I.; Spector, M.S.; Kramer, M.; McCombie, W.R. A comparative analysis of exome capture. Genome Res. 2011, 12, R91. [Google Scholar]
- Sulonen, A.M.; Ellonen, P.; Almusa, H.; Lepistö, M.; Eldfors, S.; Hannula, S.; Miettinen, T.; Tyynismaa, H.; Salo, P.; Heckman, C.; et al. Comparison of solution-based exome capture methods for next generation sequencing. Genome Biol. 2011, 12, R94. [Google Scholar] [CrossRef]
- Clark, M.J.; Chen, R.; Lam, H.Y.; Karczewski, K.J.; Chen, R.; Euskirchen, G.; Butte, A.J.; Snyder, M. Performance comparison of exome DNA sequencing technologies. Nat. Biotechnol. 2011, 29, 908–914. [Google Scholar] [CrossRef]
- Ku, C.-S.; Wu, M.; Cooper, D.N.; Naidoo, N.; Pawitan, Y.; Pang, B.; Iacopetta, B.; Soong, R. Technological advances in DNA sequence enrichment and sequencing for germline genetic diagnosis. Expert Rev. Mol. Diagn. 2012, 12, 159–173. [Google Scholar] [CrossRef]
- Zhang, W.; Cui, H.; Wong, L.-J. Application of next generation sequencing to molecular diagnosis of inherited diseases. Top. Curr. Chem. 2014, 336, 19–45. [Google Scholar] [CrossRef]
- Hui, P. Next generation sequencing: Chemistry, technology and applications. Top. Curr. Chem. 2014, 336, 1–18. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012, 2012, 251364. [Google Scholar]
- Voelkerding, K.V.; Dames, S.A.; Durtschi, J.D. Next-generation sequencing: From basic research to diagnostic. Clin. Chem. 2009, 55, 641–658. [Google Scholar] [CrossRef]
- Dressman, D.; Yan, H.; Traverso, G.; Kinzler, K.W.; Volgelstein, B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc. Natl. Acad. Sci. USA 2003, 100, 8817–8822. [Google Scholar] [CrossRef]
- Ronaghi, M.; Karamohamed, S.; Pettersson, B.; Uhlén, M.; Nyrén, P. Real-time DNA sequencing using detection of pyrophosphate release. Anal. Biochem. 1996, 242, 84–89. [Google Scholar] [CrossRef]
- Adessi, C.; Matton, G.; Ayala, G.; Turcatti, G.; Mermod, J.J.; Mayer, P.; Kawashima, E. Solid phase DNA amplification: Characterisation of primer attachment and amplifcation mechanisms. Nucleic Acids Res. 2000, 28, E87. [Google Scholar] [CrossRef]
- Turcatti, G.; Romieu, A.; Fedurco, M.; Tairi, A.P. A new class of cleavable fluorescent nucleotides: Synthesis and optimization as reversible terminators for DNA sequencing by synthesis. Nucleic Acids Res. 2008, 36, e25. [Google Scholar]
- Ewing, B.; Green, P. Base-calling of automated sequencer traces using Phred. II. Error probabilities. Genome Res. 1998, 8, 186–194. [Google Scholar]
- Rougemont, J.; Amzallag, A.; Iseli, C.; Farinelli, L.; Xenarios, I.; Naef, F. Probabilistic base calling of Solexa sequencing data. BMC Bioinforma 2008, 9, 431. [Google Scholar] [CrossRef]
- Andrews, C. FastQC. Available online: http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc (accessed on 30 January 2014).
- Allcock, R.J. Production and analytic bioinformatics for next-generation DNA sequencing. Methods Mol. Biol. 2014, 1168, 17–29. [Google Scholar]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing depth and coverage: Key considerations in genomic analysis. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef]
- Wheeler, D.A.; Srinivasan, M.; Egholm, M.; Shen, Y.; Chen, L.; McGuire, A.; He, W.; Chen, Y.J.; Makhijani, V.; Roth, G.T.; et al. The complete genome of an individual by massively parallel DNA sequencing. Nat. Biotechnol. 2008, 452, 872–876. [Google Scholar]
- Brockman, W.; Alvarez, P.; Young, S.; Garber, M.; Giannoukos, G.; Lee, W.L.; Russ, C.; Lander, E.S.; Nusbaum, C.; Jaffe, D.B. Quality scores and SNP detection in sequencing-by-synthesis systems. Genome Res. 2008, 18, 763–770. [Google Scholar] [CrossRef]
- Dohm, J.C.; Lottaz, C.; Borodina, T.; Himmelbauer, H. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res. 2008, 36, e105. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, H.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Li, R.; Li, Y.; Fang, X.; Yang, H.; Wang, J.; Kristiansen, K.; Wang, J. SNP detection for massively parallel whole-genome resequencing. Genome Res. 2009, 19, 1124–1132. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, R.; Yu, C.; Li, Y.; Lam, T.-W.; Yiu, S.-M.; Kristiansen, K.; Wang, W. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef]
- Li, H.; Ruan, J.; Durbin, R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008, 18, 1851–1858. [Google Scholar] [CrossRef]
- Novocraft.com Novoalign. Available online: http://www.novocraft.com (accessed on 30 January 2014).
- Dalca, A.V.; Rumble, S.M.; Levy, S.; Brudno, M. VARiD: A variation detection framework for color-space and letter-space platforms. Bioinformatics 2010, 26, i343–i349. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 3012, 22, 568–576. [Google Scholar]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Howells, K.; Phillips, A.D.; Thomas, N.S.; Cooper, D.N. The Human Gene Mutation Database: 2008 update. Genome Med. 2009, 1, 13. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymouse variants on protein function using the SIFT algorithm. Nat. Protocols 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- National Genetics Reference Laboratory. Available online: http://www.ngrl.org.uk/Manchester/projects/bioinformatic-tools (accessed on 30 January 2014).
- Tavtigian, S.V.; Deffenbaugh, A.M.; Yin, L.; Judkins, T.; Scholl, T.; Samollow, P.B.; de Silva, D.; Zharkikh, A.; Thomas, A. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J. Med. Genet. 2006, 43, 295–305. [Google Scholar]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Applications to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef]
- Brunham, L.R.; Singaraja, R.R.; Pape, T.D.; Kejariwal, A.; Thomas, P.D.; Hayden, M.R. Accurate prediction of the functional significance of nucleotide polymorphisms and mutations in the ABCA1 gene. PLoS Genet. 2005, 1, e83. [Google Scholar] [CrossRef]
- Stone, E.A.; Sidow, A. Physicochemical constraint violation by missense substitution mediates impairment of protein function and disease severity. Genome Res. 2005, 15, 978–986. [Google Scholar] [CrossRef]
- Choi, Y.; Sims, G.; Murphy, S. Predicting the functional effect of amino acid substitutions and indels. PLoS One 2012, 7, e46688. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Ryan, M.; Diekhans, M.; Lien, S.; Liu, Y.; Karchin, R. LS-SNP/PDB: Annotated non-synonymous SNPs mapped to protein data bank structures. Bioinformatics 2009, 25, 1431–1432. [Google Scholar] [CrossRef]
- De Baets, G.; van Durme, J.; Reumers, J.; Maurer-Stroh, S.; Vanhee, P.; Schymkowitz, J.; Rousseau, F. SNPeffect4.0: Online prediction of molecular and structural effects of protein-coding variants. Nucleic Acids Res. 2012, 40, D935–D939. [Google Scholar] [CrossRef]
- Cheng, J.; Randall, A.; Baldi, P. Prediction of Protein Stability Changes for Single-Site Mutations Using Support Vector Machines. Proteins 2005, 62, 1125–1132. [Google Scholar] [CrossRef]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef]
- Dehouck, Y.; Kwasigroch, J.M.; Gilis, D.; Rooman, M. PoPMuSiC 2.1: A web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinforma. 2011, 12, 151. [Google Scholar] [CrossRef]
- Worth, C.L.; Preissner, R.; Blundell, T.L. SDM—A server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 2011, 39, W215–W222. [Google Scholar] [CrossRef]
- Ferrer-Costa, C.; Orozco, M.; de la Cruz, X. Sequence-based prediction of pathological mutations. Proteins 2004, 57, 811–819. [Google Scholar] [CrossRef]
- Bromberg, Y.; Tachdav, G.; Rost, B. SNAP predicts effect of mutations on protein function. Bioinformatics 2008, 24, 2397–2398. [Google Scholar] [CrossRef]
- Capriotti, E.; Calabrese, R.; Casadio, R. Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics 2006, 22, 2729–2734. [Google Scholar] [CrossRef]
- Calabrese, R.; Capriotti, E.; Fariselli, P.; Martelli, P.L.; Casadio, R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum. Mutat. 2009, 30, 1237–1244. [Google Scholar] [CrossRef]
- Tian, J.; Wu, N.; Guo, X.; Guo, J.; Zhang, J.; Fan, Y. Predicting the phenotypic effects of non-synonymous single nucleotide polymorphisms based on support vector machines. BMC Bioinforma. 2007, 8, 450–464. [Google Scholar] [CrossRef]
- Kaminker, J.S.; Zhang, Y.; Waugh, A.; Haverty, P.M.; Peters, B.; Sebisanovic, D.; Stinson, J.; Forrest, W.F.; Bazan, F.; Seshagiri, S.; et al. Distinguishing cancer-associated missense mutations from common polymorphisms. Cancer Res. 2007, 67, 465–473. [Google Scholar] [CrossRef]
- Bao, L.; Cui, Y. nsSNPAnalyzer: Identifying disease-associated nonsynonymous single nucleotide polymorphisms. Nucleic Acids Res. 2005, 33, W480–W482. [Google Scholar] [CrossRef]
- Li, B.; Krishnan, V.G.; Mort, M.E.; Xin, F.; Kamati, K.K.; Cooper, D.N.; Mooney, S.D.; Radivojac, P. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics 2009, 25, 2744–2750. [Google Scholar] [CrossRef]
- Acharya, V.; Nagarajaram, H.A. Hansa: An automated method for discriminating disease and neutral human nsSNPs. Hum. Mutat. 2012, 2, 332–337. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef]
- Pertea, M.; Lin, X.; Salzberg, S.L. GeneSplicer: A new computational method for splice site prediction. Nucleic Acids Res. 2001, 29, 1185–1190. [Google Scholar] [CrossRef]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Beroud, G.; Claustres, M.; Beroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar]
- Yeo, G.; Burge, C.B. Maximum entropy modelling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef]
- Hebsgaard, S.M.; Korning, P.G.; Tolstrup, N.; Engelbrecht, J.; Rouzé, P.; Brunak, S. Splice site prediction in Arabidopsis thaliana DNA by combining local and global sequence information. Nucleic Acids Res. 1996, 24, 3439–3452. [Google Scholar] [CrossRef]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved Splice Site Detection in Genie’. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef]
- Cartegni, L.; Wang, J.; Zhu, Z.; Zhang, M.Q.; Krainer, A.R. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003, 31, 3568–3571. [Google Scholar] [CrossRef]
- Wang, M.; Marín, A. Characterization and prediction of alternative splice sites. Gene 2006, 366, 219–227. [Google Scholar] [CrossRef]
- Coordinators, N.R. Database resources of the national Center for Biotechnology Information. Nucleic Acids Res. 2007, 35, D5–D12. [Google Scholar] [CrossRef]
- Cooper, D.N.; Stenson, P.D.; Chuzhanova, N.A. The Human Gene Mutation Database (HGMD) and its exploitation in the study of mutational mechanisms. Curr. Protocols Bioinforma 2006. [Google Scholar] [CrossRef]
- Church, D.M.; Schneider, V.A.; Graves, T.; Auger, K.; Cunningham, F.; Bouk, N.; Chen, H.C.; Agarwala, R.; McLaren, W.M.; Ritchie, G.R.; et al. Modernizing reference genome assemblies. PLoS Biol. 2011, 9, e1001091. [Google Scholar] [CrossRef]
- Kidd, J.M.; Sampas, N.; Antonacci, F.; Graves, T.; Fulton, R.; Hayden, H.S.; Alkan, C.; Malig, M.; Ventura, M.; Giannuzzi, G.; et al. Characterization of missing human genome sequences and copy-number polymorphic insertions. Nat. Methods 2010, 7, 365–371. [Google Scholar] [CrossRef]
- Gargis, A.S.; Kalman, L.; Berry, M.W.; Bick, D.P.; Dimmock, D.P.; Hambuch, T.; Lu, F.; Lyon, E.; Voelkerding, K.V.; Zehnbauer, B.A.; et al. Assuring the quality of next-generation sequencing in clinical laboratory practice. Nat. Biotechnol. 2012, 30, 1033–1036. [Google Scholar] [CrossRef]
- Wong, L.-J. Challenges of bridging next generation sequencing technologies to clinical molecular diagnostics laboratories. Neurotherapeutics 2013, 10, 262–272. [Google Scholar]
- Bagnall, R.D.; Das, K.J.; Duflou, J.; Semsarian, C. Exome analysis-based molecular autopsy in cases of sudden unexplained death in the young. Heart Rhythm 2014, 11, 655–662. [Google Scholar] [CrossRef]
- Loporcaro, C.G.; Tester, D.J.; Maleszewski, J.J.; Kruisselbrink, T.; Ackerman, M.J. Confirmation of cause and manner of death via a comprehensive cardiac autopsy including whole exome next-generation sequencing. Arch. Pathol. Lab. Med. 2013. [Google Scholar] [CrossRef]
- Clarke, L.; Zheng-Bradley, X.; Smith, R.J.; Kulesha, E.; Xiao, C.; Toneva, I.; Vaughan, B.; Preuss, D.; Leinonen, R.; Shumway, M.; et al. The 1000 Genomes Project: Data management and community access. Nat. Methods 2012, 9, 459–462. [Google Scholar] [CrossRef]
- Exome Variant Server, NHLBI Exome Sequencing Project (ESP). Available online: http://evs.gs.washington.edu/EVS/ (accessed on 30 January 2014).
- Exome Chip Design. Available online: http://genome.sph.umich.edu/wiki/Exome_Chip_Design (accessed on 30 January 2014).
- Lakdawala, N.K.; Winterfield, J.R.; Funke, B.H. Arrhythmogenic disorders of genetic origin. Circ. Arrhythmia Electrophysiol. 2013, 6, 228–237. [Google Scholar]
- Berne, P. Brugada syndrome 2012. Circulation 2012, 76, 1563–1571. [Google Scholar] [CrossRef]
- Giudicessi, J.R.; Ackerman, M.J. Genotype- and phenotype-guided management of congenital long QT syndrome. Curr. Probl. Cardiol. 2013, 38, 417–455. [Google Scholar] [CrossRef]
- Perrin, M.J.; Gollob, M.H. Genetics of cardiac electrical disease. Can. J. Cardiol. 2013, 29, 89–99. [Google Scholar] [CrossRef]
- Napolitano, C.; Priori, S.G.; Bloise, R. Catecholaminergic polymorphic ventricular tachycardia. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1289/ (accessed on 28 January 2014).
- Elliott, P.; Baker, R.; Pasquale, F.; Quarta, G.; Ebrahim, H.; Mehta, A.B.; Hughes, D.A.; ACES study group. Prevalence of Anderson-Fabry disease in patients with hypertrophic cardiomyopathy: The European Anderson-Fabry Disease survey. Heart 2011, 97, 1957–1960. [Google Scholar] [CrossRef]
- Havndrup, O.; Christiansen, M.; Stoevring, B.; Jensen, M.; Hoffman-Bang, J.; Andersen, P.S.; Hasholt, L.; Nørremølle, A.; Feldt-Rasmussen, U.; Køber, L.; et al. Fabry disease mimicking hypertrophic cardiomyopathy: Genetic screening needed for establishing the diagnosis in women. Eur. Heart J. 2010, 12, 535–540. [Google Scholar] [CrossRef]
- Giudicessi, J.R.; Ackerman, M.J. Prevalence and potential genetic determinants of sensorineural deafness in KCNQ1 homozygosity and compound heterozygosity. Circ. Cardiovasc. Genet. 2013, 6, 193–200. [Google Scholar] [CrossRef]
- Keren, A.; Syrris, P.; McKenna, W.J. Hypertrophic cardiomyopathy: The genetic determinants of clinical disease expression. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 158–168. [Google Scholar] [CrossRef]
- Van Driest, S.L.; Ommen, S.R.; Tajik, A.J.; Gersh, B.J.; Ackerman, M.J. Sarcomeric genotyping in hypertrophic cardiomyopathy. Mayo Clin. Proc. 2005, 80, 463–469. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Morales, A.; Siegfried, J.D. Clinical and genetic issues in dilated cardiomyopathy: A review for genetics professionals. Genet. Med. 2010, 12, 655–667. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Norton, N.; Morales, A.; Li, D.; Siegfried, J.D.; Gonzalez-Quintana, J. Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circ. Cardiovasc. Gene. 2010, 3, 155–161. [Google Scholar]
- Tian, T.; Liu, Y.; Zhou, X.; Song, L. Progress in the molecular genetics of hypertrophic cardiomyopathy: A mini-review. Gerontology 2012, 59, 199–205. [Google Scholar]
- Møller, D.V.; Andersen, P.S.; Hedley, P.; Ersbøll, M.K.; Bundgaard, H.; Moolman-Smook, J.; Christiansen, M.; Køber, L. The role of sarcomere gene mutations in patients with idiopathic dilated cardiomyopathy. Eur. J. Hum. Gene. 2009, 17, 1241–1249. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Leong, I.U.S.; Skinner, J.R.; Love, D.R. Application of Massively Parallel Sequencing in the Clinical Diagnostic Testing of Inherited Cardiac Conditions. Med. Sci. 2014, 2, 98-126. https://doi.org/10.3390/medsci2020098
Leong IUS, Skinner JR, Love DR. Application of Massively Parallel Sequencing in the Clinical Diagnostic Testing of Inherited Cardiac Conditions. Medical Sciences. 2014; 2(2):98-126. https://doi.org/10.3390/medsci2020098
Chicago/Turabian StyleLeong, Ivone U. S., Jonathan R. Skinner, and Donald R. Love. 2014. "Application of Massively Parallel Sequencing in the Clinical Diagnostic Testing of Inherited Cardiac Conditions" Medical Sciences 2, no. 2: 98-126. https://doi.org/10.3390/medsci2020098
APA StyleLeong, I. U. S., Skinner, J. R., & Love, D. R. (2014). Application of Massively Parallel Sequencing in the Clinical Diagnostic Testing of Inherited Cardiac Conditions. Medical Sciences, 2(2), 98-126. https://doi.org/10.3390/medsci2020098