Microbial Diversity in Sub-Seafloor Sediments from the Costa Rica Margin

 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction
2.3. 16S rRNA PCR and Amplicon Sequencing
2.4. 16S rRNA Amplicon Data Analysis
2.5. Metagenomic Sequencing and Analysis
2.6. Correlations
2.7. Accession Numbers
3. Results and Discussion
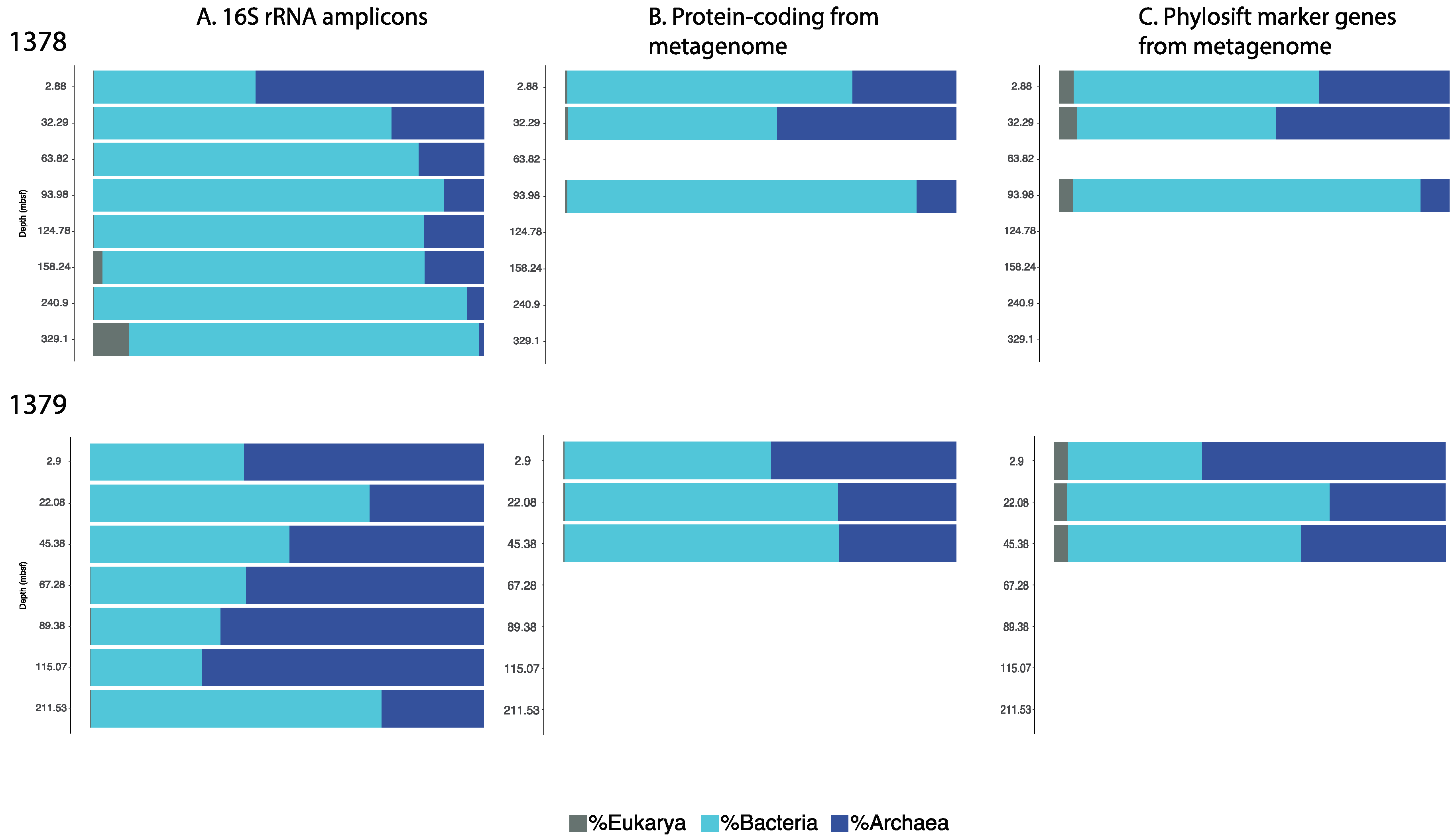
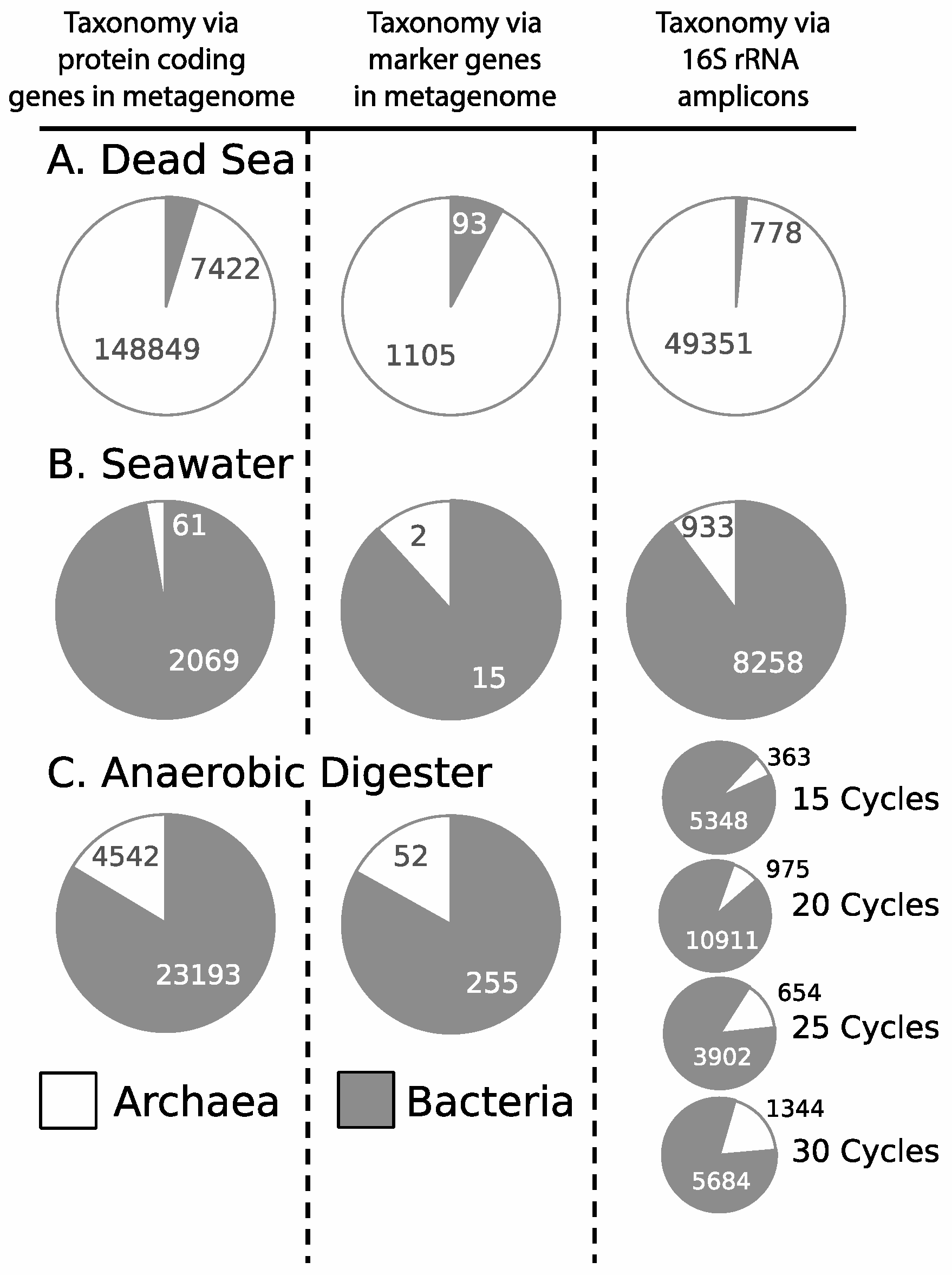
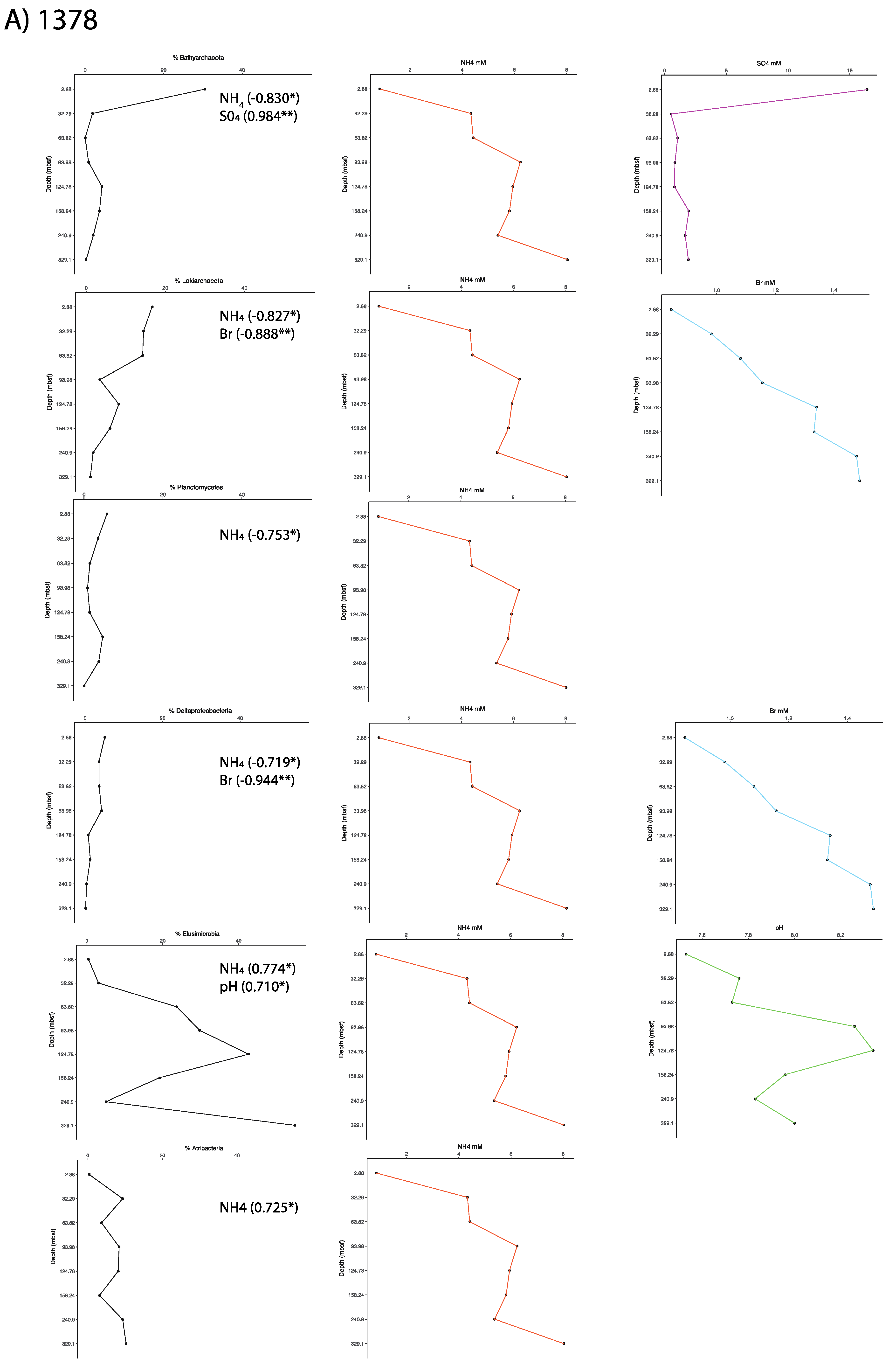
3.1. Microbial Community Analysis Reveals High Proportion Archaea
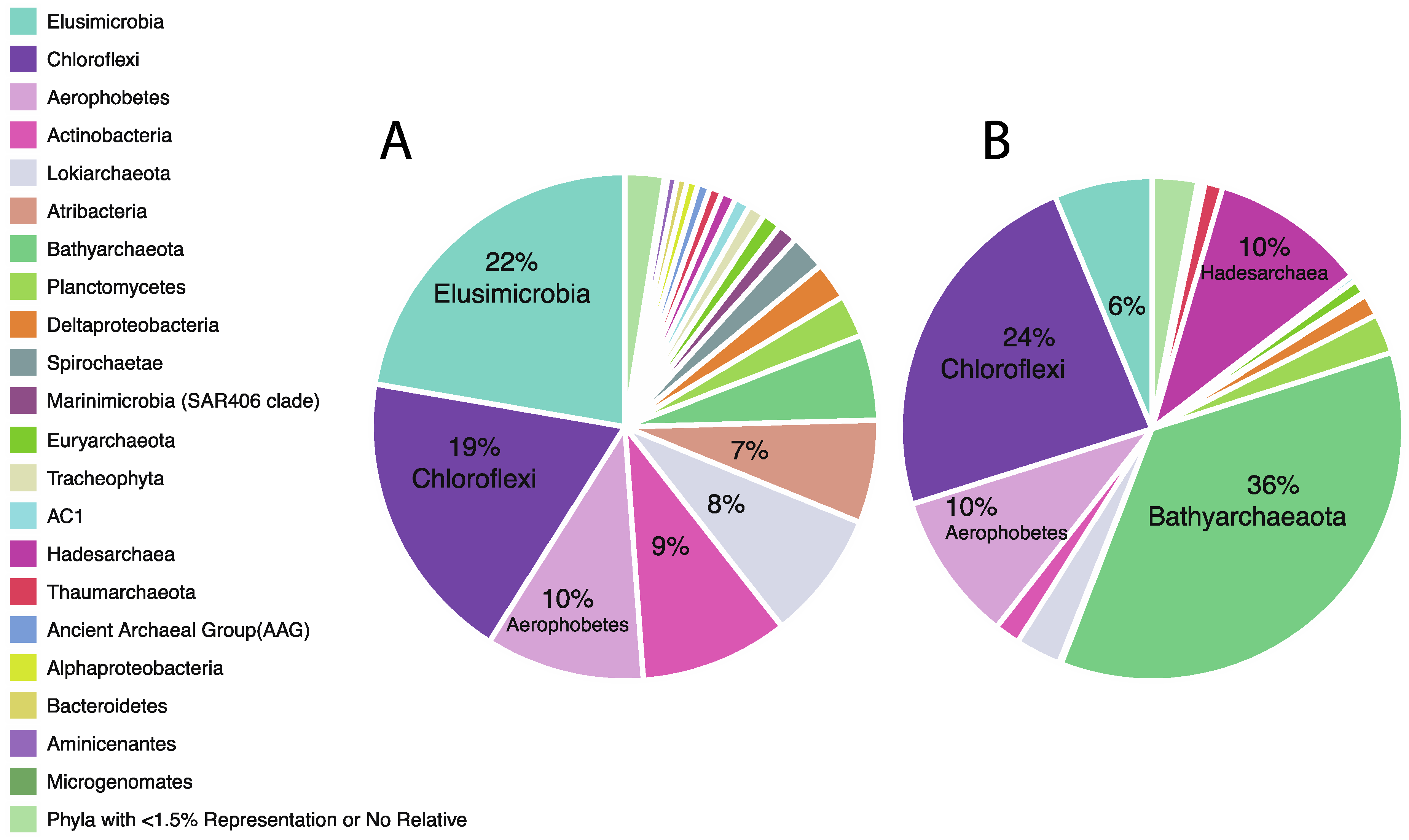
3.2. Taxonomic Analysis Beyond the Domain Level
3.3. Influence of Drilling Fluid
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- D’Hondt, S.; Jorgensen, B.B.; Miller, D.J.; Batzke, A.; Blake, R.; Cragg, B.A.; Cypionka, H.; Dickens, G.R.; Ferdelan, T.; Hinrichs, K.U.; et al. Distributions of microbial activities in deep subseafloor sediments. Science 2004, 306, 2216–2221. [Google Scholar] [CrossRef] [PubMed]
- Ciobanu, M.C.; Burgaud, G.; Dufresne, A.; Breuker, A.; Redou, V.; Maamar, S.B.; Gaboyer, F.; Vandenabeele-Trambouze, O.; Lipp, J.S.; Schippers, A.; et al. Microorganims persist at record depths in the subseafloor of the Canterbury Basin. ISMEJ 2014, 8, 1370–1380. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, F.; Nunoura, T.; Nakagawa, S.; Teske, A.; Lever, M.; Lauer, A.; Suzuki, M.; Takai, K.; Delwiche, M.; Colwell, F.S.; et al. Biogeographical distribution and diversity of microbes in methane hydrate-bearing deep marine sediments on the Pacific Ocean Margin. Proc. Natl. Acad. Sci. USA 2006, 103, 2815–2820. [Google Scholar] [CrossRef]
- Bidle, K.A.; Kastner, M.; Bartlett, D.H. A phylogenetic analysis of microbial communities associated with methane hydrate containing marine fluids and sediments in the Cascadia Margin (ODP site 892B). FEMS Microbiol. Lett. 1999, 177, 101–108. [Google Scholar]
- Vannucchi, P.; Ujiie, K.; Stroncik, N.; the Expedition 334 Scientists. Proceedings of the Integrated Ocean Drilling Program Volume 334. Available online: http://publications.iodp.org/proceedings/334/334title.htm (accessed on 31 March 2019).
- Barry, P.H.; de Moor, J.M.; Giovanelli, D.; Schrenk, M.; Hummer, D.R.; Lopez, T.; Pratt, C.A.; Alpizar Segura, Y.; Battaglia, A.; Beaudry, P.; et al. Forearc carbon sink reduces long-term volatile recycling into the mantle. Nature 2019, 568, 487–492. [Google Scholar] [CrossRef]
- Schippers, A.; Neretin, L.N.; Kallmeyer, J.; Ferdelman, T.G.; Cragg, B.A.; Parkes, R.J.; Jørgensen, B.B. Prokaryotic cells of the deep sub-seafloor biosphere identified as living bacteria. Nature 2005, 433, 861–864. [Google Scholar] [CrossRef] [PubMed]
- Briggs, B.R.; Inagaki, F.; Morono, Y.; Futagami, T.; Huguet, C.; Rosell-Mele, A.; Lorenson, T.D.; Colwell, F.S. Bacterial dominance in subseafloor sediments characterized by methane hydrates. FEMS Microbiol. Ecol. 2012, 81, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Biddle, J.F.; Fitz-Gibbon, S.; Schuster, S.C.; Brenchley, J.E.; House, C.H. Metagenomic signatures of the Peru Margin subseafloor biosphere show a genetically distinct environment. Proc. Natl. Acad. Sci. USA 2008, 105, 10583–10588. [Google Scholar] [CrossRef] [PubMed]
- Lipp, J.S.; Morono, Y.; Inagaki, F.; Hinrichs, K.-U. Significant contribution of Archaea to extant biomass in marine subsurface sediments. Nature 2008, 454, 991–994. [Google Scholar]
- Smith, D.C.; Spivack, A.J.; Fisk, M.R.; Haveman, S.A.; Staudigel, H.; ODP Leg 185 Shipboard Scientific Party. Methods for Quantifying Potential Microbial Contamination During Deep Ocean Coring. 2000. Available online: http://www-odp.tamu.edu/publications/tnotes/tn28/INDEX.HTM (accessed on 31 March 2019).
- Smith, D.C.; Spivack, A.J.; Fisk, M.R.; Haveman, S.A.; Staudigel, H.; ODP Leg 185 Shipboard Scientific Party. Tracer-Based Estimates of Drilling-Induced Microbial Contamination of Deep Sea Crust. Geomicrobiol. J. 2000, 17, 207–219. [Google Scholar]
- House, C.H.; Cragg, B.A.; Teske, A. Drilling contamination tests on ODP Leg 201 using chemical and particulate tracers. In Proceedings of the Ocean Drilling Program; Texas A&M University: College Station, TX, USA, 2003; pp. 1–19. [Google Scholar]
- Masui, N.; Morono, Y.; Inagaki, F. Microbiological assessment of circulation mud fluids during the first operation of riser drilling by the deep-earth research vessel Chikyu. Geomicrobiol. J. 2008, 25, 274–282. [Google Scholar] [CrossRef]
- Santelli, C.M.; Banerjee, N.; Bach, W.; Edwards, K.J. Tapping the subsurface ocean crust biosphere: Low biomass and drilling-related contamination calls for improved quality controls. Geomicrobiol. J. 2010, 27, 158–169. [Google Scholar] [CrossRef]
- Fry, J.C.; Parkes, R.J.; Cragg, B.C.; Weightman, A.J.; Webster, G. Prokaryotic biodiversity and activity in the deep subseafloor biosphere. FEMS Microbiol. Ecol. 2008, 66, 181–196. [Google Scholar] [CrossRef] [PubMed]
- Orcutt, B.N.; Sylvan, J.B.; Knab, N.J.; Edwards, K.J. Microbial ecology of the dark ocean above, at, and below the seafloor. Microbiol. Mol. Biol. Rev. 2011, 75, 361–422. [Google Scholar] [CrossRef]
- Inagaki, F.; Hinrichs, K.-U.; Kubo, Y.; Bowles, M.W.; Heuer, V.B.; Hong, W.-L.; Hoshino, T.; Ijiri, A.; Imachi, H.; Ito, M.; et al. Exploring deep microbial life in coal-bearing sediment down to ~2.5 km below the ocean floor. Science 2015, 349, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Yanagawa, K.; Nunoura, T.; McAllister, S.M.; Kirai, M.; Breuker, A.; Brandt, L.; House, C.H.; Moyer, C.L.; Birrien, J.L.; Aoike, K.; et al. The first microbiological contamination assessment by deep-sea drilling and coring by the D/V Chikyu at the Iheya North hydrothermal field in the Mid-Okinawa Trough (IODP Expedition 331). Frontiers Microbiol. 2013, 4, 327. [Google Scholar] [CrossRef] [PubMed]
- Stavenhagen, A.U.; Flueh, E.R.; Ranero, C.R.; McIntosh, K.D.; Shipley, T.; Leandro, G.; Schulze, A.; Dañobeitia, J.J. Seismic wide-angle investigations in Costa Rica—A crustal velocity model from the Pacific to the Caribbean. ZBL. Geol. Paläontol. 1998, 1, 393–408. [Google Scholar]
- Rhodes, M.E.; Fitz-Gibbon, S.T.; Oren, A.; House, C.H. Amino acid signatures of salinity on an environmental scale with a focus on the Dead Sea. Environ. Microbiol. 2010, 12, 2613–2623. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef]
- Pruesse, E.; Peplies, J.; Glöckner, F.O. SINA: Accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 2012, 28, 1823–1829. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Silva. Available online: http://www.arb-silva.de (accessed on 31 March 2019).
- BLAST: Basic Local Alignment Search Tool. Available online: http://blast.ncbi.blm.nih.gov/Blast.cgi (accessed on 31 March 2019).
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nature Commun. 2016, 7, 11257. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.E.; Jospin, G.; Lowe, E.; Matsen, F.A.; Bik, H.M.; Eisen, J.A. PhyloSift: Phylogenetic analysis of genomes and metagenomes. PeerJ 2014, 9, 2:e243. [Google Scholar] [CrossRef]
- Berg, R.; Solomon, E.A. Geochemical constraints on the distribution and rates of debromination in the deep subseafloor biosphere. Geochim. Cosmochim. Acta 2016, 174, 30–41. [Google Scholar] [CrossRef]
- Torres, M.E.; Muratli, J.M.; Solomon, E.A. Data Report: Minor Element Concentrations in Pore Fluids from the CRISP-A Transect Drilled During Expedition 334. Available online: http://publications.iodp.org/proceedings/334/201/201_.htm (accessed on 31 March 2019).
- Rhodes, M.E.; Oren, A.; House, C.H. Dynamics and persistence of Dead Sea microbial populations as shown by high-throughput sequencing of rRNA. Appl. Environ. Microbiol. 2012, 78, 2489–2492. [Google Scholar] [CrossRef]
- Zhou, Z.; Pan, J.; Wang, F.; Gu, J.D.; Li, M. Bathyarchaeota: Globally distributed metabolic generalists in anoxic environments. FEMS Microbiol. Lett. 2018, 42, 639–655. [Google Scholar] [CrossRef]
- Geissinger, O.; Herlemann, D.P.R.; Morschel, E.; Maier, U.G.; Brune, A. The ultramicrobacterium “Elusimicrobium minutum” gen. nov., sp. nov., the first cultivated representative of the termite group 1 phylum. Appl. Env. Microbiol. 2009, 75, 2831–2840. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Native Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Rinke, C.; Schwientek, P.; Sczyrba, A.; Ivanova, N.N.; Anderson, I.J.; Cheng, J.-F.; Darling, A.; Malfatti, S.; Swan, B.K.; Gies, E.A.; et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature 2013, 499, 431–437. [Google Scholar] [CrossRef]
- Lazar, C.S.; Biddle, J.F.; Meador, T.B.; Blair, N.; Hinrichs, K.-U.; Teske, A.P. Environmental controls on intragroup diversity of the uncultured benthic archaea of the miscellaneous Crenarchaeotal group lineage naturally enriched in anoxic sediments of the White Oak River estuary (North Caroline, USA). Env. Microbiol. 2015, 17, 2228–2238. [Google Scholar] [CrossRef] [PubMed]
- Futagami, T.; Morono, Y.; Terada, T.; Kaksonen, A.H.; Inagaki, F. Dehalogenation activities and distribution of reductive dehalogenase homologous genes in marine subsurface sediments. Appl. Environ. Microbiol. 2009, 75, 6905–6909. [Google Scholar] [CrossRef] [PubMed]
- Futagami, T.; Morono, Y.; Terada, T.; Kaksonen, A.H.; Inagaki, F. Distribution of dehalogenation activity in subseafloor sediments of the Nankai Trough subduction zone. Philos. Trans. R Soc. Lond. B Biol. Sci. 2013, 368, 20120249. [Google Scholar] [CrossRef] [PubMed]
- Kawai, M.; Futagami, T.; Toyoda, A.; Takaki, Y.; Nishi, S.; Hori, S.; Arai, W.; Tsubouchi, T.; Morono, Y.; Uchiyama, I.; et al. High frequency of phylogenetically diverse reductive dehalogenase-homologous genes in deep subseafloor sedimentary metagenomes. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
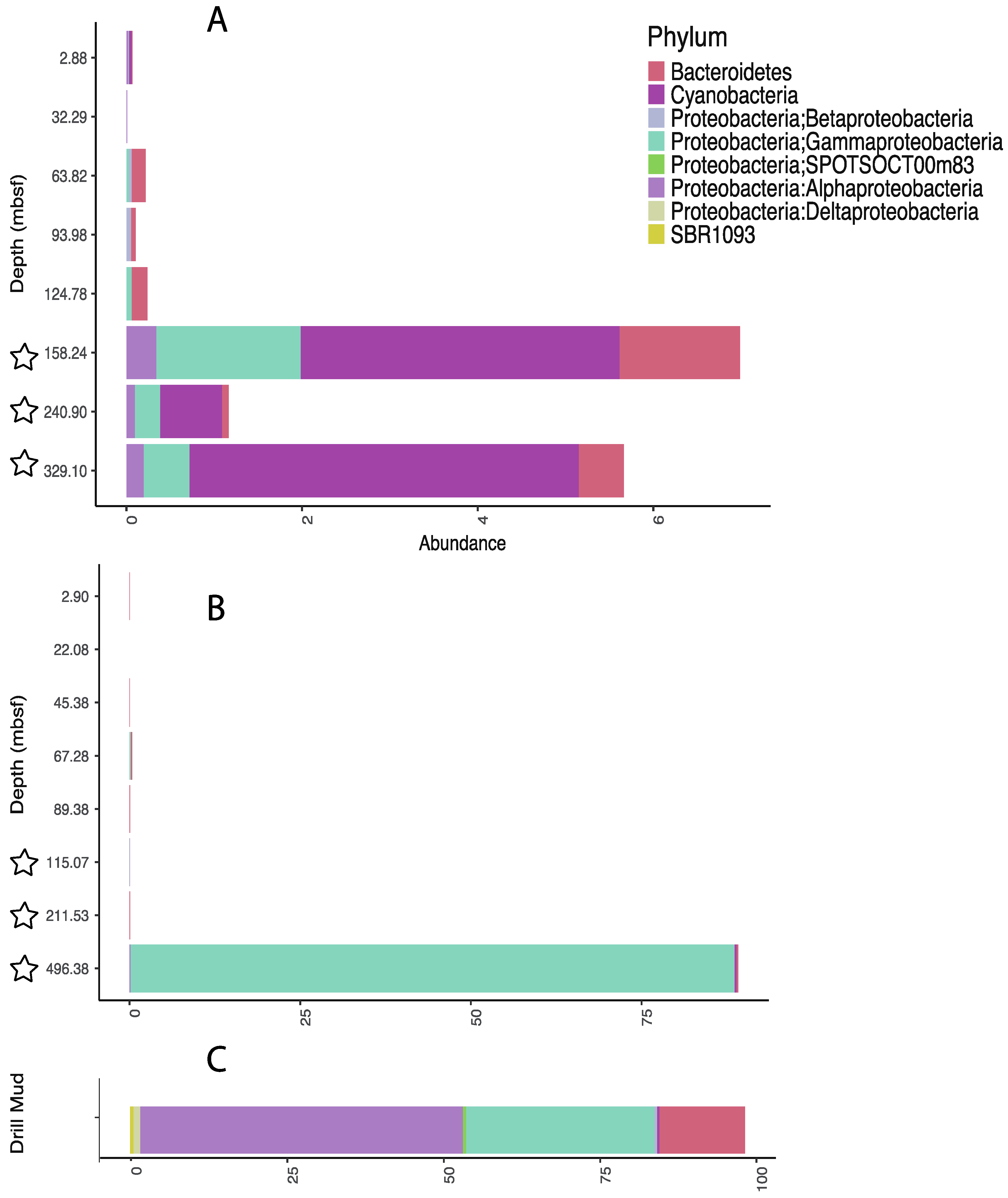{kind=link}
| 1378 | 1379 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample Depth (mbsf): | 2.88 | 32.29 | 63.82 | 93.98 | 124.78 | 158.24 | 240.90 | 329.10 | 2.90 | 22.08 | 45.38 | 67.28 | 89.38 | 115.07 | 211.53 |
| Archaea: | |||||||||||||||
| Bathyarchaeota | 30.54 | 1.88 | 0.00 | 0.88 | 4.29 | 3.66 | 2.08 | 0.24 | 34.81 | 18.21 | 28.57 | 50.86 | 51.59 | 46.30 | 20.04 |
| Lokiarchaeota | 16.65 | 14.46 | 14.29 | 3.46 | 8.22 | 6.00 | 1.76 | 1.05 | 7.51 | 3.81 | 5.27 | 0.46 | 0.04 | 0.01 | 3.39 |
| Thaumarchaeota | 4.00 | 0.91 | 0.48 | 0.21 | 0.57 | 0.33 | 0.02 | 0.00 | 1.77 | 1.74 | 1.03 | 1.59 | 1.14 | 0.69 | 0.14 |
| Euryarchaeota | 1.94 | 0.78 | 0.00 | 4.64 | 1.18 | 1.01 | 0.03 | 0.00 | 2.54 | 1.04 | 2.23 | 0.32 | 0.37 | 0.01 | 0.00 |
| Hadesarchaea | 1.34 | 1.14 | 0.12 | 0.00 | 0.52 | 4.16 | 0.16 | 0.00 | 11.30 | 3.69 | 11.56 | 5.53 | 12.84 | 24.23 | 1.22 |
| Ancient Archaeal Group (AAG) | 0.04 | 4.23 | 1.71 | 0.00 | 0.27 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Bacteria: | |||||||||||||||
| Chloroflexi | 20.61 | 27.29 | 13.19 | 19.83 | 14.52 | 25.30 | 23.64 | 5.52 | 22.18 | 47.48 | 22.30 | 28.00 | 16.93 | 13.09 | 14.71 |
| Planctomycetes | 5.83 | 3.53 | 1.48 | 0.89 | 1.43 | 4.71 | 3.74 | 0.00 | 6.00 | 2.96 | 2.30 | 1.87 | 1.36 | 1.40 | 2.05 |
| Deltaproteobacteria | 5.07 | 3.56 | 3.59 | 4.25 | 0.82 | 1.32 | 0.38 | 0.12 | 2.53 | 2.69 | 1.61 | 0.61 | 0.09 | 0.06 | 2.31 |
| Aminicenantes | 2.09 | 0.77 | 1.61 | 0.10 | 0.00 | 0.26 | 0.01 | 0.00 | 0.34 | 0.23 | 0.37 | 0.50 | 0.19 | 0.13 | 0.10 |
| Microgenomates | 1.63 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 1.01 | 0.00 | 0.00 | 0.11 | 0.00 | 0.00 | 0.00 |
| Aerophobetes | 1.32 | 10.35 | 14.72 | 20.05 | 10.66 | 19.08 | 4.46 | 0.38 | 2.40 | 4.18 | 5.06 | 2.56 | 8.68 | 7.14 | 36.96 |
| AC1 | 0.68 | 2.13 | 2.24 | 1.75 | 0.85 | 0.26 | 0.06 | 0.00 | 0.14 | 0.35 | 0.75 | 0.92 | 0.13 | 0.03 | 0.39 |
| Spirochaetae | 0.64 | 0.61 | 2.37 | 0.90 | 1.20 | 0.61 | 0.44 | 11.56 | 0.23 | 0.20 | 0.27 | 0.00 | 0.00 | 0.00 | 0.00 |
| Elusimicrobia | 0.40 | 3.07 | 23.65 | 29.72 | 42.60 | 19.16 | 5.03 | 54.81 | 1.96 | 1.95 | 14.66 | 1.75 | 2.89 | 4.99 | 15.98 |
| Atribacteria | 0.37 | 9.34 | 3.65 | 8.39 | 8.12 | 3.10 | 9.35 | 10.22 | 0.08 | 0.28 | 0.07 | 0.12 | 0.04 | 0.03 | 0.53 |
| Marinimicrobia (SAR406 clade) | 0.32 | 2.17 | 2.70 | 0.58 | 0.38 | 0.91 | 1.58 | 1.61 | 0.13 | 0.08 | 0.32 | 0.08 | 0.03 | 0.05 | 0.00 |
| Actinobacteria | 0.23 | 10.17 | 12.53 | 0.51 | 2.17 | 4.94 | 44.58 | 0.51 | 0.20 | 7.85 | 1.44 | 0.40 | 0.59 | 0.93 | 0.00 |
| Bacteroidetes | 0.05 | 1.60 | 1.09 | 0.12 | 0.34 | 0.23 | 0.71 | 0.93 | 0.05 | 0.02 | 0.08 | 0.02 | 0.00 | 0.02 | 0.06 |
| Alphaproteobacteria | 0.00 | 0.00 | 0.00 | 0.06 | 0.06 | 1.97 | 0.57 | 3.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.01 | 0.09 | 0.00 |
| Eukaryota: | |||||||||||||||
| Tracheophyta | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 8.61 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Phyla with <1.5% Representation or No Relative | 6.25 | 1.98 | 0.58 | 3.69 | 1.79 | 2.98 | 1.42 | 1.44 | 4.81 | 3.23 | 2.12 | 4.31 | 3.08 | 0.79 | 2.11 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martino, A.; Rhodes, M.E.; León-Zayas, R.; Valente, I.E.; Biddle, J.F.; House, C.H. Microbial Diversity in Sub-Seafloor Sediments from the Costa Rica Margin. Geosciences 2019, 9, 218. https://doi.org/10.3390/geosciences9050218
Martino A, Rhodes ME, León-Zayas R, Valente IE, Biddle JF, House CH. Microbial Diversity in Sub-Seafloor Sediments from the Costa Rica Margin. Geosciences. 2019; 9(5):218. https://doi.org/10.3390/geosciences9050218
Chicago/Turabian StyleMartino, Amanda, Matthew E. Rhodes, Rosa León-Zayas, Isabella E. Valente, Jennifer F. Biddle, and Christopher H. House. 2019. "Microbial Diversity in Sub-Seafloor Sediments from the Costa Rica Margin" Geosciences 9, no. 5: 218. https://doi.org/10.3390/geosciences9050218
APA StyleMartino, A., Rhodes, M. E., León-Zayas, R., Valente, I. E., Biddle, J. F., & House, C. H. (2019). Microbial Diversity in Sub-Seafloor Sediments from the Costa Rica Margin. Geosciences, 9(5), 218. https://doi.org/10.3390/geosciences9050218