
Monitoring the Organic Matter Quality Highlights the Ways in Which Organic Matter Is Removed from Wetland Soil

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Soil Sampling and Soil Characteristics
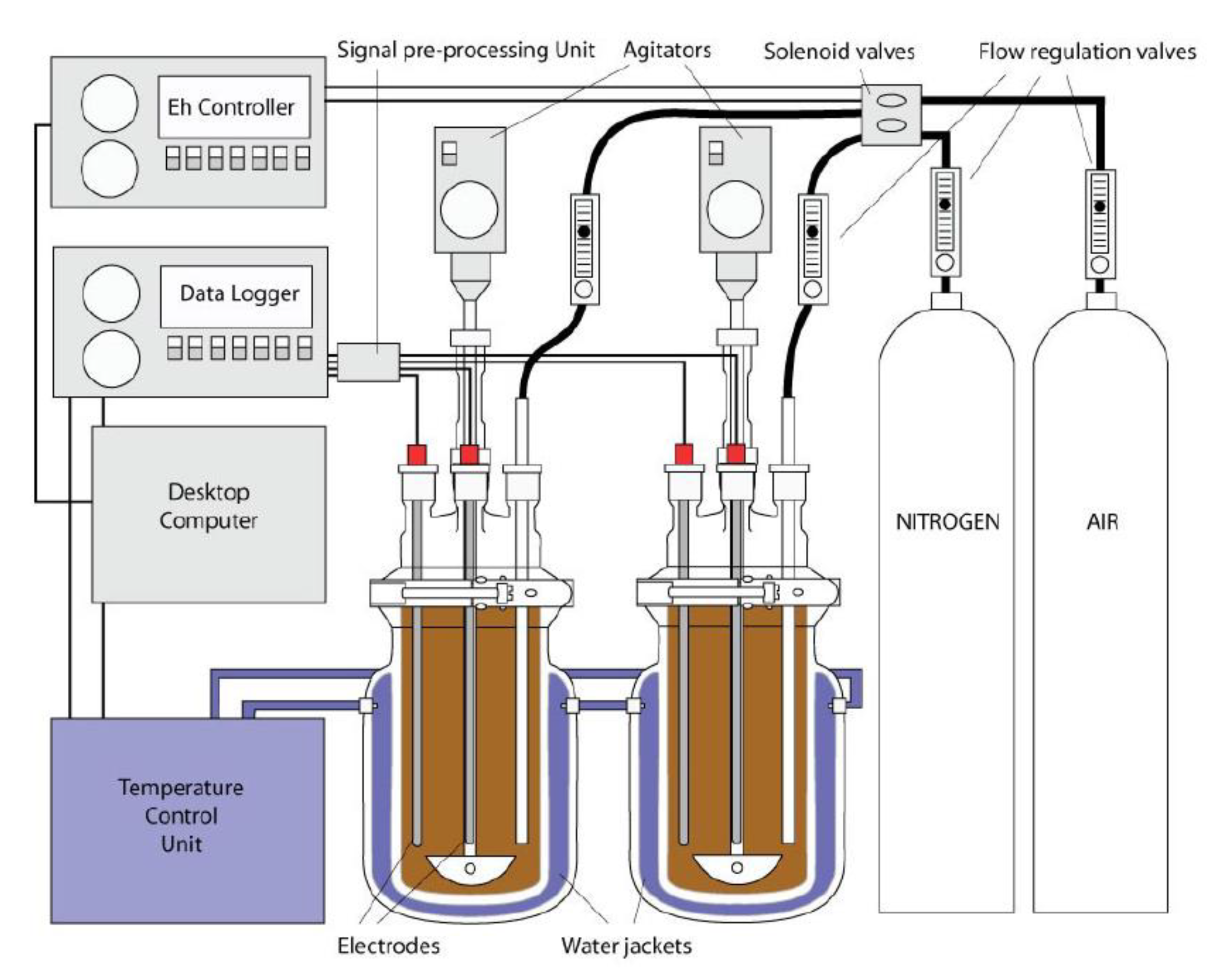
2.2. Experimental Setup
2.3. Analytical Procedures
2.3.1. Elemental Concentrations
2.3.2. Stable Carbon and Nitrogen Isotopic Compositions
2.3.3. Spectroscopic Analyses
2.4. Speciation Modeling
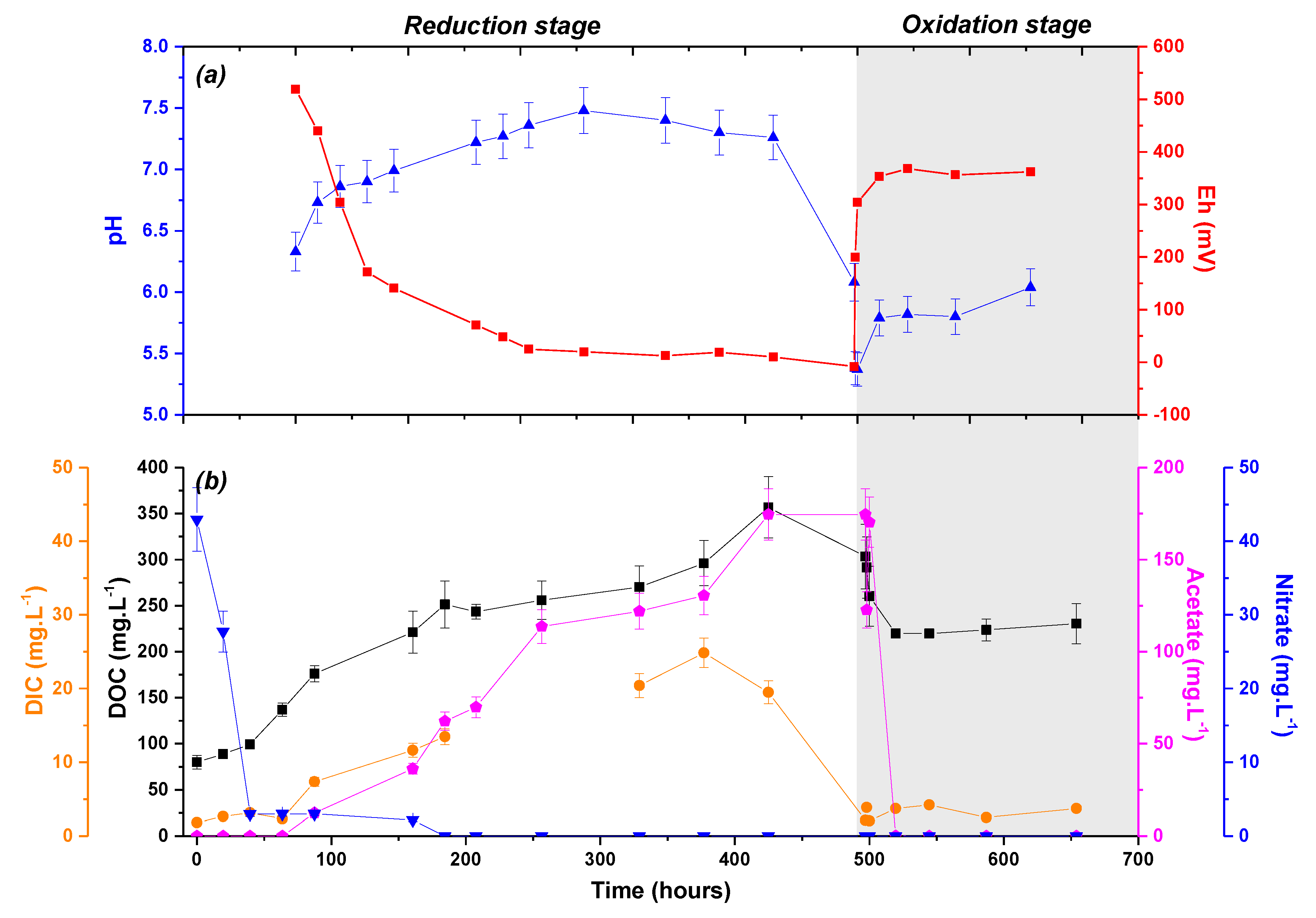
3. Results
3.1. Evolution of the Soil Solution Chemistry
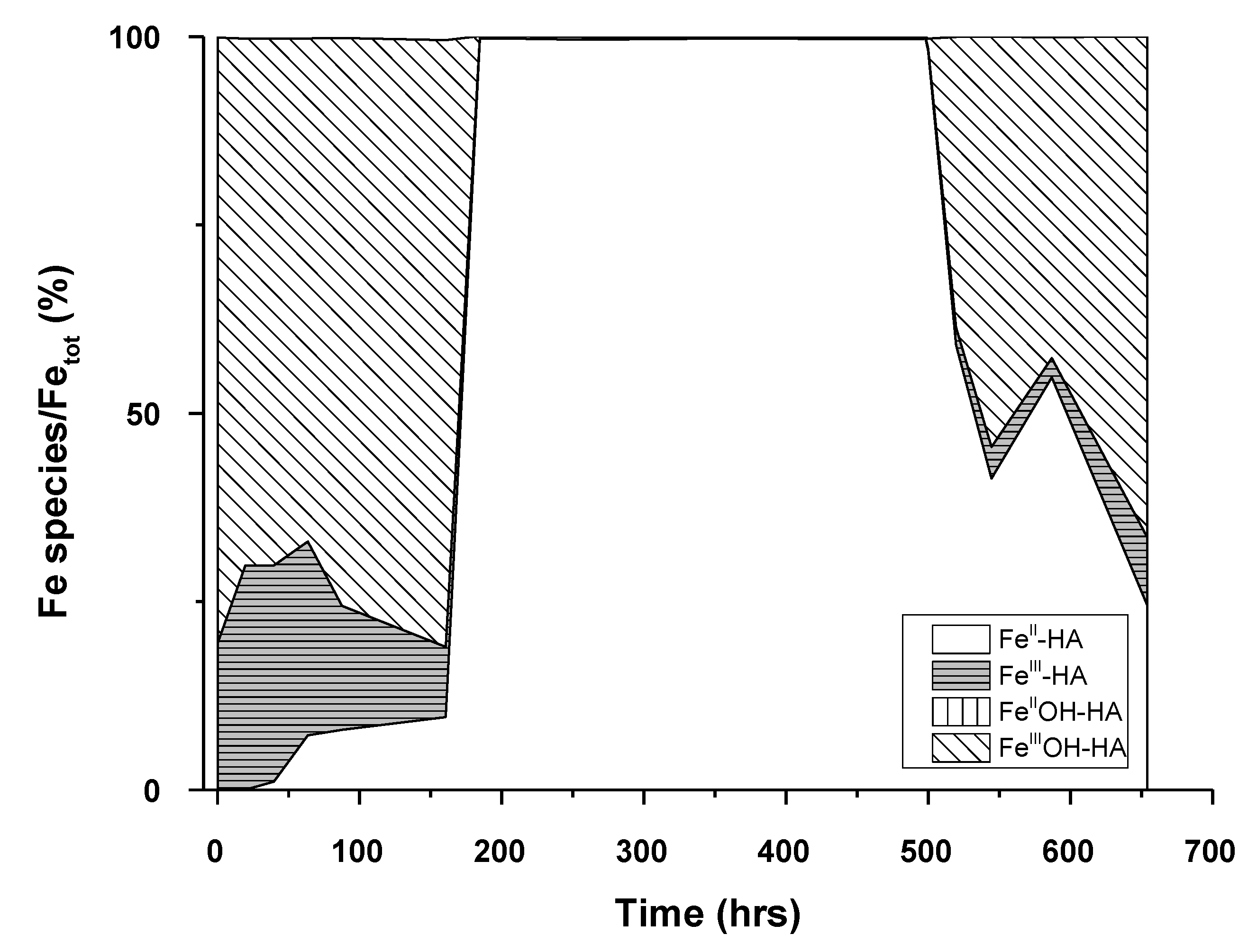
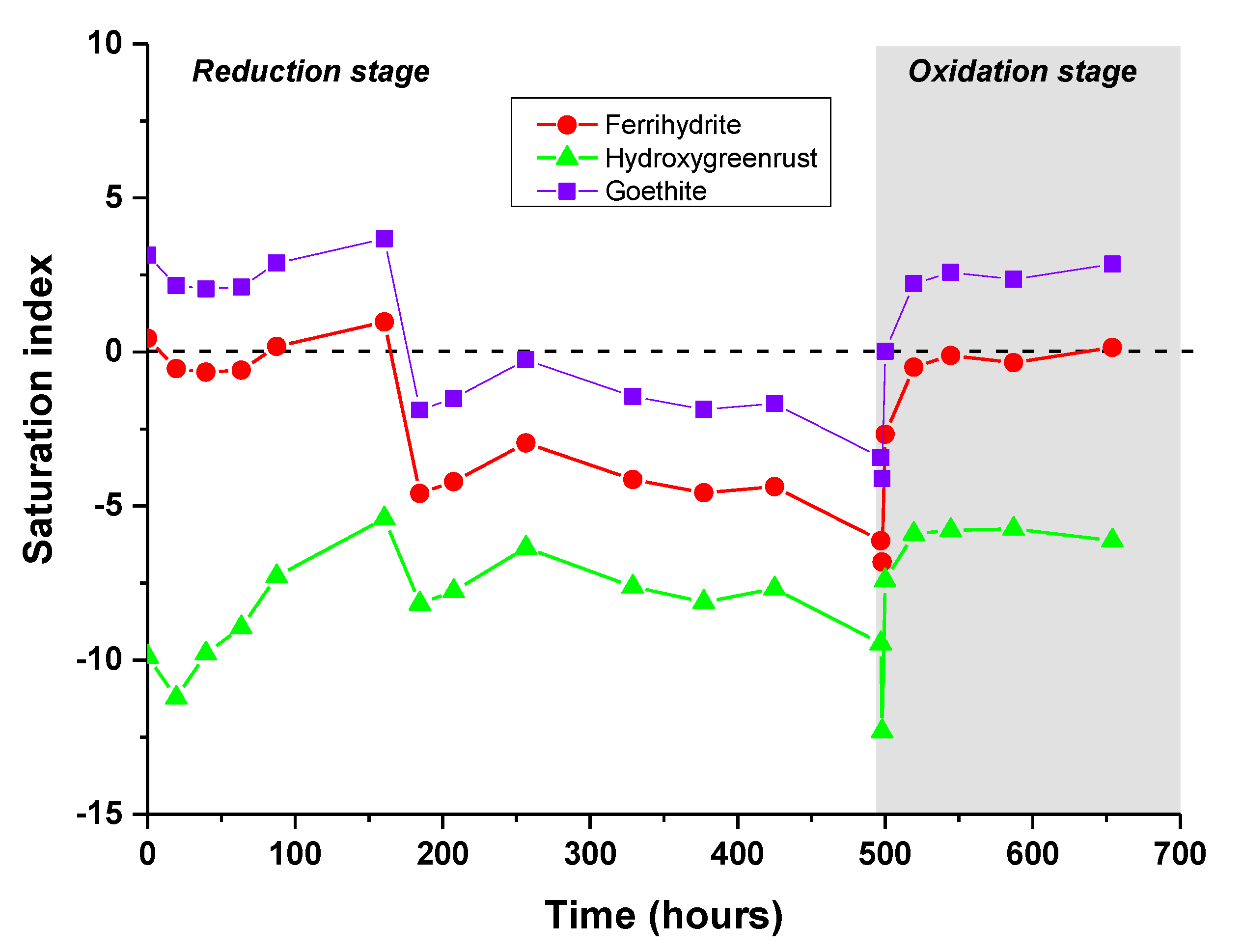
3.2. PHREEQC-Model VI Modeling
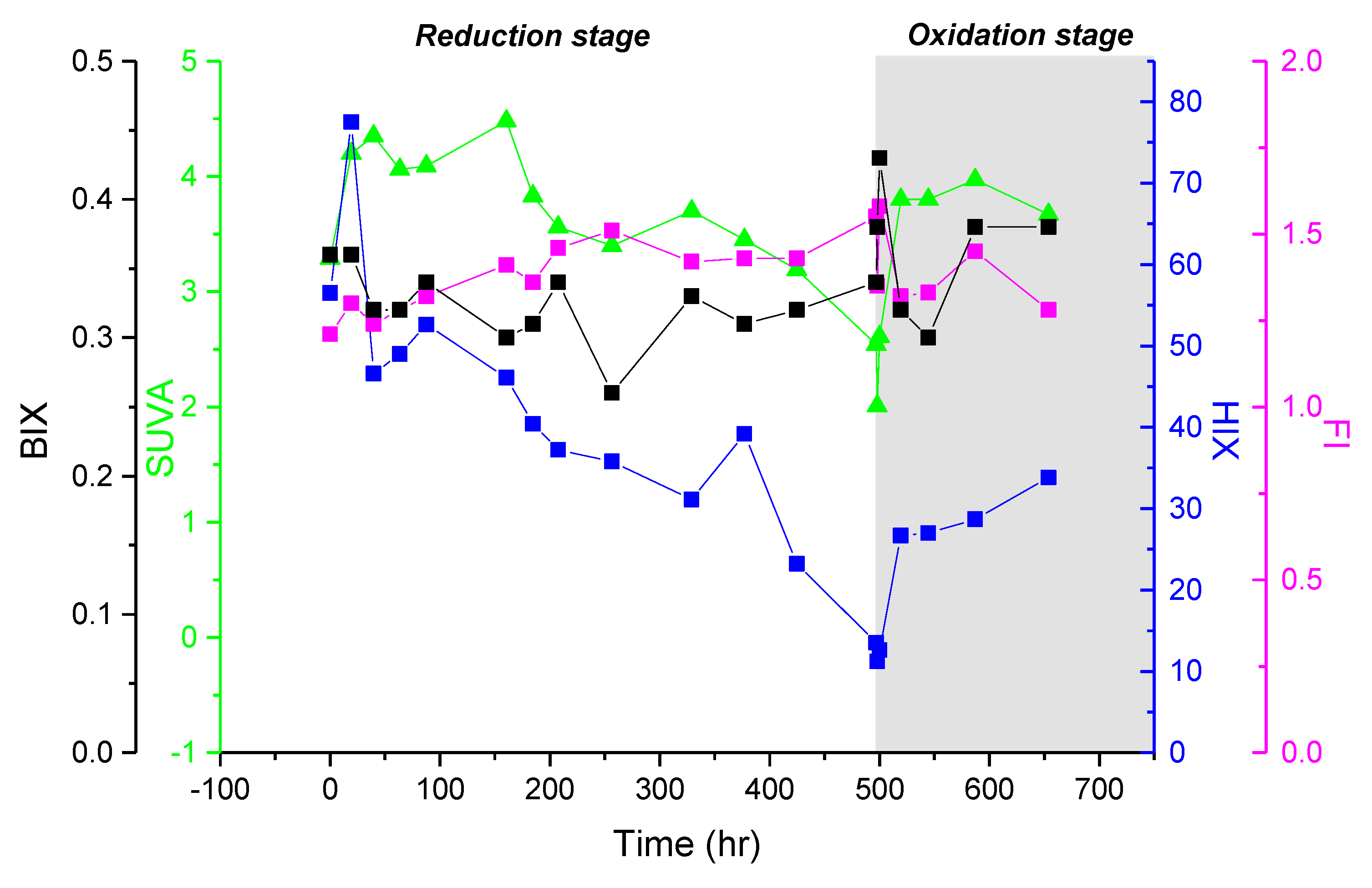
3.3. Spectroscopic Properties of Bulk DOM
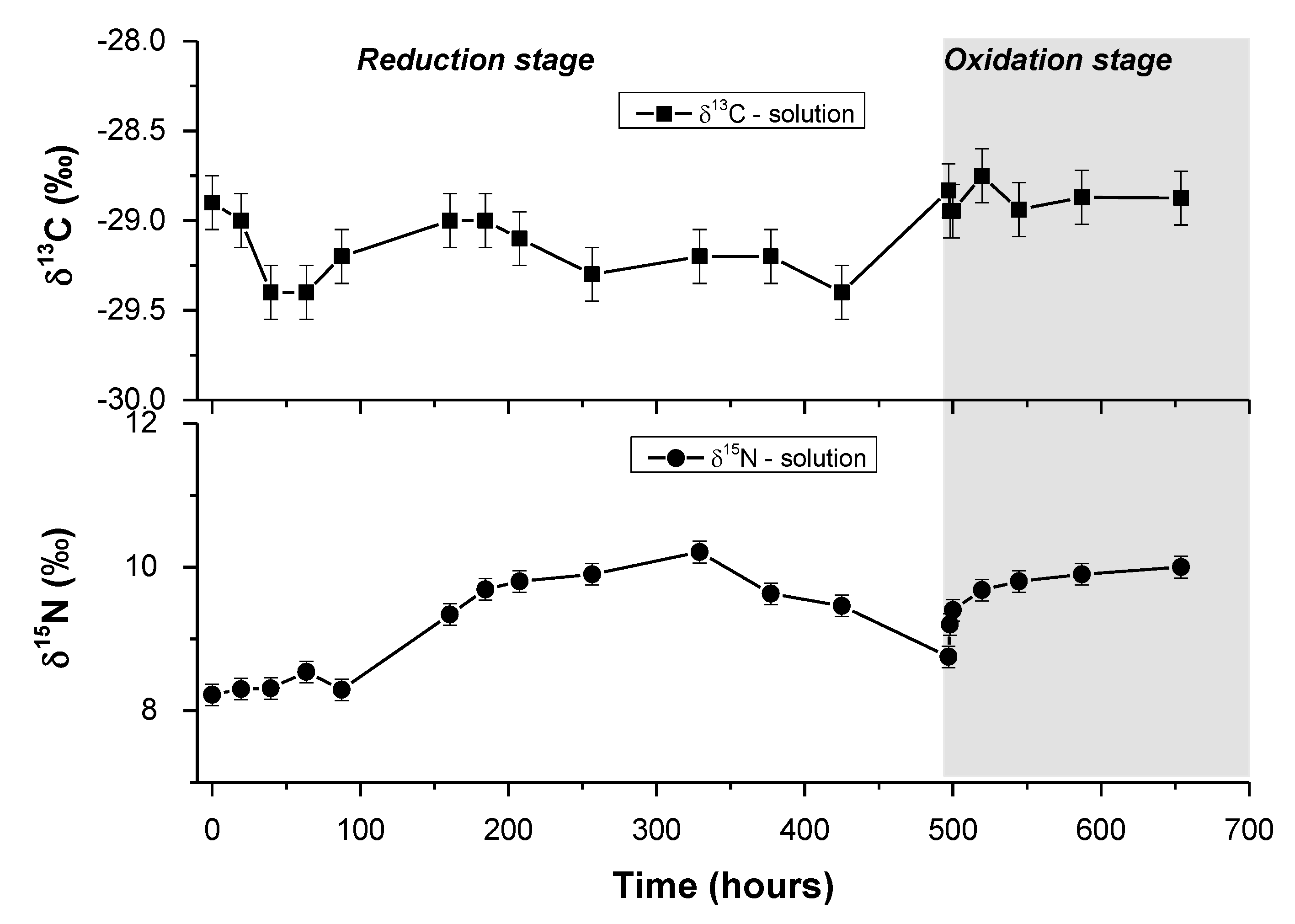
3.4. Carbon Isotopic Composition of DOC during the Experiment
4. Discussion
4.1. Evolution of the DOM Quality during the Redox Cycle
4.2. Impact on Soil OM Degradation and Preservation
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SiO2 (wt. %) | Al2O3 (wt. %) | Fe2O3 (wt. %) | MnO (wt. %) | MgO (wt. %) | CaO (wt. %) | Na2O (wt. %) | K2O (wt. %) | TiO2 (wt. %) | P2O5 (wt. %) | PF (wt. %) | Total | Organic Carbon (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 58.39 | 11.51 | 2.85 | 0.01 | 0.44 | 0.25 | 0.37 | 1.98 | 0.71 | 0.12 | 22.56 | 99.19 | 6.64 |
| As (ppm) | Cd (ppm) | Co (ppm) | Cr (ppm) | Cu (ppm) | Ni (ppm) | Pb (ppm) | Zn (ppm) | U (ppm) | - | - | - | - |
| 7.64 | 7.65 | 4.80 | 74.88 | 10.51 | 21.26 | 25.13 | 40.96 | 2.34 | - | - | - | - |
References
- Torn, M.S.; Trumbore, S.E.; Chadwick, O.A.; Vitousek, P.M.; Hendricks, D.M. Mineral control of soil organic carbon storage and turnover. Nature 1997, 389, 170–173. [Google Scholar] [CrossRef]
- Masiello, C.A.; Chadwick, O.A.; Southon, J.; Torn, M.S.; Harden, J.W. Weathering controls on mechanisms of carbon storage in grassland soils. Glob. Biogeochem. Cycles 2004, 18, 1–9. [Google Scholar] [CrossRef]
- Kramer, M.G.; Chadwick, O.A. Climate-driven thresholds in reactive mineral retention of soil carbon at the global scale. Nat. Clim. Chang. 2018, 8, 1104–1108. [Google Scholar] [CrossRef]
- Guénet, H.; Davranche, M.; Vantelon, D.; Pédrot, M.; Al-Sid-Cheikh, M.; Dia, A.; Jestin, J. Evidence of organic matter control on As oxidation by iron oxides in riparian wetlands. Chem. Geol. 2016, 439, 161–172. [Google Scholar] [CrossRef]
- Herndon, E.; Al Bashaireh, A.; Singer, D.; Chowdhury, T.R.; Gu, B.; Graham, D. Influence of iron redox cycling on organo-mineral associations in Arctic tundra soil. Geochim. Cosmochim. Acta 2017, 207, 210–231. [Google Scholar] [CrossRef]
- Hall, S.J.; Berhe, A.A.; Thompson, A. Order from disorder: Do soil organic matter composition and turnover co-vary with iron phase crystallinity? Biogeochemistry 2018, 140, 93–110. [Google Scholar] [CrossRef]
- Chen, C.; Hall, S.J.; Coward, E.; Thompson, A. Iron-mediated organic matter decomposition in humid soils can counteract protection. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Henneberry, Y.K.; Kraus, T.E.C.; Nico, P.S.; Horwath, W.R. Structural stability of coprecipitated natural organic matter and ferric iron under reducing conditions. Org. Geochem. 2012, 48, 81–89. [Google Scholar] [CrossRef]
- Lambert, T.; Pierson-Wickmann, A.C.; Gruau, G.; Jaffrezic, A.; Petitjean, P.; Thibault, J.N.; Jeanneau, L. DOC sources and DOC transport pathways in a small headwater catchment as revealed by carbon isotope fluctuation during storm events. Biogeosciences 2014, 11, 3043–3056. [Google Scholar] [CrossRef]
- Riedel, T.; Zak, D.; Biester, H.; Dittmar, T. Iron traps terrestrially derived dissolved organic matter at redox interfaces. Proc. Natl. Acad. Sci. USA 2013, 110, 10101–10105. [Google Scholar] [CrossRef]
- Curmi, P.; Durand, P.; Gascuel-Odoux, C.; Merot, P.; Walter, C.; Taha, A. Hydromorphic soils, hydrology and water quality: Spatial distribution and functional modelling at different scales. Nutr. Cycl. Agroecosys. 1998, 50, 127–142. [Google Scholar] [CrossRef]
- Grybos, M.; Davranche, M.; Gruau, G.; Petitjean, P. Is trace metal release in wetland soils controlled by organic matter mobility or Fe-oxyhydroxides reduction? J. Colloid Interface Sci. 2007, 314, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Lambert, T.; Pierson-Wickmann, A.C.; Gruau, G.; Jaffrezic, A.; Petitjean, P.; Thibault, J.N.; Jeanneau, L. Hydrologically driven seasonal changes in the sources and production mechanisms of dissolved organic carbon in a small lowland catchment. Water Resour. Res. 2013, 49, 5792–5803. [Google Scholar] [CrossRef]
- Merot, P.; Durand, P.; Morisson, C. Four-component hydrograph separation using isotopic and chemical determinations in an agricultural catchment in western France. Phys. Chem. Earth 1995, 20, 415–425. [Google Scholar] [CrossRef]
- Al-Sid-Cheikh, M.; Pédrot, M.; Dia, A.; Guenet, H.; Vantelon, D.; Davranche, M.; Gruau, G.; Delhaye, T. Interactions between natural organic matter, sulfur, arsenic and iron oxides in re-oxidation compounds within riparian wetlands: NanoSIMS and X-ray adsorption spectroscopy evidences. Sci. Total Environ. 2015, 515–516, 118–128. [Google Scholar] [CrossRef]
- Dia, A.; Gruau, G.; Olivié-Lauquet, G.; Riou, C.; Molénat, J.; Curmi, P. The distribution of rare earth elements in groundwaters: Assessing the role of source-rock composition, redox changes and colloidal particles. Geochim. Cosmochim. Acta 2000, 64, 4131–4148. [Google Scholar] [CrossRef]
- Gruau, G.; Dia, A.; Olivié-Lauquet, G.; Davranche, M.; Pinay, G. Controls on the distribution of rare earth elements in shallow groundwaters. Water Res. 2004, 38, 3576–3586. [Google Scholar] [CrossRef]
- Pauwels, H.; Kloppmann, W.; Foucher, J.C.; Martelat, A.; Fritsche, V. Field tracer test for denitrification in a pyrite-bearing schist aquifer. Appl. Geochem. 1998, 13, 767–778. [Google Scholar] [CrossRef]
- AFNOR. Evaluation De La Qualité des Sols. Méthodes D’analyse Chimique; AFNOR: La Plaine Saint Denis, France, 2004; Volume 1. (In French) [Google Scholar]
- Weishaar, J.L.; Aiken, G.R.; Bergamaschi, B.A.; Fram, M.S.; Fujii, R.; Mopper, K. Evaluation of specific ultraviolet absorbance as an indicator of the chemical composition and reactivity of dissolved organic carbon. Environ. Sci. Technol. 2003, 37, 4702–4708. [Google Scholar] [CrossRef] [PubMed]
- Chin, Y.-P.; Aiken, G.; O’Loughlin, E. Molecular weight, polydispersity, and spectroscopic properties of aquatic humic substances. Environ. Sci. Technol. 1994, 28, 1853–1858. [Google Scholar] [CrossRef] [PubMed]
- Leenheer, J.A.; Croué, J.-P. Peer Reviewed: Characterizing aquatic dissolved organic matter. Environ. Sci. Technol. 2003, 37, 18A–26A. [Google Scholar] [CrossRef]
- Ohno, T. Fluorescence inner-filtering correction for determining the humification index of dissolved organic matter. Environ. Sci. Technol. 2002, 36, 742–746. [Google Scholar] [CrossRef]
- Lawaetz, A.J.; Stedmon, C.A. Fluorescence intensity calibration using the Raman scatter peak of water. Appl. Spectrosc. 2009, 63, 936–940. [Google Scholar] [CrossRef]
- Marsac, R.; Davranche, M.; Gruau, G.; Bouhnik-Le Coz, M.; Dia, A. An improved description of the interactions between rare earth elements and humic acids by modeling: PHREEQC-Model VI coupling. Geochim. Cosmochim. Acta 2011, 75, 5625–5637. [Google Scholar] [CrossRef]
- Rickard, D.; Luther, G.W. Chemistry of iron sulfides. Chem. Rev. 2007, 107, 514–562. [Google Scholar] [CrossRef] [PubMed]
- Tipping, E. Humic ion-binding model VI: An improved description of the interactions of protons and metal ions with humic substances. Aquat. Geochem. 1998, 4, 3–48. [Google Scholar] [CrossRef]
- Marsac, R.; Davranche, M.; Gruau, G.; Dia, A.; Bouhnik-Le Coz, M. Aluminium competitive effect on rare earth elements binding to humic acid. Geochim. Cosmochim. Acta 2012, 89, 1–9. [Google Scholar] [CrossRef]
- Catrouillet, C.; Davranche, M.; Dia, A.; Bouhnik-Le Coz, M.; Marsac, R.; Pourret, O.; Gruau, G. Geochemical modeling of Fe(II) binding to humic and fulvic acids. Chem. Geol. 2014, 372, 109–118. [Google Scholar] [CrossRef]
- Ponnamperuma, F.N. The Chemistry of submerged soils. Adv. Agron. 1972, 24, 29–96. [Google Scholar] [CrossRef]
- Thompson, A.; Chadwick, O.A.; Boman, S.; Chorover, J. Colloid mobilization during soil iron redox oscillations. Environ. Sci. Technol. 2006, 40, 5743–5749. [Google Scholar] [CrossRef] [PubMed]
- Lotfi-Kalahroodi, E.; Pierson-Wickmann, A.-C.A.-C.; Guénet, H.; Rouxel, O.; Ponzevera, E.; Bouhnik-Le Coz, M.; Vantelon, D.; Dia, A.; Davranche, M.; LeCoz-Bouhnik, M.; et al. Iron isotope fractionation in iron-organic matter associations: Experimental evidence using filtration and ultrafiltration. Geochim. Cosmochim. Acta 2019, 250, 98–116. [Google Scholar] [CrossRef]
- Wood, P.M. Pathways for production of Fenton’s reagent by wood-rotting fungi. FEMS Microbiol. Rev. 1994, 13, 313–320. [Google Scholar] [CrossRef]
- Hammel, K.E.; Kapich, A.N.; Jensen, K.A.; Ryan, Z.C. Reactive oxygen species as agents of wood decay by fungi. Enzyme Microb. Technol. 2002, 30, 445–453. [Google Scholar] [CrossRef]
- Hall, S.J.; Silver, W.L. Iron oxidation stimulates organic matter decomposition in humid tropical forest soils. Glob. Chang. Biol. 2013, 19, 2804–2813. [Google Scholar] [CrossRef]
- Cismasu, A.C.; Michel, F.M.; Tcaciuc, A.P.; Tyliszczak, T.; Brown, G.E. Composition et propriétés structurales des ferrihydrites naturelles. Comptes Rendus Geosci. 2011, 343, 210–218. [Google Scholar] [CrossRef]
- Guénet, H.; Davranche, M.; Vantelon, D.; Bouhnik-Le Coz, M.; Jardé, E.; Pierson-Wickmann, A.C.; Dorcet, V.; Demangeat, E.; Jestin, J. Highlighting the wide variability in arsenic speciation in wetlands: A new insight into the control of the behavior of arsenic. Geochim. Cosmochim. Acta 2017, 203, 284–302. [Google Scholar] [CrossRef]
- Keiluweit, M.; Bougoure, J.J.; Nico, P.S.; Pett-Ridge, J.; Weber, P.K.; Kleber, M. Mineral protection of soil carbon counteracted by root exudates. Nat. Clim. Chang. 2015, 5, 588–595. [Google Scholar] [CrossRef]
- Gabor, R.S.; McKnight, D.M.; Miller, M.P. Fluorescence indices and their interpretation. In Aquatic Organic Matter Fluorescence; Cambridge University Press: Cambridge, UK, 2014; pp. 303–338. [Google Scholar]
- Lambert, T.; Pierson-Wickmann, A.C.; Gruau, G.; Thibault, J.N.; Jaffrezic, A. Carbon isotopes as tracers of dissolved organic carbon sources and water pathways in headwater catchments. J. Hydrol. 2011, 402, 228–238. [Google Scholar] [CrossRef]
- Lipson, D.A.; Jha, M.; Raab, T.K.; Oechel, W.C. Reduction of iron (III) and humic substances plays a major role in anaerobic respiration in an Arctic peat soil. J. Geophys. Res. Biogeosci. 2010, 115. [Google Scholar] [CrossRef]
- Lovley, D.R.; Phillips, E.J.P. Availability of ferric iron for microbial reduction in bottom sediments of the freshwater tidal Potomac River. Appl. Environ. Microbiol. 1986, 52, 751–757. [Google Scholar] [CrossRef]
- Roden, E.E.; Wetzel, R.G. Kinetics of microbial Fe(III) oxide reduction in freshwater wetland sediments. Limnol. Oceanogr. 2002, 47, 198–211. [Google Scholar] [CrossRef]
- Davranche, M.; Dia, A.; Fakih, M.; Nowack, B.; Gruau, G.; Ona-nguema, G.; Petitjean, P.; Martin, S.; Hochreutener, R. Organic matter control on the reactivity of Fe(III)-oxyhydroxides and associated As in wetland soils: A kinetic modeling study. Chem. Geol. 2013, 335, 24–35. [Google Scholar] [CrossRef]
- Dia, A.; Lauga, B.; Davranche, M.; Fahy, A.; Duran, R.; Nowack, B.; Petitjean, P.; Henin, O.; Martin, S.; Marsac, R.; et al. Bacteria-mediated reduction of As(V)-doped lepidocrocite in a flooded soil sample. Chem. Geol. 2015, 406, 34–44. [Google Scholar] [CrossRef]
- Beauvois, A.; Vantelon, D.; Jestin, J.; Rivard, C.; Bouhnik-Le Coz, M.; Dupont, A.; Briois, V.; Bizien, T.; Sorrentino, A.; Wu, B.; et al. How does calcium drive the structural organization of iron-organic matter aggregates? A multiscale investigation. Environ. Sci. Nano 2020, 7, 2833–2849. [Google Scholar] [CrossRef]
- Macko, S.A.; Fogel, M.L.; Hare, P.E.; Hoering, T.C. Isotopic fractionation of nitrogen and carbon in the synthesis of amino acids by microorganisms. Chem. Geol. Isot. Geosci. Sect. 1987, 65, 79–92. [Google Scholar] [CrossRef]
- Lehmann, M.F.; Bernasconi, S.M.; Barbieri, A.; McKenzie, J.A. Preservation of organic matter and alteration of its carbon and nitrogen isotope composition during simulated and in situ early sedimentary diagenesis. Geochim. Cosmochim. Acta 2002, 66, 3573–3584. [Google Scholar] [CrossRef]
- Kohzu, A.; Imai, A.; Miyajima, T.; Fukushima, T.; Matsushige, K.; Komatsu, K.; Kawasaki, N.; Miura, S.; Sato, T. Direct evidence for nitrogen isotope discrimination during sedimentation and early diagenesis in Lake Kasumigaura, Japan. Org. Geochem. 2011, 42, 173–183. [Google Scholar] [CrossRef]
- Guénet, H.; Davranche, M.; Vantelon, D.; Gigault, J.; Prévost, S.; Taché, O.; Jaksch, S.; Pédrot, M.; Dorcet, V.; Boutier, A.; et al. Characterization of iron-organic matter nano-aggregate networks through a combination of SAXS/SANS and XAS analyses: Impact on As binding. Environ. Sci. Nano 2017, 4, 938–954. [Google Scholar] [CrossRef]
- Adhikari, D.; Yang, Y. Selective stabilization of aliphatic organic carbon by iron oxide. Sci. Rep. 2015, 5, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mikutta, R.; Kleber, M.; Torn, M.S.; Jahn, R. Stabilization of soil organic matter: Association with minerals or chemical recalcitrance? Biogeochemistry 2006, 77, 25–56. [Google Scholar] [CrossRef]
- Kögel-Knabner, I.; Guggenberger, G.; Kleber, M.; Kandeler, E.; Kalbitz, K.; Scheu, S.; Eusterhues, K.; Leinweber, P. Organo-mineral associations in temperate soils: Integrating biology, mineralogy, and organic matter chemistry. J. Plant Nutr. Soil Sci. 2008, 171, 61–82. [Google Scholar] [CrossRef]
- Eusterhues, K.; Neidhardt, J.; Hädrich, A.; Küsel, K.; Totsche, K.U. Biodegradation of ferrihydrite-associated organic matter. Biogeochemistry 2014, 119, 45–50. [Google Scholar] [CrossRef]
- Newcomb, C.J.; Qafoku, N.P.; Grate, J.W.; Bailey, V.L.; De Yoreo, J.J. Developing a molecular picture of soil organic matter–mineral interactions by quantifying organo–mineral binding. Nat. Commun. 2017, 8, 396. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pierson-Wickmann, A.-C.; Davranche, M.; Kerloc'h, J.; Catrouillet, C.; Lotfi-Kalahroodi, E. Monitoring the Organic Matter Quality Highlights the Ways in Which Organic Matter Is Removed from Wetland Soil. Geosciences 2021, 11, 134. https://doi.org/10.3390/geosciences11030134
Pierson-Wickmann A-C, Davranche M, Kerloc'h J, Catrouillet C, Lotfi-Kalahroodi E. Monitoring the Organic Matter Quality Highlights the Ways in Which Organic Matter Is Removed from Wetland Soil. Geosciences. 2021; 11(3):134. https://doi.org/10.3390/geosciences11030134
Chicago/Turabian StylePierson-Wickmann, Anne-Catherine, Mélanie Davranche, Julien Kerloc'h, Charlotte Catrouillet, and Elaheh Lotfi-Kalahroodi. 2021. "Monitoring the Organic Matter Quality Highlights the Ways in Which Organic Matter Is Removed from Wetland Soil" Geosciences 11, no. 3: 134. https://doi.org/10.3390/geosciences11030134
APA StylePierson-Wickmann, A.-C., Davranche, M., Kerloc'h, J., Catrouillet, C., & Lotfi-Kalahroodi, E. (2021). Monitoring the Organic Matter Quality Highlights the Ways in Which Organic Matter Is Removed from Wetland Soil. Geosciences, 11(3), 134. https://doi.org/10.3390/geosciences11030134