Evaluation of Linkage Disequilibrium, Effective Population Size and Haplotype Block Structure in Chinese Cattle

Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Animal Samples and Quality Control
2.2. Linkage Disequilibrium
2.3. Haplotype Blocks
2.4. Persistence of Phase Across Groups
2.5. Ancestral Effective Population Size
3. Results
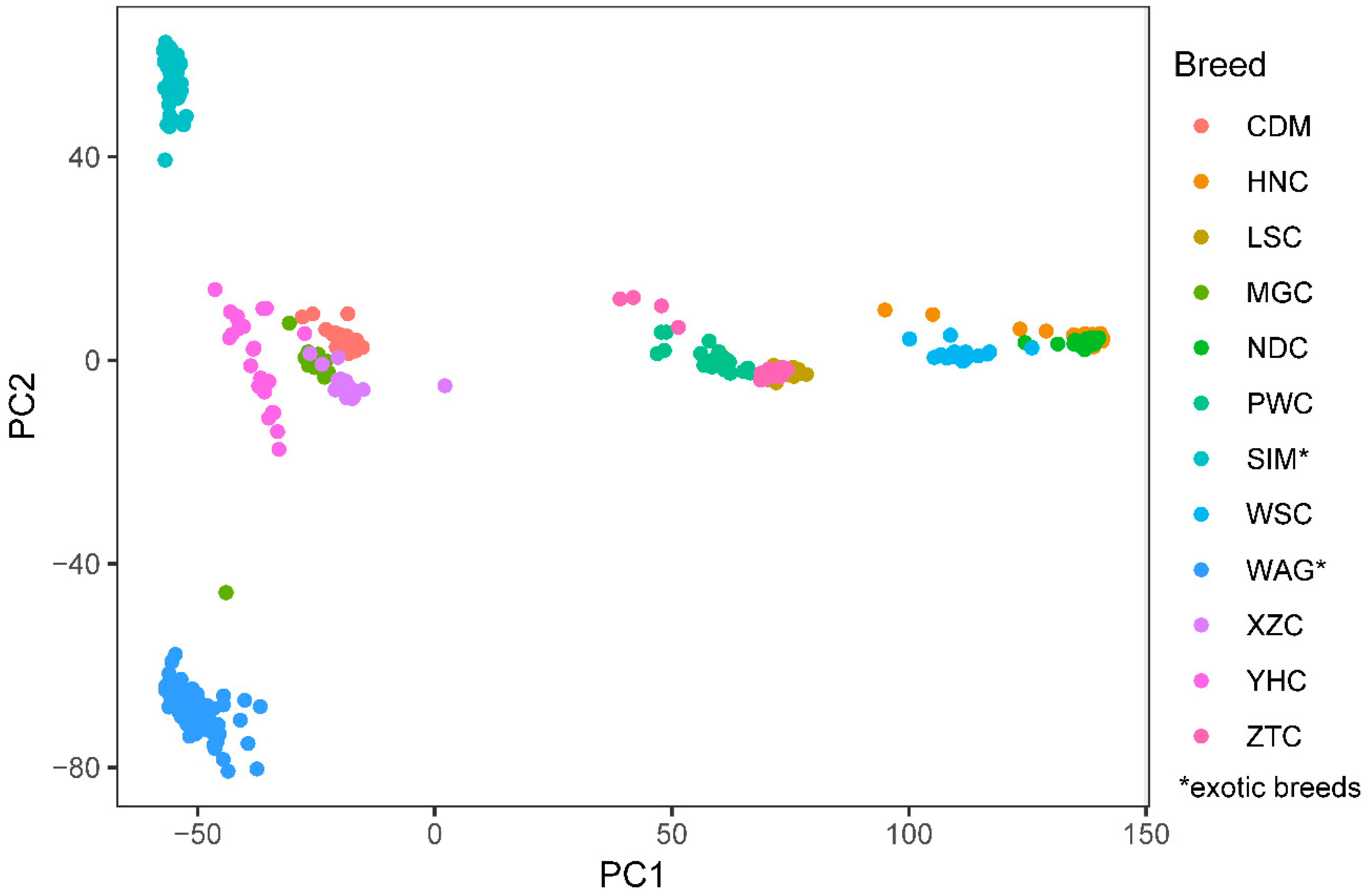
3.1. Population Structure and SNP Distribution
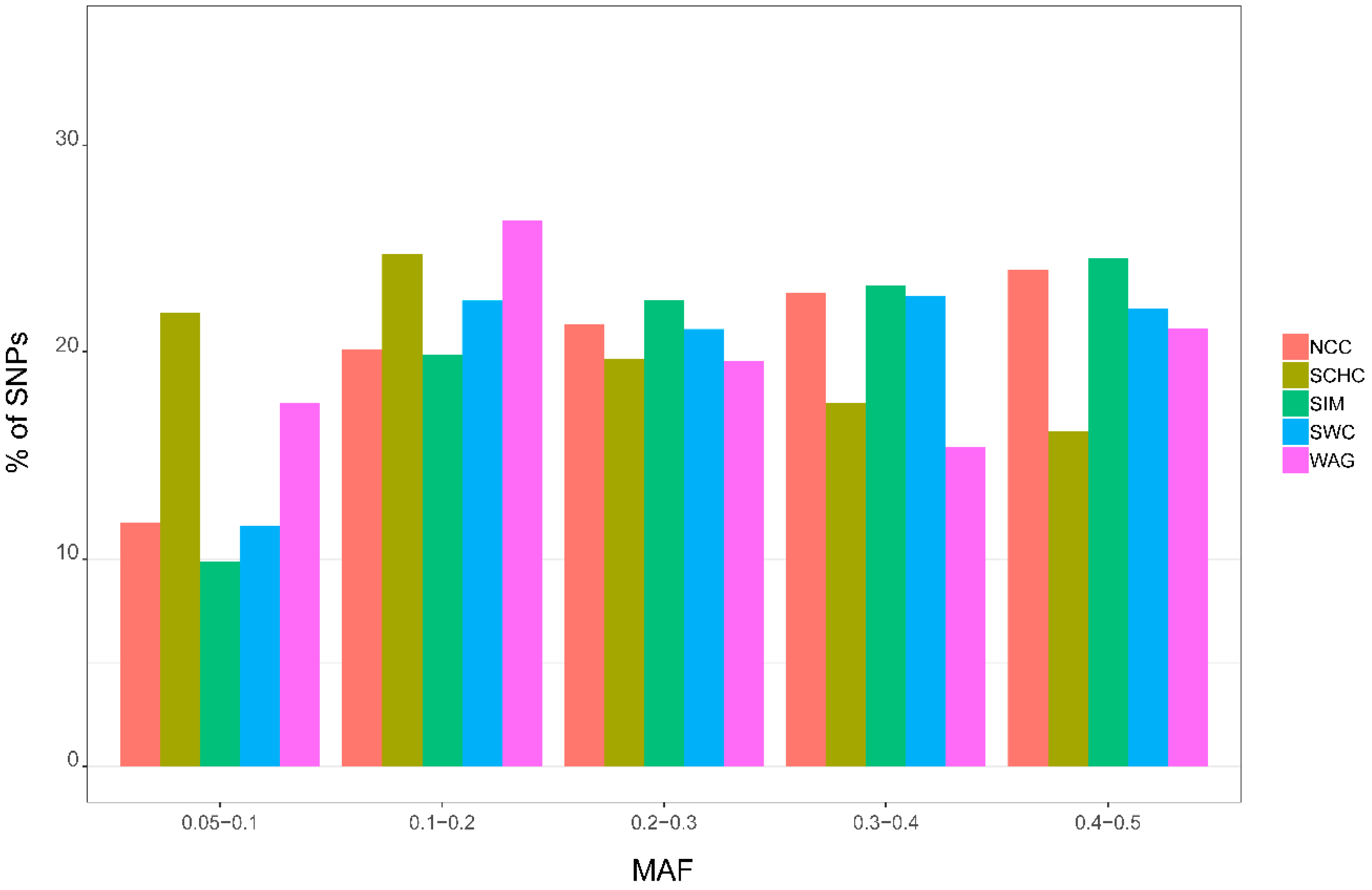
3.2. Minor Allele Frequency
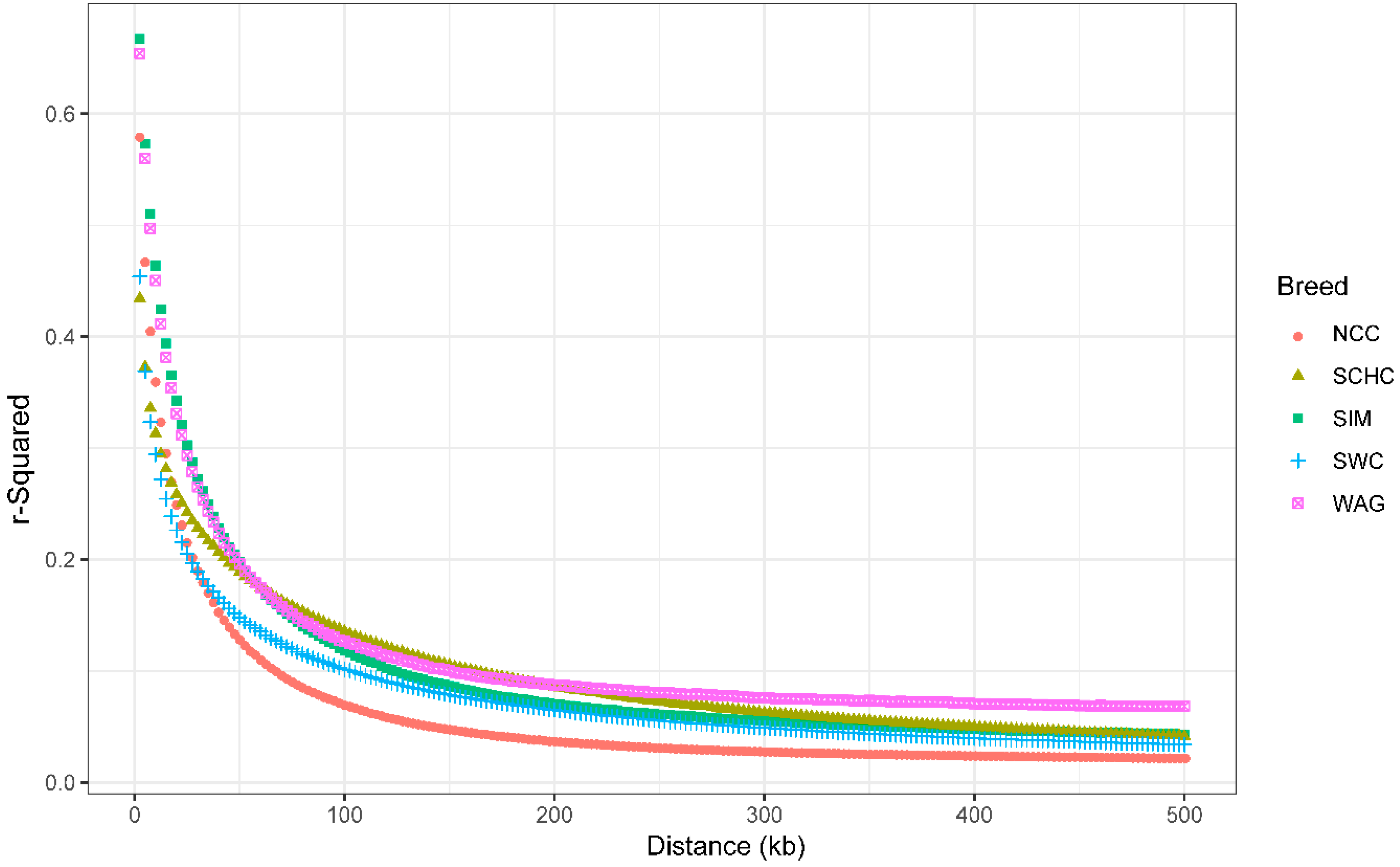
3.3. Extent of Linkage Disequilibrium
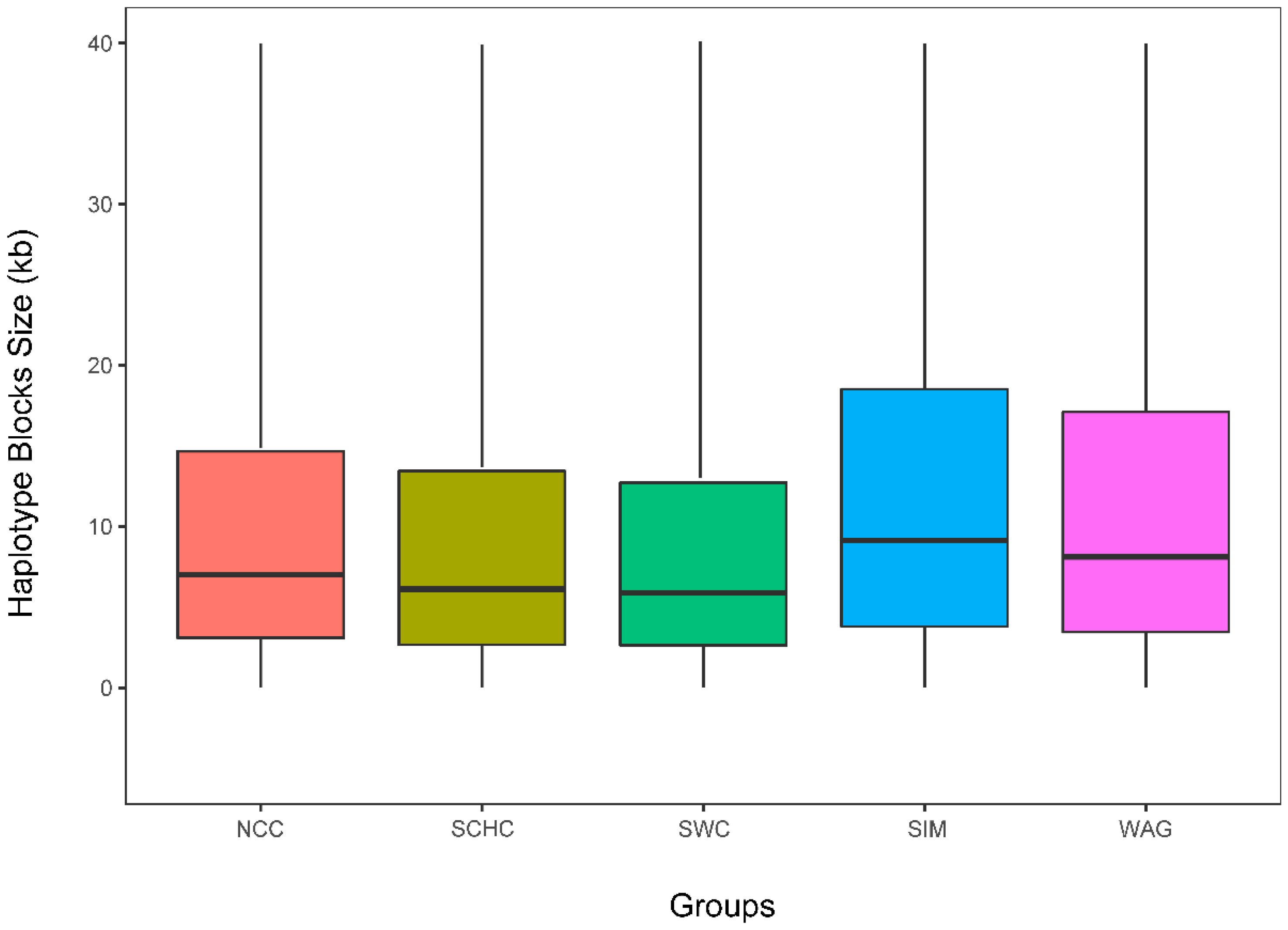
3.4. Haplotype Block Structure
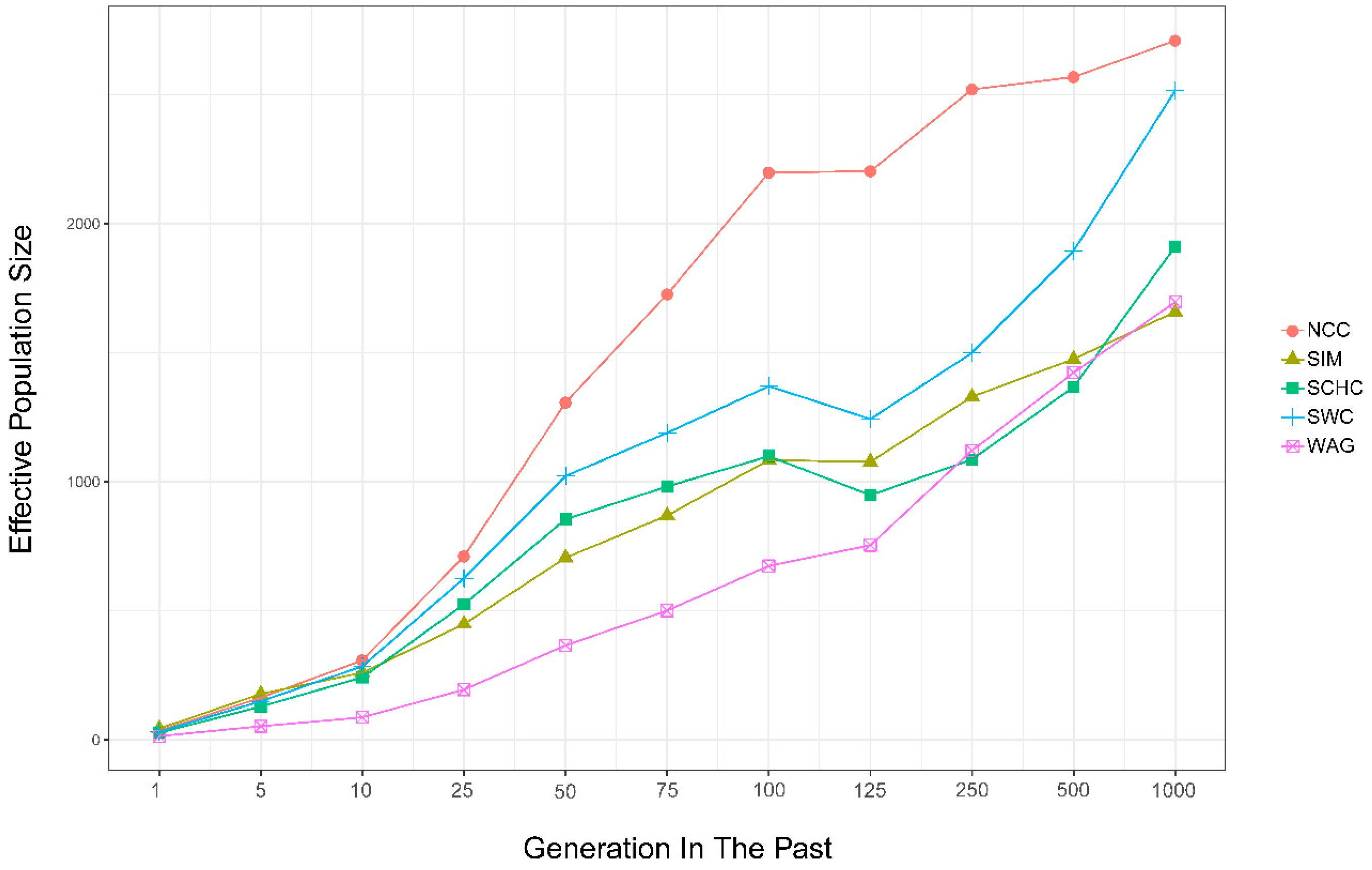
3.5. Effective Population Size
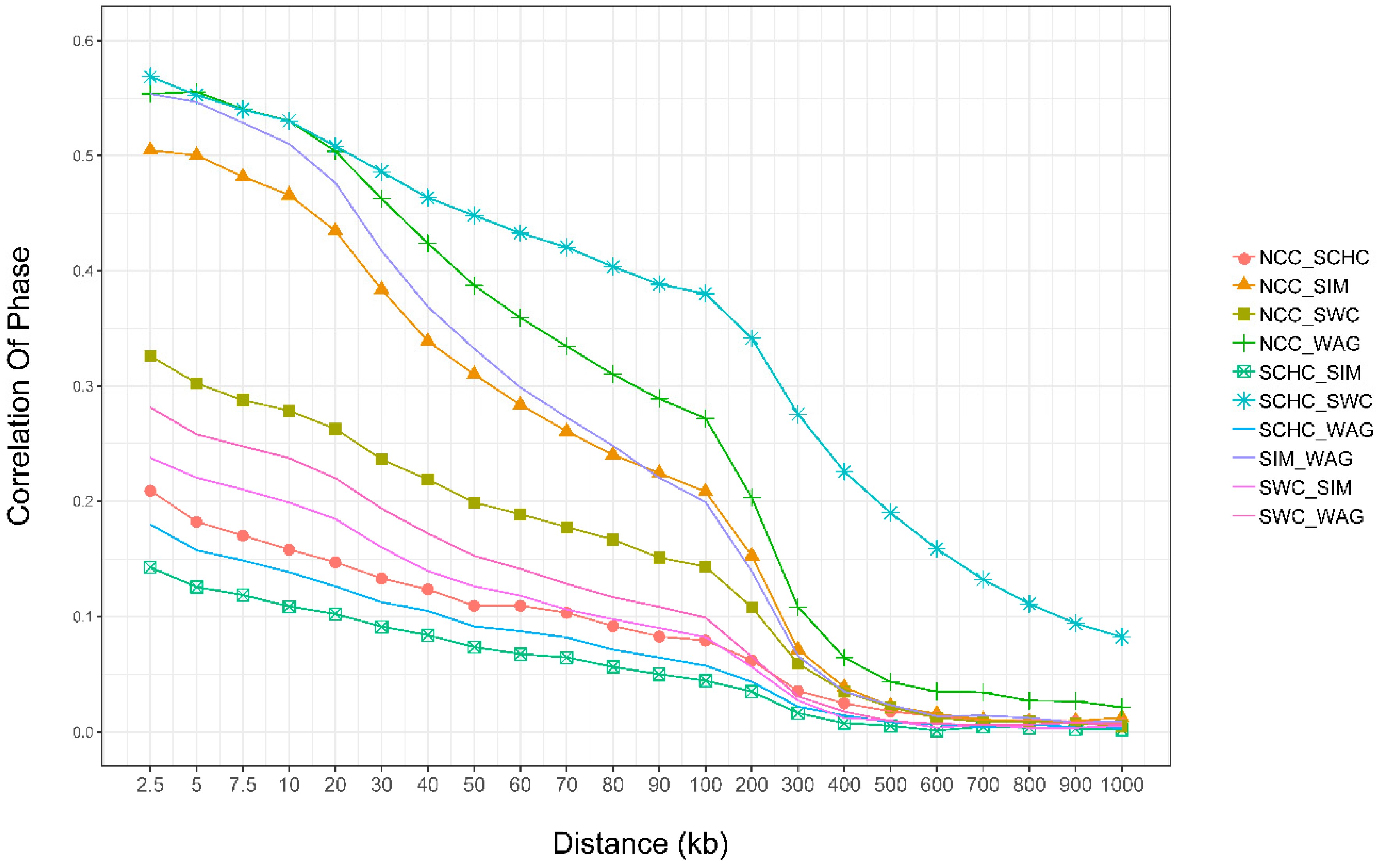
3.6. Persistence of Phase
4. Discussion
4.1. Animals and Grouping of Chinese Indigenous Cattle
4.2. Minor Allele Frequencies (MAF) and Linkage Disequilibrium (LD)
4.3. Haplotype Blocks
4.4. Effective Population Size
4.5. Persistence of Phase
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Jasielczuk, I.; Gurgul, A.; Szmatoła, T.; Ząbek, T.; Pawlina, K.; Semik, E.; Bugno-Poniewierska, M. Linkage disequilibrium and haplotype block structure in Limousin, Simmental and native Polish Red cattle. Livest. Sci. 2016, 191, 57–63. [Google Scholar] [CrossRef]
- Espigolan, R.; Baldi, F.; Boligon, A.A.; Souza, F.R.P.; Gordo, D.G.M.; Tonussi, R.L.; Cardoso, D.F.D.F.; Oliveira, H.N.; Tonhati, H.; Sargolzaei, M.; et al. Study of whole genome linkage disequilibrium in Nellore cattle. BMC Genom. 2013, 14, 305. [Google Scholar] [CrossRef] [PubMed]
- Gurgul, A.; Semik, E.; Pawlina, K.; Szmatoła, T.; Jasielczuk, I.; Bugno-Poniewierska, M. The application of genome-wide SNP genotyping methods in studies on livestock genomes. J. Appl. Genet. 2014, 55, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Pérez O’Brien, A.M.; Mészáros, G.; Utsunomiya, Y.T.; Sonstegard, T.S.; Garcia, J.F.; Van Tassell, C.P.; Carvalheiro, R.; da Silva, M.V.B.; Sölkner, J. Linkage disequilibrium levels in Bos indicus and Bos taurus cattle using medium and high density SNP chip data and different minor allele frequency distributions. Livest. Sci. 2014, 166, 121–132. [Google Scholar] [CrossRef]
- Bohmanova, J.; Sargolzaei, M.; Schenkel, F.S. Characteristics of linkage disequilibrium in North American Holsteins. BMC Genom. 2010, 11, 421. [Google Scholar] [CrossRef] [PubMed]
- Flury, C.; Tapio, M.; Sonstegard, T.; Drögemüller, C.; Leeb, T.; Simianer, H.; Hanotte, O.; Rieder, S. Effective population size of an indigenous Swiss cattle breed estimated from linkage disequilibrium. J. Anim. Breed. Genet. 2010, 127, 339–347. [Google Scholar] [CrossRef] [PubMed]
- McKay, S.D.; Schnabel, R.D.; Murdoch, B.M.; Matukumalli, L.K.; Aerts, J.; Coppieters, W.; Crews, D.; Neto, E.; Gill, C.A.; Gao, C.; et al. Whole genome linkage disequilibrium maps in cattle. BMC Genet. 2007, 8, 74. [Google Scholar] [CrossRef] [PubMed]
- Hayes, B.J.; Bowman, P.J.; Chamberlain, A.J.; Goddard, M.E. Invited review: Genomic selection in dairy cattle: Progress and challenges. J. Dairy Sci. 2009, 92, 433–443. [Google Scholar] [CrossRef] [PubMed]
- De Roos, A.P.W.W.; Hayes, B.J.; Spelman, R.J.; Goddard, M.E. Linkage disequilibrium and persistence of phase in Holstein-Friesian, Jersey and Angus cattle. Genetics 2008, 179, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Makina, S.O.; Taylor, J.F.; Van Marle-Köster, E.; Muchadeyi, F.C.; Makgahlela, M.L.; MacNeil, M.D.; Maiwashe, A. Extent of Linkage Disequilibrium and Effective Population Size in Four South African Sanga Cattle Breeds. Front. Genet. 2015, 6, 337. [Google Scholar] [CrossRef] [PubMed]
- Grossi, D.A.; Jafarikia, M.; Brito, L.F.; Buzanskas, M.E.; Sargolzaei, M.; Schenkel, F.S. Genetic diversity, extent of linkage disequilibrium and persistence of gametic phase in Canadian pigs. BMC Genet. 2017, 18, 6. [Google Scholar] [CrossRef] [PubMed]
- National Bureau of Statistics of China. China Statistical Yearbook; National Bureau of Statistics of China: Beijing, China, 2017; ISBN 0106279181.
- Cañas-Álvarez, J.J.; Mouresan, E.F.; Varona, L.; Díaz, C.; Molina, A.; Baro, J.A.; Altarriba, J.; Carabaño, M.J.; Casellas, J.; Piedrafita, J. Linkage disequilibrium, persistence of phase, and effective population size in Spanish local beef cattle breeds assessed through a high-density single nucleotide polymorphism chip. J. Anim. Sci. 2016, 94, 2779–2788. [Google Scholar] [CrossRef] [PubMed]
- Biegelmeyer, P.; Gulias-Gomes, C.C.; Caetano, A.R.; Steibel, J.P.; Cardoso, F.F. Linkage disequilibrium, persistence of phase and effective population size estimates in Hereford and Braford cattle. BMC Genet. 2016, 17, 32. [Google Scholar] [CrossRef] [PubMed]
- Karimi, K.; Esmailizadeh Koshkoiyeh, A.; Gondro, C. Comparison of linkage disequilibrium levels in Iranian indigenous cattle using whole genome SNPs data. J. Anim. Sci. Technol. 2015, 57, 47. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Cho, Y.M.; Lim, D.; Kim, H.C.; Choi, B.H.; Park, H.S.; Kim, O.H.; Kim, S.; Kim, T.H.; Yoon, D.; et al. Linkage disequilibrium and effective population size in Hanwoo Korean cattle. Asian-Australas. J. Anim. Sci. 2011, 34, 1660–1665. [Google Scholar] [CrossRef]
- Khatkar, M.S.; Nicholas, F.W.; Collins, A.R.; Zenger, K.R.; Cavanagh, J.A.L.L.; Barris, W.; Schnabel, R.D.; Taylor, J.F.; Raadsma, H.W. Extent of genome-wide linkage disequilibrium in Australian Holstein-Friesian cattle based on a high-density SNP panel. BMC Genom. 2008, 9, 187. [Google Scholar] [CrossRef] [PubMed]
- Hartigan, J.A.; Wong, M.A. Algorithm AS 136: A K-Means Clustering Algorithm. Appl. Stat. 1979, 28, 100–108. [Google Scholar] [CrossRef]
- Jombart, T.; Ahmed, I. adegenet 1.3-1: New tools for the analysis of genome-wide SNP data. Bioinformatics 2011, 27, 3070–3071. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Badke, Y.M.; Bates, R.O.; Ernst, C.W.; Schwab, C.; Steibel, J.P. Estimation of linkage disequilibrium in four US pig breeds. BMC Genom. 2012, 13, 24. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, S.B.; Schaffner, S.F.; Nguyen, H.; Moore, J.M.; Roy, J.; Blumenstiel, B.; Higgins, J.; DeFelice, M.; Lochner, A.; Faggart, M.; et al. The structure of haplotype blocks in the human genome. Science 2002, 296, 2225–2229. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.H.S.; Niu, T.H.; Liu, J.S.S. Partition-ligation-expectation-maximization algorithm for haplotype inference with single-nucleotide polymorphisms. Am. J. Hum. Genet. 2002, 71, 1242–1247. [Google Scholar] [CrossRef] [PubMed]
- Tenesa, A.; Navarro, P.; Hayes, B.J.; Duffy, D.L.; Clarke, G.M.; Goddard, M.E.; Visscher, P.M. Recent human effective population size estimated from linkage disequilibrium. Genome Res. 2007, 17, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Gautier, M.; Faraut, T.; Moazami-Goudarzi, K.; Navratil, V.; Foglio, M.; Grohs, C.; Boland, A.; Garnier, J.G.; Boichard, D.; Lathrop, G.M.; et al. Genetic and haplotypic structure in 14 European and African cattle breeds. Genetics 2007, 177, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
- Sved, J.A. Linkage disequilibrium and homozygosity of chromosome segments in finite populations. Theor. Popul. Biol. 1971, 2, 125–141. [Google Scholar] [CrossRef]
- R Core Team R Development Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2017; Volume Voulme 55, pp. 275–286. [Google Scholar]
- Porto-Neto, L.R.; Kijas, J.W.; Reverter, A. The extent of linkage disequilibrium in beef cattle breeds using high-density SNP genotypes. Genet. Sel. Evol. 2014, 46, 22. [Google Scholar] [CrossRef] [PubMed]
- Saatchi, M.; McClure, M.C.; McKay, S.D.; Rolf, M.M.; Kim, J.; Decker, J.E.; Taxis, T.M.; Chapple, R.H.; Ramey, H.R.; Northcutt, S.L.; et al. Accuracies of genomic breeding values in American Angus beef cattle using K-means clustering for cross-validation. Genet. Sel. Evol. 2011, 43, 40. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Cai, Y.; Chen, Q.; Li, R.; Wang, K.; Huang, Y.; Hu, S.; Huang, S.; Zhang, H.; Zheng, Z.; et al. Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia. Nat. Commun. 2018, 9, 2337. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Gautier, M.; Ding, X.; Zhang, H.; Wang, Y.; Wang, X.; Omar Faruque, M.D.; Li, J.; Ye, S.; Gou, X.; et al. Species composition and environmental adaptation of indigenous Chinese cattle. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xu, L.; Zhu, B.; Niu, H.; Zhang, W.; Miao, J.; Shi, X.; Zhang, M.; Chen, Y.; Zhang, L.; et al. Genome-wide analysis reveals differential selection involved with copy number variation in diverse Chinese Cattle. Sci. Rep. 2017, 7, 14299. [Google Scholar] [CrossRef] [PubMed]
- Pryce, J.E.E.; Gredler, B.; Bolormaa, S.; Bowman, P.J.J.; Egger-Danner, C.; Fuerst, C.; Emmerling, R.; Sölkner, J.; Goddard, M.E.E.; Hayes, B.J.J. Short communication: Genomic selection using a multi-breed, across-country reference population. J. Dairy Sci. 2011, 94, 2625–2630. [Google Scholar] [CrossRef] [PubMed]
- Qwabe, S.O.; vanMarle-Köster, E.; Maiwashe, A.; Muchadeyi, F.C. Evaluation of the bovineSNP50 genotyping array in four south african cattle populations. S. Afr. J. Anim. Sci. 2013, 43, 64–67. [Google Scholar]
- Sudrajad, P.; Seo, D.W.; Choi, T.J.; Park, B.H.; Roh, S.H.; Jung, W.Y.; Lee, S.S.; Lee, J.H.; Kim, S.; Lee, S.H. Genome-wide linkage disequilibrium and past effective population size in three Korean cattle breeds. Anim. Genet. 2017, 48, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Sargolzaei, M.; Kelly, M.; Li, C.; Voort, G.V.; Wang, Z.; Plastow, G.; Moore, S.; Miller, S.P. Linkage disequilibrium in Angus, Charolais, and Crossbred beef cattle. Front. Genet. 2012, 3, 152. [Google Scholar] [CrossRef] [PubMed]
- Edea, Z.; Dadi, H.; Kim, S.W.; Park, J.H.; Shin, G.H.; Dessie, T.; Kim, K.S. Linkage disequilibrium and genomic scan to detect selective loci in cattle populations adapted to different ecological conditions in Ethiopia. J. Anim. Breed. Genet. 2014, 131. [Google Scholar] [CrossRef] [PubMed]
- Decker, J.E.; Vasco, D.A.; McKay, S.D.; McClure, M.C.; Rolf, M.M.; Kim, J.W.; Northcutt, S.L.; Bauck, S.; Woodward, B.W.; Schnabel, R.D.; et al. A novel analytical method, Birth Date Selection Mapping, detects response of the Angus (Bos taurus) genome to selection on complex traits. BMC Genom. 2012, 13, 606. [Google Scholar] [CrossRef] [PubMed]
- Sargolzaei, M.; Schenkel, F.S.; Jansen, G.B.; Schaeffer, L.R. Extent of Linkage Disequilibrium in Holstein Cattle in North America. J. Dairy Sci. 2008, 91, 2106–2117. [Google Scholar] [CrossRef] [PubMed]
- McClure, M.C.; Morsci, N.S.; Schnabel, R.D.; Kim, J.W.; Yao, P.; Rolf, M.M.; McKay, S.D.; Gregg, S.J.; Chapple, R.H.; Northcutt, S.L.; et al. A genome scan for quantitative trait loci influencing carcass, post-natal growth and reproductive traits in commercial Angus cattle. Anim. Genet. 2010, 41, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Lindholm-Perry, A.K.; Sexten, A.K.; Kuehn, L.A.; Smith, T.P.L.; King, D.A.; Shackelford, S.D.; Wheeler, T.L.; Ferrell, C.L.; Jenkins, T.G.; Snelling, W.M.; et al. Association, effects and validation of polymorphisms within the NCAPG—LCORL locus located on BTA6 with feed intake, gain, meat and carcass traits in beef cattle. BMC Genet. 2011. [Google Scholar] [CrossRef] [PubMed]
- Snelling, W.M.; Allan, M.F.; Keele, J.W.; Kuehn, L.A.; Thallman, R.M.; Bennett, G.L.; Ferrell, C.L.; Jenkins, T.G.; Freetly, H.C.; Nielsen, M.K.; et al. Partial-genome evaluation of postweaning feed intake and efficiency of crossbred beef cattle. J. Anim. Sci. 2011, 89, 1731–1741. [Google Scholar] [CrossRef] [PubMed]
- Ardlie, K.G.; Kruglyak, L.; Seielstad, M. Patterns of linkage disequilibrium in the human genome. Nat. Rev. Genet. 2002, 3, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Hayes, B.J.; Lien, S.; Nilsen, H.; Olsen, H.G.; Berg, P.; Maceachern, S.; Potter, S.; Meuwissen, T.H.E. The origin of selection signatures on bovine chromosome 6. Anim. Genet. 2008, 39, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [PubMed]
- Villa-Angulo, R.; Matukumalli, L.K.; Gill, C.A.; Choi, J.; Van Tassell, C.P.; Grefenstette, J.J. High-resolution haplotype block structure in the cattle genome. BMC Genet. 2009, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Mokry, F.B.; Buzanskas, M.E.; de Alvarenga Mudadu, M.; do Amaral Grossi, D.; Higa, R.H.; Ventura, R.V.; de Lima, A.O.; Sargolzaei, M.; Meirelles, S.L.C.; Schenkel, F.S.; et al. Linkage disequilibrium and haplotype block structure in a composite beef cattle breed. BMC Genom. 2014, 15, S6. [Google Scholar] [CrossRef] [PubMed]
- Caballero, A.; Toro, M.A.; López-Fanjul, C. The response to artificial selection from new mutations in Drosophila melanogaster. Genetics 1991, 128, 89–102. [Google Scholar] [PubMed]
- Wang, J. Estimation of effective population sizes from data on genetic markers. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 1395–1409. [Google Scholar] [CrossRef] [PubMed]
- Hill, W.G. Estimation of effective population size from data on linkage disequilibrium. Genet. Res. 1981, 38, 209–216. [Google Scholar] [CrossRef]
- Nomura, T.; Honda, T.; Mukai, F. Inbreeding and effective population size of Japanese Black cattle. J. Anim. Sci. 2001, 79, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Uimari, P.; Tapio, M. Extent of linkage disequilibrium and effective population size in finnish landrace and finnish yorkshire pig breeds. J. Anim. Sci. 2011, 89, 609–614. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | NCC | SCHC | SWC | SIM | WAG |
|---|---|---|---|---|---|
| Blocks (n) | 88,829 | 61,241 | 86,957 | 72,287 | 68,627 |
| Total block length a (Mb) | 1237.49 | 933.96 | 1111.53 | 1597.17 | 1358.55 |
| Mean block length (kb) | 13.93 | 15.25 | 12.78 | 22.09 | 19.8 |
| SNP% in blocks | 74.55% | 57.88% | 63.98% | 86.67% | 80.68% |
| Coverage rate of autosomes | 47.59% | 35.92% | 42.75% | 61.43% | 52.25% |
| Mean num of SNPs in blocks | 5.29 | 4.45 | 4.64 | 7.16 | 6.4 |
| Max num of SNPs in blocks | 145 | 122 | 99 | 225 | 162 |
| Number of Generations Ago | NCC | SCHC | SWC | SIM | WAG | Average |
|---|---|---|---|---|---|---|
| 1 | 34 | 27 | 31 | 43 | 14 | 30 |
| 5 | 163 | 129 | 149 | 178 | 52 | 134 |
| 10 | 308 | 241 | 284 | 260 | 87 | 236 |
| 25 | 710 | 525 | 625 | 448 | 194 | 500 |
| 50 | 1307 | 854 | 1022 | 705 | 366 | 851 |
| 75 | 1726 | 981 | 1189 | 869 | 500 | 1053 |
| 100 | 2198 | 1099 | 1370 | 1083 | 673 | 1285 |
| 125 | 2204 | 948 | 1243 | 1077 | 753 | 1245 |
| 250 | 2521 | 1086 | 1499 | 1329 | 1121 | 1511 |
| 500 | 2569 | 1367 | 1895 | 1476 | 1422 | 1746 |
| 1000 | 2710 | 1912 | 2517 | 1658 | 1696 | 2099 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, L.; Zhu, B.; Wang, Z.; Xu, L.; Liu, Y.; Chen, Y.; Zhang, L.; Gao, X.; Gao, H.; Zhang, S.; et al. Evaluation of Linkage Disequilibrium, Effective Population Size and Haplotype Block Structure in Chinese Cattle. Animals 2019, 9, 83. https://doi.org/10.3390/ani9030083
Xu L, Zhu B, Wang Z, Xu L, Liu Y, Chen Y, Zhang L, Gao X, Gao H, Zhang S, et al. Evaluation of Linkage Disequilibrium, Effective Population Size and Haplotype Block Structure in Chinese Cattle. Animals. 2019; 9(3):83. https://doi.org/10.3390/ani9030083
Chicago/Turabian StyleXu, Lei, Bo Zhu, Zezhao Wang, Ling Xu, Ying Liu, Yan Chen, Lupei Zhang, Xue Gao, Huijiang Gao, Shengli Zhang, and et al. 2019. "Evaluation of Linkage Disequilibrium, Effective Population Size and Haplotype Block Structure in Chinese Cattle" Animals 9, no. 3: 83. https://doi.org/10.3390/ani9030083
APA StyleXu, L., Zhu, B., Wang, Z., Xu, L., Liu, Y., Chen, Y., Zhang, L., Gao, X., Gao, H., Zhang, S., Xu, L., & Li, J. (2019). Evaluation of Linkage Disequilibrium, Effective Population Size and Haplotype Block Structure in Chinese Cattle. Animals, 9(3), 83. https://doi.org/10.3390/ani9030083